
Pro-Inflammatory Cytokines Trigger the Overexpression of Tumour-Related Splice Variant RAC1B in Polarized Colorectal Cells


Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. THP-1 Cell Differentiation
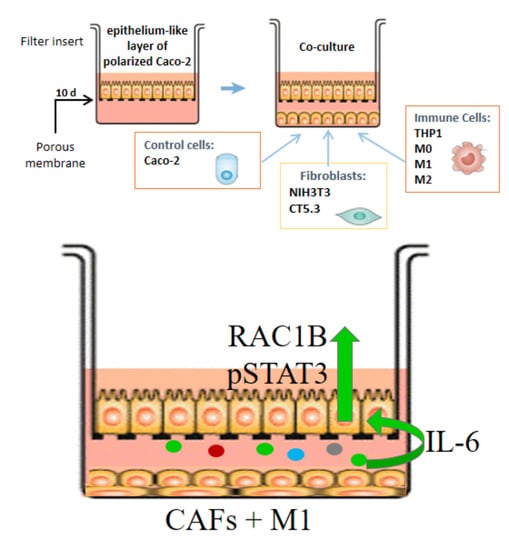
2.3. Cell Polarization and Co-Culture Assays
2.4. Cell Treatments
2.5. Qualitative and Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.6. Western Blot (WB) Procedures
2.7. Cytokine Array Procedure
2.8. Confocal Immunofluorescence Microscopy
2.9. Statistical Analysis
3. Results
3.1. Expression of RAC1B in Caco-2 Cells Is Modulated by Co-Cultured Stromal Cells
3.2. Cytokines from the Co-Culture Medium Trigger the Changes in RAC1B Protein Levels
3.3. Interleukin-6 Is a Key Factor Stimulating RAC1B Expression in Polarized Caco-2 Cells
3.4. Interleukin-6 also Stimulates RAC1B Expression in Polarized T84 Colorectal Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAF | Carcinoma-associated fibroblasts |
| ctrl | control |
| DAPI | 40,6-diamidine-20-phenylindole dihydrochloride |
| IBD | inflammatory bowel disease |
| IL | interleukin |
| qRT-PCR | quantitative reverse-transcription polymerase chain reaction |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| SDS-PAGE | sodium dodecyl sulphate-polyacrylamide gel electrophoresis |
| STAT3 | signal transducer and activator of transcription 3 |
| TEER | transepithelial electrical resistance |
| WB | Western blot |
References
- Zhang, K.; Hornef, M.W.; Dupont, A. The intestinal epithelium as guardian of gut barrier integrity: The epithelium as a barrier to infection. Cell. Microbiol. 2015, 17, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Velho, S.; Moutinho, C.; Cirnes, L.; Albuquerque, C.; Hamelin, R.; Schmitt, F.; Carneiro, F.; Oliveira, C.; Seruca, R. BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: Primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer 2008, 8, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Jordan, P. Colorectal Cancer Subtypes—The Current Portrait. In Targeted Therapy of Colorectal Cancer Subtypes; Jordan, P., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; Volome 1110, pp. 1–6. ISBN 978-3-030-02770-4. [Google Scholar]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Dyson, J.K.; Rutter, M.D. Colorectal cancer in inflammatory bowel disease: What is the real magnitude of the risk? World J. Gastroenterol. 2012, 18, 3839–3848. [Google Scholar] [CrossRef]
- Yalchin, M.; Baker, A.-M.; Graham, T.A.; Hart, A. Predicting Colorectal Cancer Occurrence in IBD. Cancers 2021, 13, 2908. [Google Scholar] [CrossRef]
- Clevers, H. At the crossroads of inflammation and cancer. Cell 2004, 118, 671–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.F.S.; Jordan, P.; Matos, P. A Signaling View into the Inflammatory Tumor Microenvironment. Immuno 2021, 1, 91–118. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Prenen, H.; Mazzone, M. Tumor-associated macrophages: A short compendium. Cell. Mol. Life Sci. 2019, 76, 1447–1458. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016, 17, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Prados, A.; Armaka, M.; Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 2020, 21, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Kotelevets, L.; Goncalves, V.; Henriques, A.; Zerbib, P.; Moyer, M.P.; Chastre, E.; Jordan, P. Ibuprofen inhibits colitis-induced overexpression of tumor-related Rac1b. Neoplasia 2013, 15, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Matos, P.; Oliveira, C.; Velho, S.; Gonçalves, V.; da Costa, L.T.; Moyer, M.P.; Seruca, R.; Jordan, P. B-Raf(V600E) cooperates with alternative spliced Rac1b to sustain colorectal cancer cell survival. Gastroenterology 2008, 135, 899–906. [Google Scholar] [CrossRef]
- Matos, P.; Gonçalves, V.; Jordan, P. Targeting the serrated pathway of colorectal cancer with mutation in BRAF. Biochim. Biophys. Acta Rev. Cancer 2016, 1866, 51–63. [Google Scholar] [CrossRef]
- Matos, P.; Collard, J.G.; Jordan, P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J. Biol. Chem. 2003, 278, 50442–50448. [Google Scholar] [CrossRef] [Green Version]
- Matos, P.; Jordan, P. Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp. Cell Res. 2005, 305, 292–299. [Google Scholar] [CrossRef]
- Matos, P.; Jordan, P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol. Cancer Res. 2008, 6, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Barros, P.; Moyer, M.P.; Matos, P.; Jordan, P. Expression of tumour-related Rac1b antagonizes B-Raf-induced senescence in colorectal cells. Cancer Lett. 2015, 369, 368–375. [Google Scholar] [CrossRef]
- De Boeck, A.; Hendrix, A.; Maynard, D.; Van Bockstal, M.; Daniëls, A.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics 2013, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Delie, F.; Rubas, W. A human colonic cell line sharing similarities with enterocytes as a model to examine oral absorption: Advantages and limitations of the Caco-2 model. Crit. Rev. Ther. Drug Carr. Syst. 1997, 14, 221–286. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Chen, Q.K.; Lui, C.; Cichon, M.A.; Radisky, D.C.; Nelson, C.M. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial–mesenchymal transition. MBoC 2012, 23, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Mehner, C.; Miller, E.; Khauv, D.; Nassar, A.; Oberg, A.L.; Bamlet, W.R.; Zhang, L.; Waldmann, J.; Radisky, E.S.; Crawford, H.C.; et al. Tumor Cell–Derived MMP3 Orchestrates Rac1b and Tissue Alterations That Promote Pancreatic Adenocarcinoma. Mol. Cancer Res. 2014, 12, 1430–1439. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T. Regulation of intestinal epithelial permeability by tight junctions. Cell. Mol. Life Sci. 2013, 70, 631–659. [Google Scholar] [CrossRef]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 103–111. ISBN 978-3-319-15791-7. [Google Scholar]
- Moyer, M.P.; Manzano, L.A.; Merriman, R.L.; Stauffer, J.S.; Tanzer, L.R. NCM460, a normal human colon mucosal epithelial cell line. In Vitro Cell. Dev. Biol. Anim. 1996, 32, 315–317. [Google Scholar] [CrossRef]
- Jordan, P.; Brazão, R.; Boavida, M.G.; Gespach, C.; Chastre, E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999, 18, 6835–6839. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Espinaco, V.; Cuatrecasas, M.; Alonso, V.; Escudero, P.; Marmol, M.; Horndler, C.; Ortego, J.; Gallego, R.; Codony-Servat, J.; Garcia-Albeniz, X.; et al. RAC1b overexpression correlates with poor prognosis in KRAS/BRAF WT metastatic colorectal cancer patients treated with first-line FOLFOX/XELOX chemotherapy. Eur. J. Cancer 2014, 50, 1973–1981. [Google Scholar] [CrossRef]
- Vitkus, S.J.D.; Hanifin, S.A.; McGee, D.W. Factors affecting Caco-2 intestinal epithelial cell interleukin-6 secretion. In Vitro Cell. Dev. Biol.-Anim. 1998, 34, 660–664. [Google Scholar] [CrossRef]
- Oh, K.; Lee, O.-Y.; Park, Y.; Seo, M.W.; Lee, D.-S. IL-1β induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer 2016, 16, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Gehlot, P. Inflammation and cancer: How friendly is the relationship for cancer patients? Curr. Opin. Pharmacol. 2009, 9, 351–369. [Google Scholar] [CrossRef] [Green Version]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Devriese, S.; Van den Bossche, L.; Van Welden, S.; Holvoet, T.; Pinheiro, I.; Hindryckx, P.; De Vos, M.; Laukens, D. T84 monolayers are superior to Caco-2 as a model system of colonocytes. Histochem. Cell Biol. 2017, 148, 85–93. [Google Scholar] [CrossRef]
- Liang, G.H.; Weber, C.R. Molecular aspects of tight junction barrier function. Curr. Opin. Pharmacol. 2014, 19, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Fath, K.R.; Mamajiwalla, S.N.; Burgess, D.R. The cytoskeleton in development of epithelial cell polarity. J. Cell Sci. 1993, 1993, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Massey-Harroche, D. Epithelial cell polarity as reflected in enterocytes. Microsc. Res. Tech. 2000, 49, 353–362. [Google Scholar] [CrossRef]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Rahman, S.; Ghiboub, M.; Donkers, J.M.; van de Steeg, E.; van Tol, E.A.F.; Hakvoort, T.B.M.; de Jonge, W.J. The Progress of Intestinal Epithelial Models from Cell Lines to Gut-On-Chip. Int. J. Mol. Sci. 2021, 22, 13472. [Google Scholar] [CrossRef]
- Navabi, N.; McGuckin, M.A.; Lindén, S.K. Gastrointestinal Cell Lines Form Polarized Epithelia with an Adherent Mucus Layer when Cultured in Semi-Wet Interfaces with Mechanical Stimulation. PLoS ONE 2013, 8, e68761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubashkin, M.G.; Ou, G.; Weaver, V.M. Deconstructing signaling in three dimensions. Biochemistry 2014, 53, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rios, E.; Durães, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [Green Version]
- Panja, A.; Goldberg, S.; Eckmann, L.; Krishen, P.; Mayer, L. The Regulation and Functional Consequence of Proinflammatory Cytokine Binding on Human Intestinal Epithelial Cells. J. Immunol. 1998, 161, 3675. [Google Scholar]
- Li, A.; Varney, M.L.; Singh, R.K. Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin. Cancer Res. 2001, 7, 3298–3304. [Google Scholar] [PubMed]
- Suzuki, T.; Yoshinaga, N.; Tanabe, S. Interleukin-6 (IL-6) Regulates Claudin-2 Expression and Tight Junction Permeability in Intestinal Epithelium. J. Biol. Chem. 2011, 286, 31263–31271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, C.; McLean, M.H.; Durum, S.K. Cytokine Tuning of Intestinal Epithelial Function. Front. Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.-Y.; Li, Y.-Y.; Hsieh, L.-L.; Lu, C.-H.; Chou, W.-C.; Liaw, C.-C.; Tang, R.-P.; Liao, S.-K. Analysis of the effect of serum interleukin-6 (IL-6) and soluble IL-6 receptor levels on survival of patients with colorectal cancer. Jpn. J. Clin. Oncol. 2010, 40, 580–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, T.; Shimada, E.; Urakawa, T. Serum levels of cytokines in patients with colorectal cancer: Possible involvement of interleukin-6 and interleukin-8 in hematogenous metastasis. J. Gastroenterol. 1994, 29, 423–429. [Google Scholar] [CrossRef]
- Kinoshita, T.; Ito, H.; Miki, C. Serum interleukin-6 level reflects the tumor proliferative activity in patients with colorectal carcinoma. Cancer 1999, 85, 2526–2531. [Google Scholar] [CrossRef]
- Chung, Y.-C.; Chang, Y.-F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef]
- Esfandi, F.; Mohammadzadeh Ghobadloo, S.; Basati, G. Interleukin-6 level in patients with colorectal cancer. Cancer Lett. 2006, 244, 76–78. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. Onco Targets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Atreya, R.; Neurath, M.F. Involvement of IL-6 in the Pathogenesis of Inflammatory Bowel Disease and Colon Cancer. Clin. Rev. Allergy Immunol. 2005, 28, 187–196. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.R.; Hoeflich, A.; Fischer, J.R.; Wolf, E.; Sordat, B.; Lahm, H. Interleukin-6 stimulates clonogenic growth of primary and metastatic human colon carcinoma cells. Cancer Lett. 2000, 151, 31–38. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Ye, D.; Boivin, M.; Guo, S.; Hashimi, M.; Ereifej, L.; Ma, T.Y. Interleukin-6 Modulation of Intestinal Epithelial Tight Junction Permeability Is Mediated by JNK Pathway Activation of Claudin-2 Gene. PLoS ONE 2014, 9, e85345. [Google Scholar] [CrossRef] [Green Version]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufman, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 Activation in Colon Cancer Is Associated with Enhanced Cell Proliferation and Tumor Growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Mahida, Y.R.; Wu, K.; Jewell, D.P. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut 1989, 30, 835–838. [Google Scholar] [CrossRef] [Green Version]
- Reimund, J.-M.; Wittersheim, C.; Dumont, S.; Muller, C.D.; Baumann, R.; Poindron, P.; Duclos, B. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J. Clin. Immunol. 1996, 16, 144–150. [Google Scholar] [CrossRef]
- McAlindon, M.E.; Hawkey, C.J.; Mahida, Y.R. Expression of interleukin 1β and interleukin 1β converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 1998, 42, 214–219. [Google Scholar] [CrossRef]
- Reinecker, H.-C.; Steffen, M.; Witthoeft, T.; Pflueger, I.; Schreiber, S.; MAcDERMOTT, R.P.; Raedler, A. Enhand secretion of tumour necrosis factor-alpha, IL-6, and IL-1β by isolated lamina ropria monouclear cells from patients with ulcretive cilitis and Crohn’s disease. Clin. Exp. Immunol. 2008, 94, 174–181. [Google Scholar] [CrossRef]
- Voronov, E.; Apte, R.N. IL-1 in Colon Inflammation, Colon Carcinogenesis and Invasiveness of Colon Cancer. Cancer Microenviron. 2015, 8, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef]
- Landström, M. The TAK1–TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Strickson, S.; Emmerich, C.H.; Goh, E.T.H.; Zhang, J.; Kelsall, I.R.; Macartney, T.; Hastie, C.J.; Knebel, A.; Peggie, M.; Marchesi, F.; et al. Roles of the TRAF6 and Pellino E3 ligases in MyD88 and RANKL signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E3481–E3489. [Google Scholar] [CrossRef] [Green Version]
- Visvikis, O.; Lorès, P.; Boyer, L.; Chardin, P.; Lemichez, E.; Gacon, G. Activated Rac1, but not the tumorigenic variant Rac1b, is ubiquitinated on Lys 147 through a JNK-regulated process: Ubiquitination of Rac1 is regulated by JNK. FEBS J. 2008, 275, 386–396. [Google Scholar] [CrossRef]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Majolée, J.; Podieh, F.; Hordijk, P.L.; Kovačević, I. The interplay of Rac1 activity, ubiquitination and GDI binding and its consequences for endothelial cell spreading. PLoS ONE 2021, 16, e0254386. [Google Scholar] [CrossRef]
- Gonçalves, V.; Matos, P.; Jordan, P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum. Mol. Genet. 2009, 18, 3696–3707. [Google Scholar] [CrossRef] [PubMed]
- Fiegen, D.; Haeusler, L.-C.; Blumenstein, L.; Herbrand, U.; Dvorsky, R.; Vetter, I.R.; Ahmadian, M.R. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J. Biol. Chem. 2004, 279, 4743–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, V.; Matos, P.; Jordan, P. The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA 2008, 14, 2538–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, V.; Henriques, A.; Pereira, J.; Neves Costa, A.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 2014, 20, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.W.; Valcárcel, J. Alternative pre-mRNA splicing: The logic of combinatorial control. Trends Biochem. Sci. 2000, 25, 381–388. [Google Scholar] [CrossRef]
- Shin, C.; Manley, J.L. Cell signalling and the control of pre-mRNA splicing. Nat. Rev. Mol. Cell Biol. 2004, 5, 727–738. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, J.F.S.; Bessa, C.; Matos, P.; Jordan, P. Pro-Inflammatory Cytokines Trigger the Overexpression of Tumour-Related Splice Variant RAC1B in Polarized Colorectal Cells. Cancers 2022, 14, 1393. https://doi.org/10.3390/cancers14061393
Pereira JFS, Bessa C, Matos P, Jordan P. Pro-Inflammatory Cytokines Trigger the Overexpression of Tumour-Related Splice Variant RAC1B in Polarized Colorectal Cells. Cancers. 2022; 14(6):1393. https://doi.org/10.3390/cancers14061393
Chicago/Turabian StylePereira, Joana F. S., Cláudia Bessa, Paulo Matos, and Peter Jordan. 2022. "Pro-Inflammatory Cytokines Trigger the Overexpression of Tumour-Related Splice Variant RAC1B in Polarized Colorectal Cells" Cancers 14, no. 6: 1393. https://doi.org/10.3390/cancers14061393