
The Occurrence of MET Ectodomain Shedding in Oral Cancer and Its Potential Impact on the Use of Targeted Therapies


 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction: Receptor Tyrosine Kinase MET, a Suitable Target for Therapy in Oral Cancer?
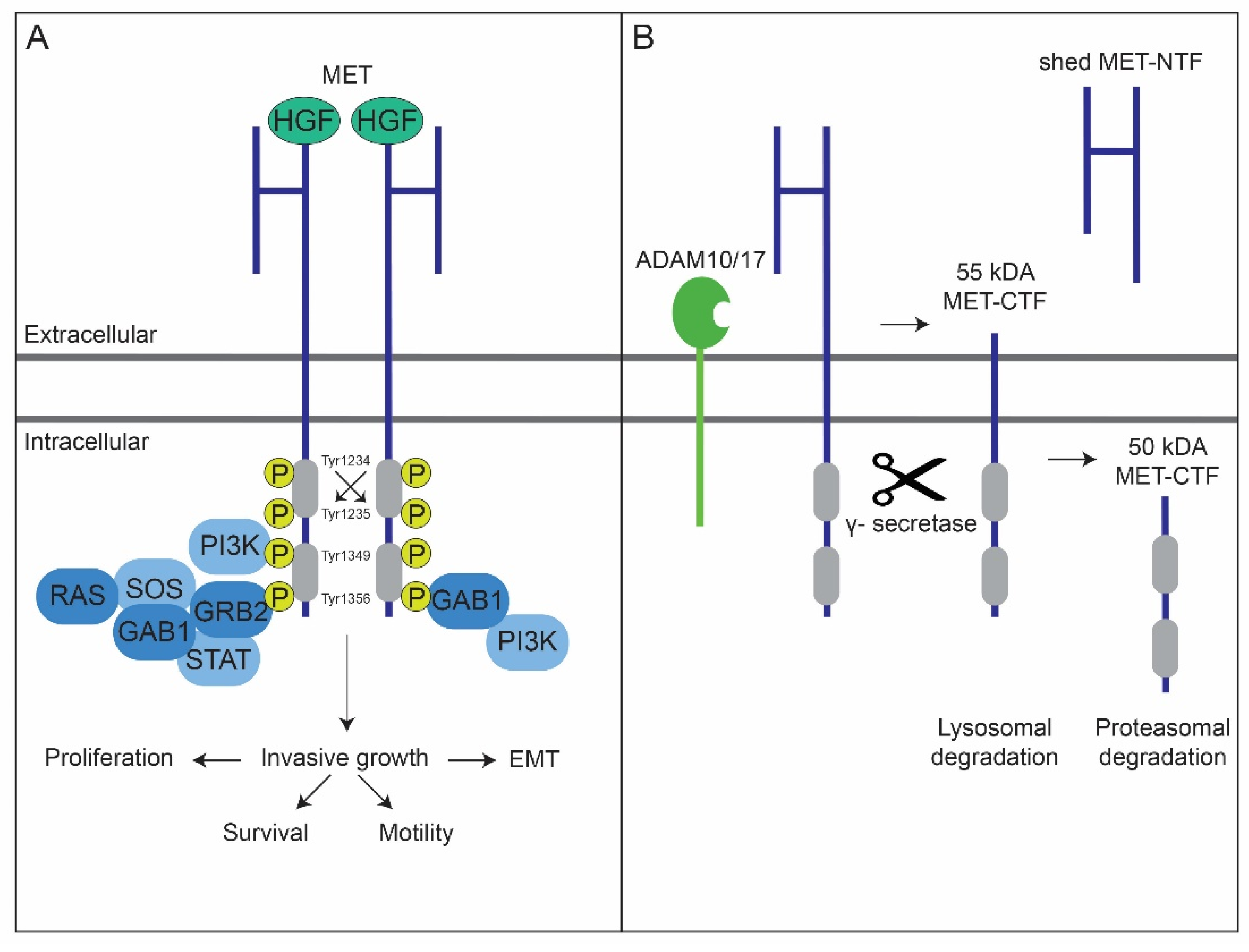
2. MET’s Protein Structure and Its Degradation
3. MET Ectodomain Shedding in the Tumor Suppressive and Oncogenic Context
4. Biochemical Evidence for MET Ectodomain Shedding in Oral Squamous Cell Carcinoma
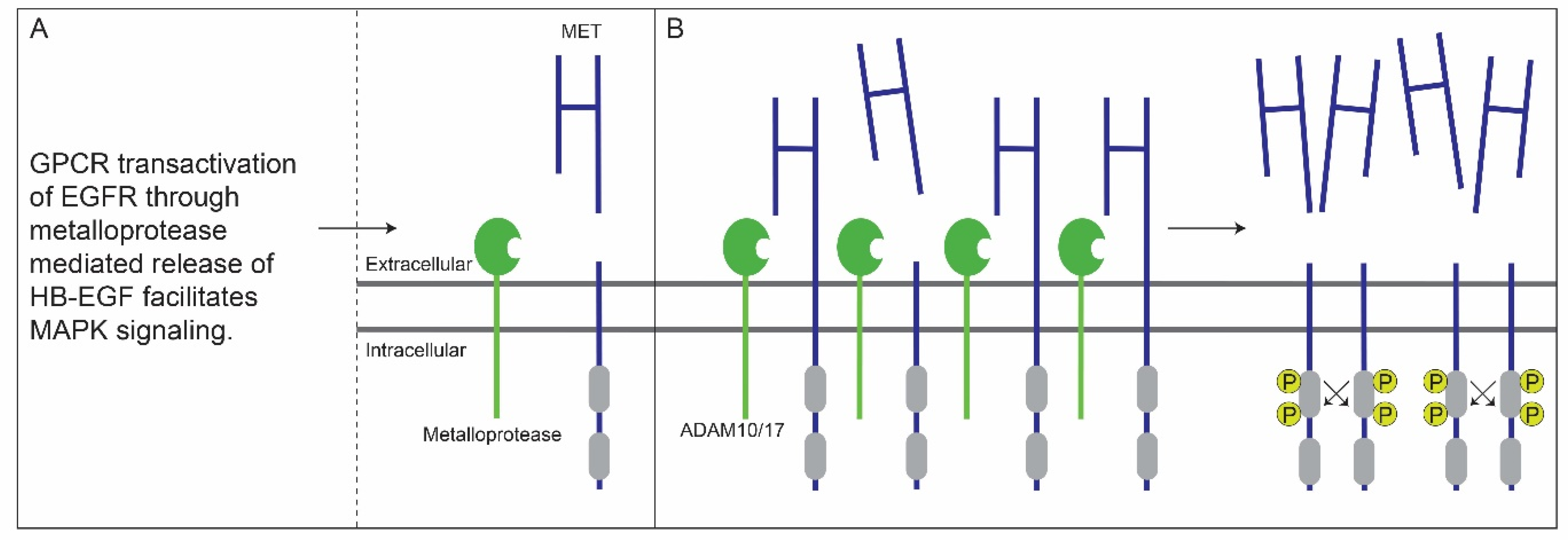
5. Crosstalk between G-Protein-Coupled and EGF Receptors Enhances MET Ectodomain Shedding
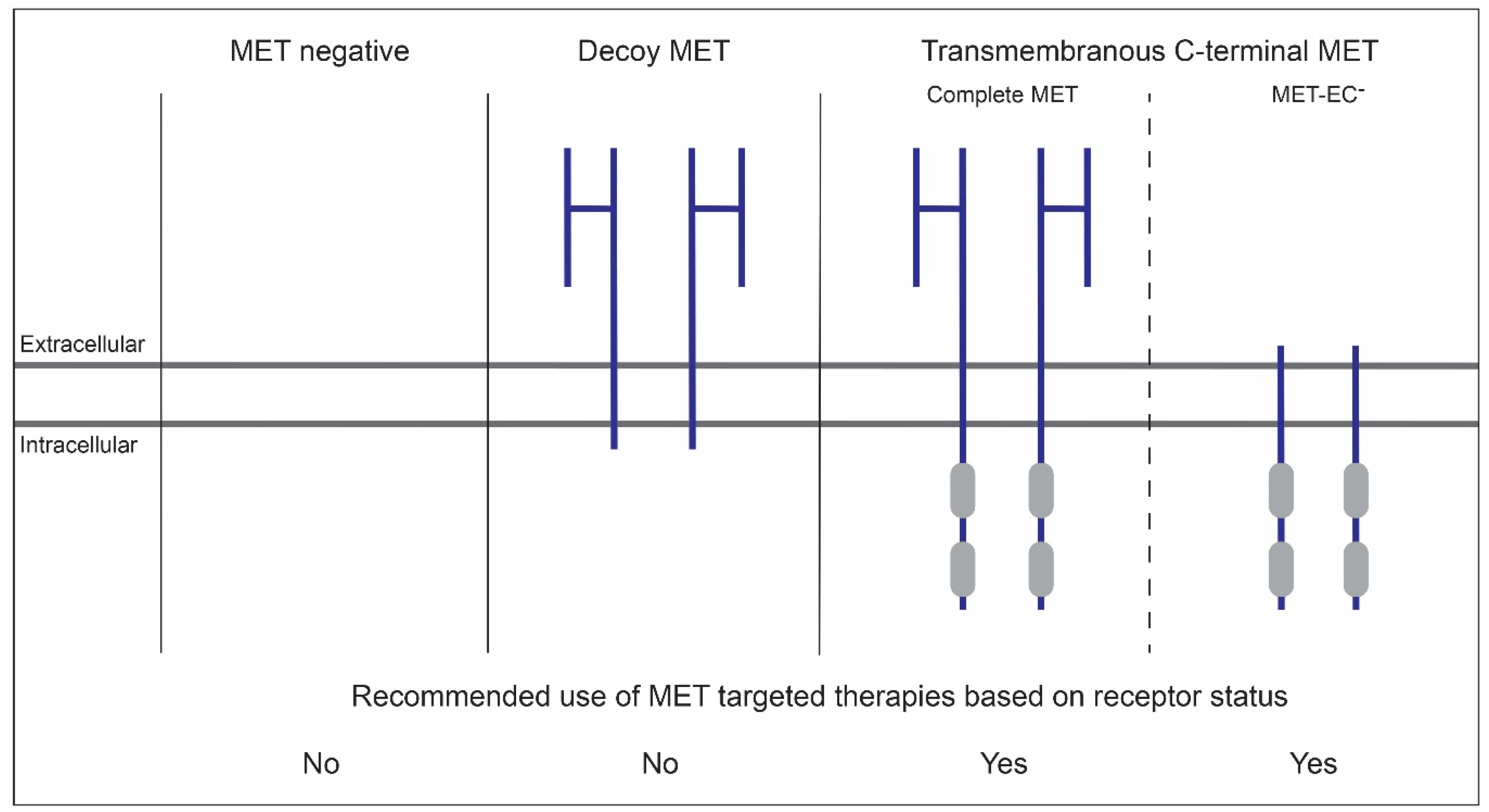
6. Membranous MET-EC- in OSCC
7. MET-EC- as Biomarker in OSCC
8. Discussion
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. 2021. Available online: https://gco.iarc.fr/today.2020 (accessed on 10 June 2021).
- SEER. Cancer Statistics Review (CSR) 1975—2018. 2021. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 10 June 2021).
- Hoesseini, A.; van Leeuwen, N.; Offerman, M.P.J.; Zhang, J.; Dronkers, E.A.C.; Sewnaik, A.; Lingsma, H.F.; Baatenburg de Jong, R.J. Predicting survival in head and neck cancer: External validation and update of the prognostic model OncologIQ in 2189 patients. Head Neck 2021, 43, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Ponzetto, C.; Di Renzo, M.F.; Cooper, C.S.; Comoglio, P.M. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 1989, 339, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Szturz, P.; Raymond, E.; Abitbol, C.; Albert, S.; de Gramont, A.; Faivre, S. Understanding c-MET signalling in squamous cell carcinoma of the head & neck. Crit. Rev. Oncol. Hematol. 2017, 111, 39–51. [Google Scholar] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991, 6, 501–504. [Google Scholar] [PubMed]
- Kong-Beltran, M.; Stamos, J.; Wickramasinghe, D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004, 6, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelter, F.; Kobuch, J.; Moss, M.L.; Becherer, J.D.; Comoglio, P.M.; Boccaccio, C.; Kruger, A. A disintegrin and metalloproteinase-10 (ADAM-10) mediates DN30 antibody-induced shedding of the met surface receptor. J. Biol. Chem. 2010, 285, 26335–26340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, M.; Duplaquet, L.; Tulasne, D. Proteolytic cleavages of MET: The divide-and-conquer strategy of a receptor tyrosine kinase. BMB Rep. 2019, 52, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Szturz, P.; Budikova, M.; Vermorken, J.B.; Horova, I.; Gal, B.; Raymond, E.; de Gramont, A.; Faivre, S. Prognostic value of c-MET in head and neck cancer: A systematic review and meta-analysis of aggregate data. Oral Oncol. 2017, 74, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 2017, 49, e307. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Ma, Z.; Pollan, S.; Yuan, X.; Swartwood, S.; Gertych, A.; Rodriguez, M.; Mallick, J.; Bhele, S.; Guindi, M.; et al. Quantitative imaging for development of companion diagnostics to drugs targeting HGF/MET. J. Pathol. Clin. Res. 2016, 2, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Administration of USFD. Companion Diagnostics. 2018. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/companion-diagnostics (accessed on 14 January 2022).
- Lefebvre, J.; Ancot, F.; Leroy, C.; Muharram, G.; Lemiere, A.; Tulasne, D. Met degradation: More than one stone to shoot a receptor down. FASEB J. 2012, 26, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Prat, M.; Crepaldi, T.; Gandino, L.; Giordano, S.; Longati, P.; Comoglio, P. C-terminal truncated forms of Met, the hepatocyte growth factor receptor. Mol. Cell Biol. 1991, 11, 5954–5962. [Google Scholar] [PubMed] [Green Version]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Park, M.; Le Beau, M.M.; Robins, T.S.; Diaz, M.O.; Rowley, J.D.; Blair, D.G.; Vande Woude, G.F. The human met oncogene is related to the tyrosine kinase oncogenes. Nature 1985, 318, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; Vande Woude, G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc. Natl. Acad. Sci. USA 1987, 84, 6379–6383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, S.; Di Renzo, M.F.; Narsimhan, R.P.; Cooper, C.S.; Rosa, C.; Comoglio, P.M. Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 1989, 4, 1383–1388. [Google Scholar] [PubMed]
- Giordano, S.; Di Renzo, M.F.; Ferracini, R.; Chiado-Piat, L.; Comoglio, P.M. p145, a protein with associated tyrosine kinase activity in a human gastric carcinoma cell line. Mol. Cell Biol. 1988, 8, 3510–3517. [Google Scholar]
- Tempest, P.R.; Cooper, C.S.; Major, G.N. The activated human met gene encodes a protein tyrosine kinase. FEBS Lett. 1986, 209, 357–361. [Google Scholar] [CrossRef] [Green Version]
- Hammond, D.E.; Urbe, S.; Vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef] [Green Version]
- Foveau, B.; Ancot, F.; Leroy, C.; Petrelli, A.; Reiss, K.; Vingtdeux, V.; Giordano, S.; Fafeur, V.; Tulasne, D. Down-regulation of the met receptor tyrosine kinase by presenilin-dependent regulated intramembrane proteolysis. Mol. Biol. Cell. 2009, 20, 2495–2507. [Google Scholar] [CrossRef] [Green Version]
- Landman, N.; Kim, T.W. Got RIP? Presenilin-dependent intramembrane proteolysis in growth factor receptor signaling. Cytokine Growth Factor Rev. 2004, 15, 337–351. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Ancot, F.; Leroy, C.; Muharram, G.; Lefebvre, J.; Vicogne, J.; Lemiere, A.; Kherrouche, Z.; Foveau, B.; Pourtier, A.; Melnyk, O.; et al. Shedding-generated Met receptor fragments can be routed to either the proteasomal or the lysosomal degradation pathway. Traffic 2012, 13, 1261–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangemi, R.; Amaro, A.; Gino, A.; Barisione, G.; Fabbi, M.; Pfeffer, U.; Brizzolara, A.; Queirolo, P.; Salvi, S.; Boccardo, S.; et al. ADAM10 correlates with uveal melanoma metastasis and promotes in vitro invasion. Pigment Cell Melanoma Res. 2014, 27, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Chalupsky, K.; Kanchev, I.; Zbodakova, O.; Buryova, H.; Jirouskova, M.; Korinek, V.; Gregor, M.; Sedlacek, R. ADAM10/17-dependent release of soluble c-Met correlates with hepatocellular damage. Folia Biol. 2013, 59, 76–86. [Google Scholar]
- Prat, M.; Crepaldi, T.; Pennacchietti, S.; Bussolino, F.; Comoglio, P.M. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J. Cell Sci. 1998, 111 Pt 2, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Athauda, G.; Giubellino, A.; Coleman, J.A.; Horak, C.; Steeg, P.S.; Lee, M.J.; Trepel, J.; Wimberly, J.; Sun, J.; Coxon, A.; et al. c-Met ectodomain shedding rate correlates with malignant potential. Clin. Cancer Res. 2006, 12, 4154–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crepaldi, T.; Prat, M.; Giordano, S.; Medico, E.; Comoglio, P.M. Generation of a truncated hepatocyte growth factor receptor in the endoplasmic reticulum. J. Biol. Chem. 1994, 269, 1750–1755. [Google Scholar] [CrossRef]
- De Herdt, M.J.; Koljenovic, S.; van der Steen, B.; Willems, S.M.; Noorlag, R.; Nieboer, D.; Hardillo, J.A.; Gruver, A.M.; Zeng, W.; Liu, L.; et al. MET ectodomain shedding is associated with poor disease-free survival of patients diagnosed with oral squamous cell carcinoma. Mod. Pathol. 2020, 33, 1015–1032. [Google Scholar] [CrossRef] [PubMed]
- Deheuninck, J.; Foveau, B.; Goormachtigh, G.; Leroy, C.; Ji, Z.; Tulasne, D.; Fafeur, V. Caspase cleavage of the MET receptor generates an HGF interfering fragment. Biochem. Biophys. Res. Commun. 2008, 367, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Dolled-Filhart, M.; Ocal, I.T.; Singh, B.; Lin, C.Y.; Dickson, R.B.; Rimm, D.L.; Camp, R.L. Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res. 2003, 63, 1101–1105. [Google Scholar] [PubMed]
- Merlin, S.; Pietronave, S.; Locarno, D.; Valente, G.; Follenzi, A.; Prat, M. Deletion of the ectodomain unleashes the transforming, invasive, and tumorigenic potential of the MET oncogene. Cancer Sci. 2009, 100, 633–638. [Google Scholar] [CrossRef] [PubMed]
- De Herdt, M.J.; Willems, S.M.; van der Steen, B.; Noorlag, R.; Verhoef, E.I.; van Leenders, G.J.; van Es, R.J.; Koljenovi, A.S.; Baatenburg de Jong, R.J.; Looijenga, L.H. Absent and abundant MET immunoreactivity is associated with poor prognosis of patients with oral and oropharyngeal squamous cell carcinoma. Oncotarget 2016, 7, 13167–13181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Herdt, M.J.; Koljenovic, S.; van der Steen, B.; Willems, S.M.; Wieringa, M.H.; Nieboer, D.; Hardillo, J.A.; Gruver, A.M.; Zeng, W.; Liu, L.; et al. A novel immunohistochemical scoring system reveals associations of C-terminal MET, ectodomain shedding, and loss of E-cadherin with poor prognosis in oral squamous cell carcinoma. Hum. Pathol. 2020, 104, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.; Williamson, N.J.; Jarvis, R.; Murphy, G. Shedding of c-Met is regulated by crosstalk between a G-protein coupled receptor and the EGF receptor and is mediated by a TIMP-3 sensitive metalloproteinase. J. Cell Sci. 2001, 114, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Thomas, S.M.; Zhang, Q.; Wentzel, A.L.; Siegfried, J.M.; Li, J.Y.; Grandis, J.R. Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene 2003, 22, 6183–6193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornfeld, J.W.; Meder, S.; Wohlberg, M.; Friedrich, R.E.; Rau, T.; Riethdorf, L.; Loning, T.; Pantel, K.; Riethdorf, S. Overexpression of TACE and TIMP3 mRNA in head and neck cancer: Association with tumour development and progression. Br. J. Cancer 2011, 104, 138–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Sha, X.Y.; Bai, Y.X.; Quan, F.; Wu, S.L. Effect of A disintegrin and metalloproteinase 10 gene silencing on the proliferation, invasion and migration of the human tongue squamous cell carcinoma cell line TCA8113. Mol. Med. Rep. 2015, 11, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, L.; Enders, J.; Thomas, S.M. Activated HGF-c-Met Axis in Head and Neck Cancer. Cancers 2017, 9, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, U.; Sharma, S.V.; Dowell, L.; Greninger, P.; Montagut, C.; Lamb, J.; Archibald, H.; Raudales, R.; Tam, A.; Lee, D.; et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. USA 2007, 104, 19936–19941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuti, S.; Gentile, A.; Lazzari, L.; Arnesano, A.; Trusolino, L.; Comoglio, P.M. An ‘in-cell trial’ to assess the efficacy of a monovalent anti-MET antibody as monotherapy and in association with standard cytotoxics. Mol. Oncol. 2014, 8, 378–388. [Google Scholar] [CrossRef]
- Michieli, P.; Mazzone, M.; Basilico, C.; Cavassa, S.; Sottile, A.; Naldini, L.; Comoglio, P.M. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell 2004, 6, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madoz-Gurpide, J.; Zazo, S.; Chamizo, C.; Casado, V.; Carames, C.; Gavin, E.; Cristobal, I.; Garcia-Foncillas, J.; Rojo, F. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J. Transl. Med. 2015, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Ilangumaran, S.; Villalobos-Hernandez, A.; Bobbala, D.; Ramanathan, S. The hepatocyte growth factor (HGF)-MET receptor tyrosine kinase signaling pathway: Diverse roles in modulating immune cell functions. Cytokine 2016, 82, 125–139. [Google Scholar] [CrossRef]
- Hubel, J.; Hieronymus, T. HGF/Met-Signaling Contributes to Immune Regulation by Modulating Tolerogenic and Motogenic Properties of Dendritic Cells. Biomedicines 2015, 3, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, G.E.; Hockings, H.; Hilton, D.M.; Kermorgant, S. The role of MET in chemotherapy resistance. Oncogene 2021, 40, 1927–1941. [Google Scholar] [CrossRef] [PubMed]
- Joffre, C.; Barrow, R.; Menard, L.; Calleja, V.; Hart, I.R.; Kermorgant, S. A direct role for Met endocytosis in tumorigenesis. Nat. Cell Biol. 2011, 13, 827–837. [Google Scholar] [CrossRef]
- Hughes, V.S.; Siemann, D.W. Have Clinical Trials Properly Assessed c-Met Inhibitors? Trends Cancer 2018, 4, 94–97. [Google Scholar] [CrossRef]
- Watermann, I.; Schmitt, B.; Stellmacher, F.; Muller, J.; Gaber, R.; Kugler, C.; Reinmuth, N.; Huber, R.M.; Thomas, M.; Zabel, P.; et al. Improved diagnostics targeting c-MET in non-small cell lung cancer: Expression, amplification and activation? Diagn. Pathol. 2015, 10, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copin, M.C.; Lesaffre, M.; Berbon, M.; Doublet, L.; Leroy, C.; Tresch, E.; Porte, H.; Vicogne, J.; Cortot, A.B.; Dansin, E.; et al. High-MET status in non-small cell lung tumors correlates with receptor phosphorylation but not with the serum level of soluble form. Lung Cancer 2016, 101, 59–67. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| MET N-Terminal Fragments (MET-NTFs) Observed in the Culture Medium of a Breast Cancer Progression Model [40] | MET C-Terminal Fragments (MET-CTFs) Observed in OSCC Cell Lines and Fresh Frozen Tissues [42] |
|---|---|
| p50β | p95β |
| p55β | p90β |
| p75β | p70β |
| p85β | p60β |
| * p100β | p55β |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Herdt, M.J.; van der Steen, B.; Baatenburg de Jong, R.J.; Looijenga, L.H.J.; Koljenović, S.; Hardillo, J.A. The Occurrence of MET Ectodomain Shedding in Oral Cancer and Its Potential Impact on the Use of Targeted Therapies. Cancers 2022, 14, 1491. https://doi.org/10.3390/cancers14061491
De Herdt MJ, van der Steen B, Baatenburg de Jong RJ, Looijenga LHJ, Koljenović S, Hardillo JA. The Occurrence of MET Ectodomain Shedding in Oral Cancer and Its Potential Impact on the Use of Targeted Therapies. Cancers. 2022; 14(6):1491. https://doi.org/10.3390/cancers14061491
Chicago/Turabian StyleDe Herdt, Maria J., Berdine van der Steen, Robert J. Baatenburg de Jong, Leendert H. J. Looijenga, Senada Koljenović, and Jose A. Hardillo. 2022. "The Occurrence of MET Ectodomain Shedding in Oral Cancer and Its Potential Impact on the Use of Targeted Therapies" Cancers 14, no. 6: 1491. https://doi.org/10.3390/cancers14061491
APA StyleDe Herdt, M. J., van der Steen, B., Baatenburg de Jong, R. J., Looijenga, L. H. J., Koljenović, S., & Hardillo, J. A. (2022). The Occurrence of MET Ectodomain Shedding in Oral Cancer and Its Potential Impact on the Use of Targeted Therapies. Cancers, 14(6), 1491. https://doi.org/10.3390/cancers14061491