
MitoQ Inhibits Human Breast Cancer Cell Migration, Invasion and Clonogenicity

 , , , , ,
, , , , ,  , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Dosage of MitoQ in Mouse Plasma
2.3. Cells and Cell Culture
2.4. Metabolic Assays
2.5. Mitochondrial Potential
2.6. Mitochondrial Superoxide
2.7. Cell Cycle
2.8. Apoptosis and Necrosis Assays
2.9. Immunocytochemistry
2.10. Cell Numbers
2.11. Electron Microscopy
2.12. Real-Time Quantitative PCR
2.13. Western Blotting
2.14. Cell Migration
2.15. Cell Invasion
2.16. Clonogenic Assays
2.17. Spheroids
2.18. Statistics
3. Results
3.1. Determination of a Biologically Relevant Dose Range of MitoQ
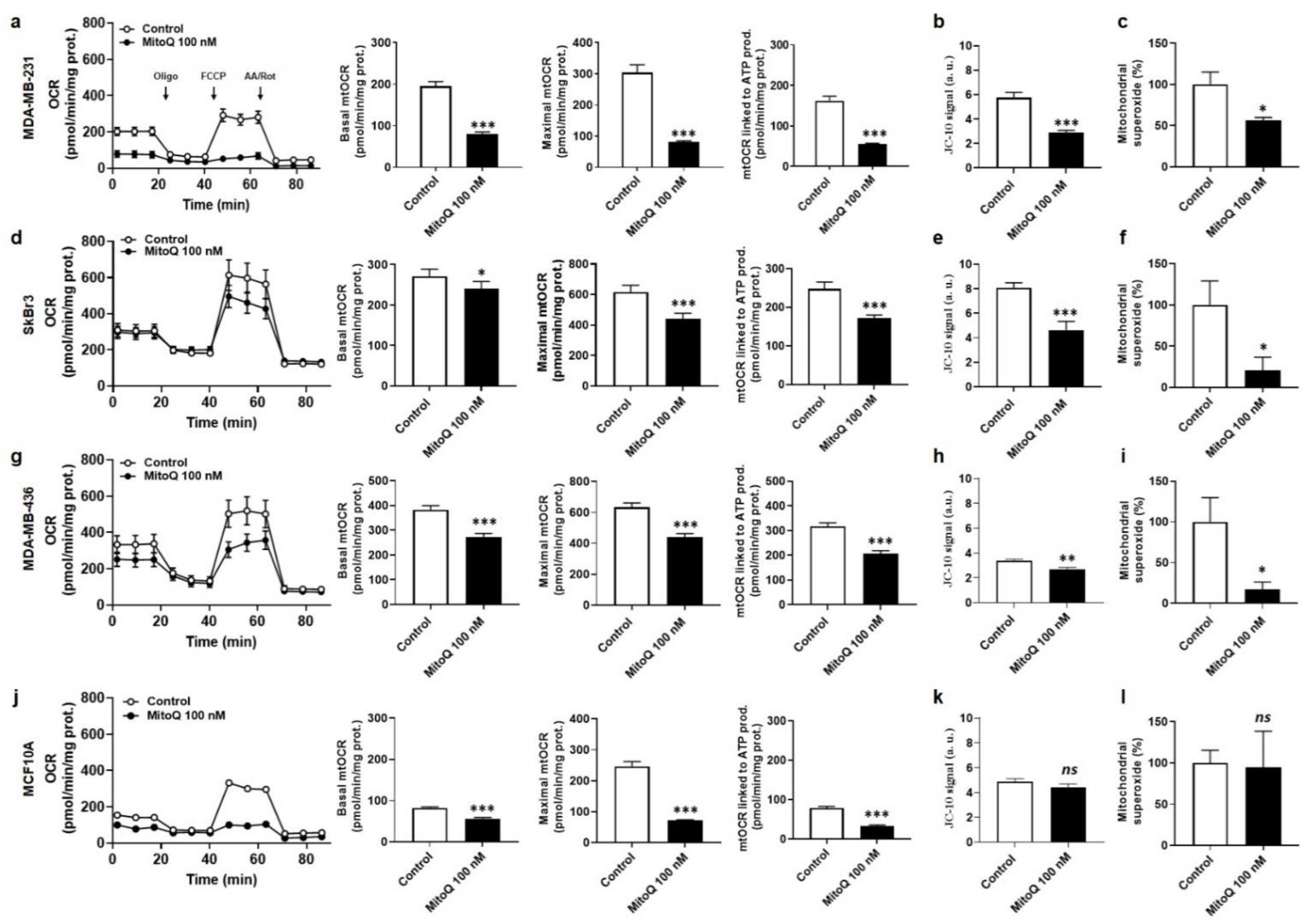
3.2. MitoQ Represses Oxidative Phosphorylation and Mitochondrial Superoxide Production by Metastatic Human Breast Cancer Cells
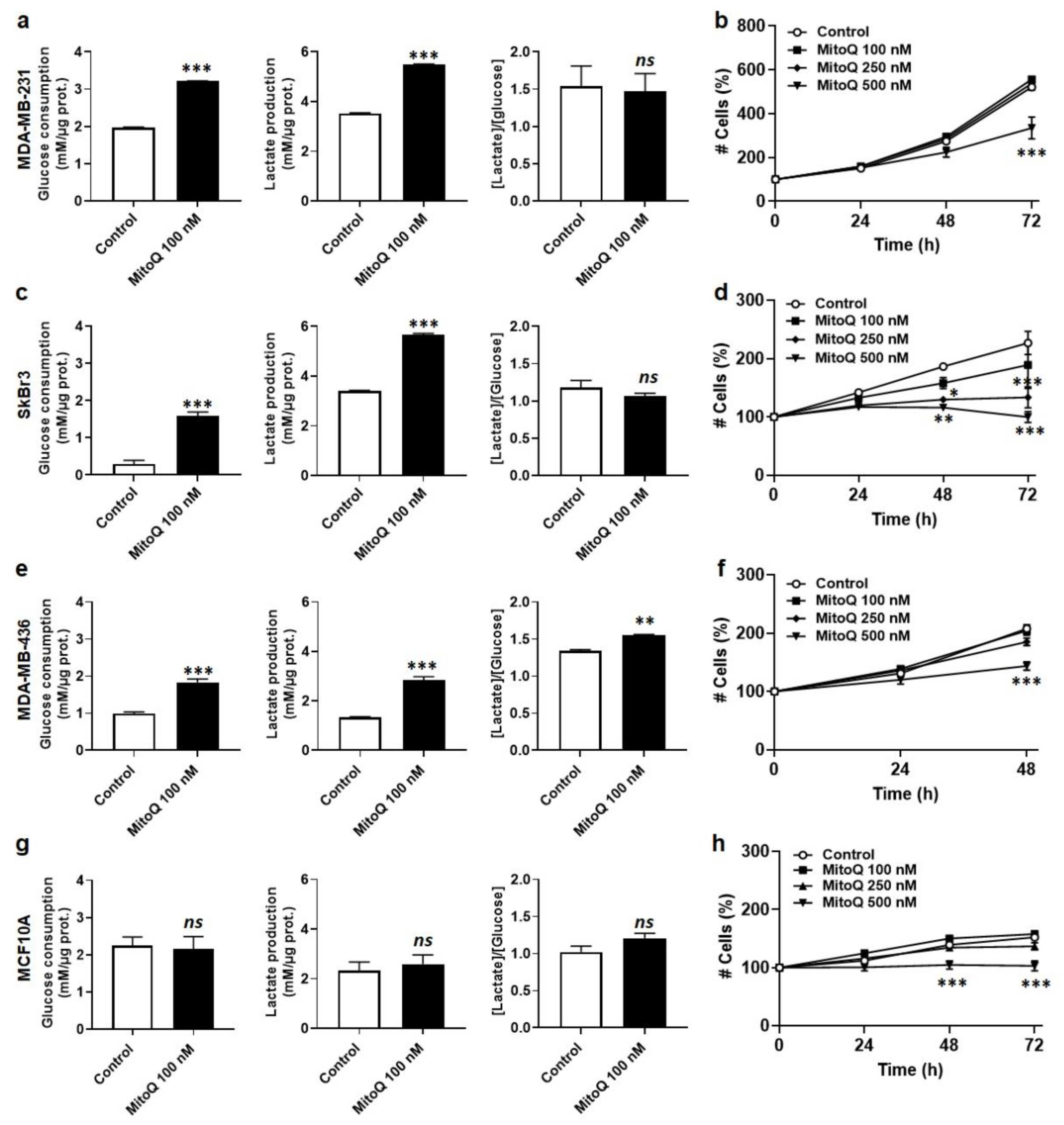
3.3. MitoQ Is Cytostatic for Human Breast Cancer Cells
3.4. MitoQ Partially Represses the Expression of Mesenchymal Marks by Human Breast Cancer Cells
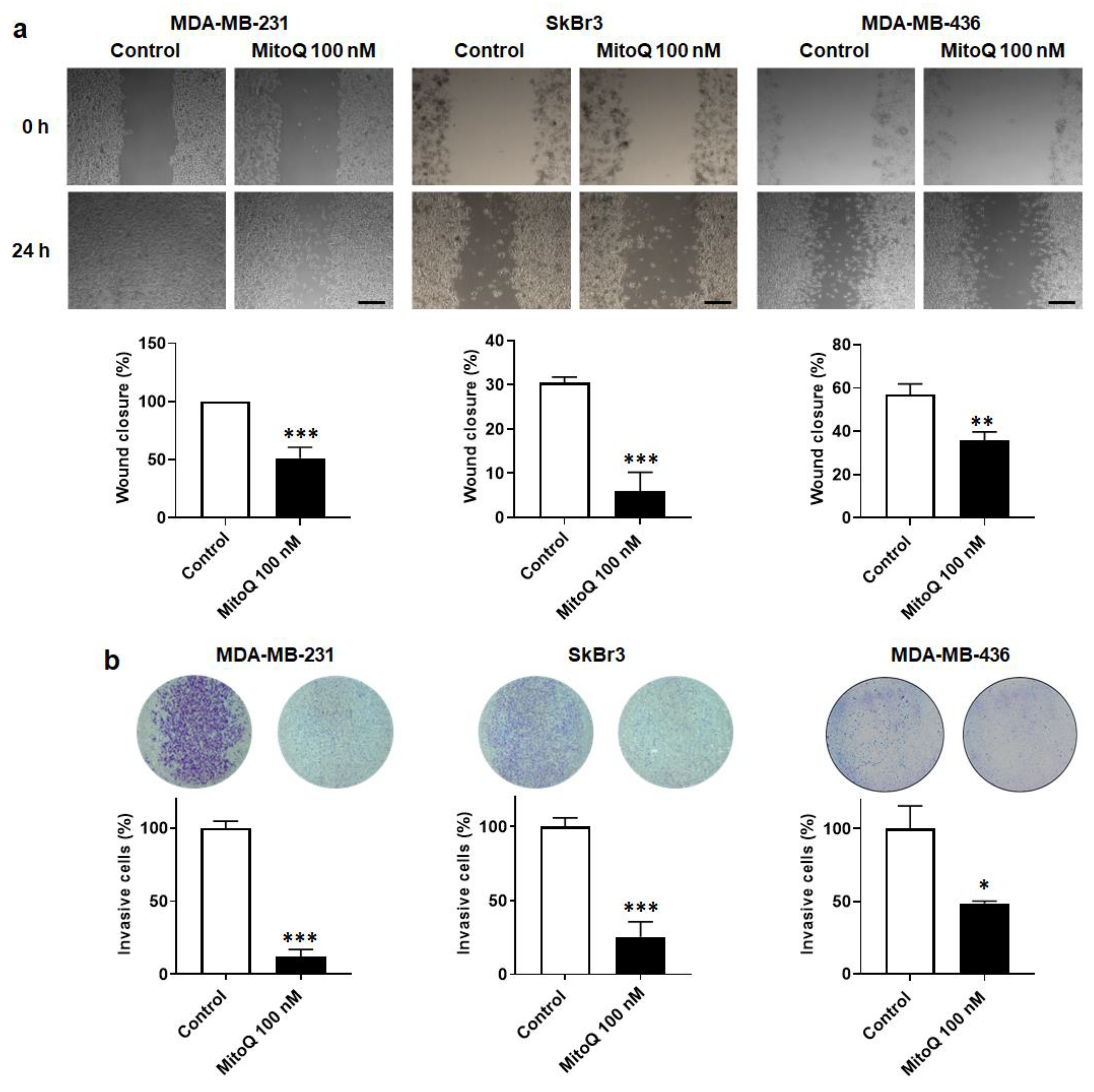
3.5. MitoQ Inhibits Human Breast Cancer Cell Migration and Invasion
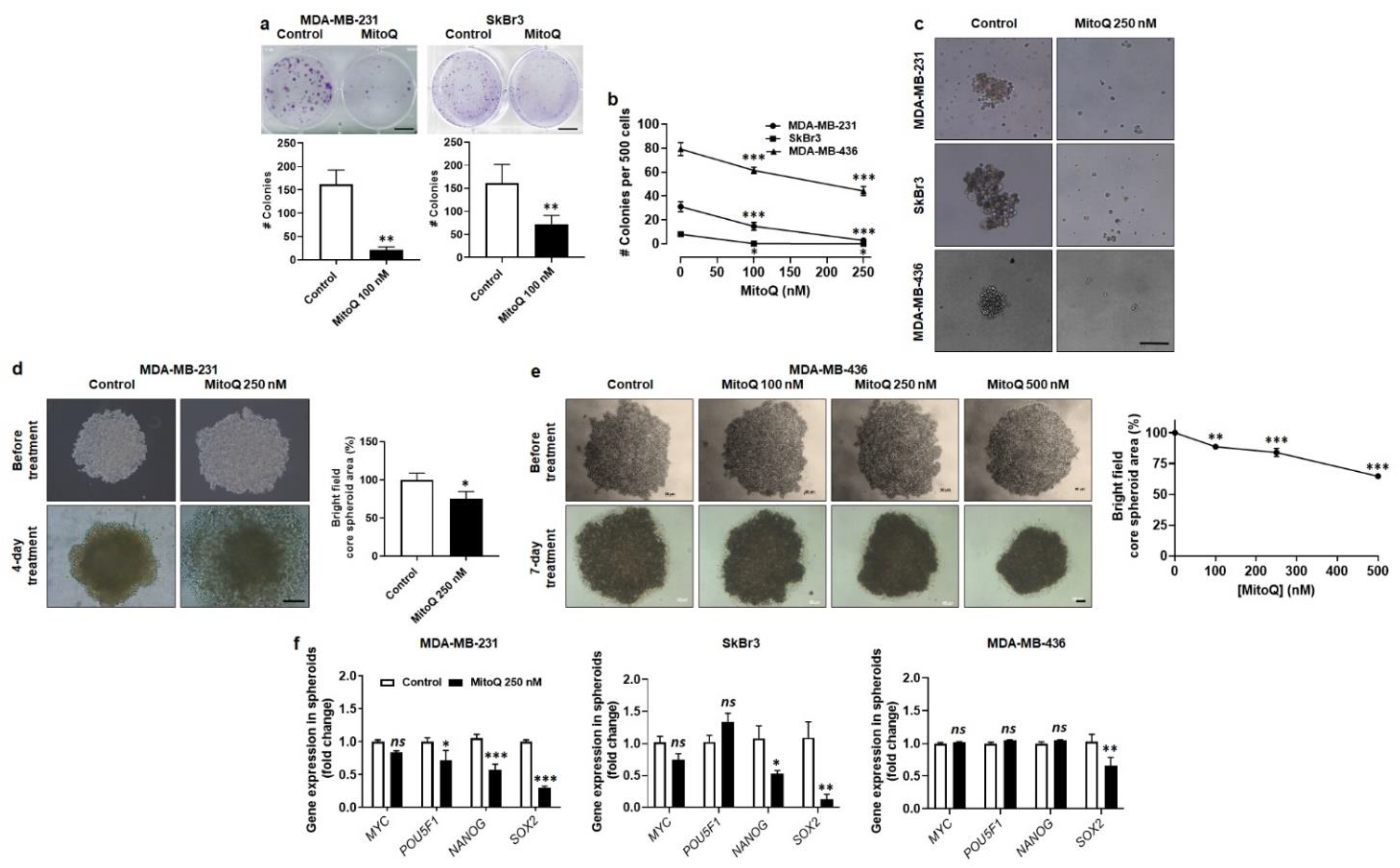
3.6. MitoQ Represses Human Breast Cancer Cell Clonogenicity, Sphere Formation and Spheroid Stability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Detailed Materials and Methods
Appendix A.1. Seahorse Oximetry
Appendix A.2. Mitochondrial Superoxide
References
- Pietila, M.; Ivaska, J.; Mani, S.A. Whom to blame for metastasis, the epithelial-mesenchymal transition or the tumor microenvironment? Cancer Lett. 2016, 380, 359–568. [Google Scholar] [CrossRef]
- Foroni, C.; Broggini, M.; Generali, D.; Damia, G. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat. Rev. 2012, 38, 689–697. [Google Scholar] [CrossRef]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brezillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol. 2020, 62, 125–133. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Hamza, B.; Miller, A.B.; Meier, L.; Stockslager, M.; Ng, S.R.; King, E.M.; Lin, L.; DeGouveia, K.L.; Mulugeta, N.; Calistri, N.L.; et al. Measuring kinetics and metastatic propensity of CTCs by blood exchange between mice. Nat. Commun. 2021, 12, 5680. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bauerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Ramundo, V.; Giribaldi, G.; Aldieri, E. Transforming growth factor-beta and oxidative stress in cancer: A crosstalk in driving tumor transformation. Cancers 2021, 13, 3093. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capeloa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cell. Mol. Life Sci. 2016, 73, 1349–1363. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Payen, V.L.; Zampieri, L.X.; Porporato, P.E.; Sonveaux, P. Pro- and antitumor effects of mitochondrial reactive oxygen species. Cancer Metastasis Rev. 2019, 38, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Sonveaux, P. Paving the way for therapeutic prevention of tumor metastasis with agents targeting mitochondrial superoxide. Mol. Cell. Oncol. 2015, 2, e968043. [Google Scholar] [CrossRef] [Green Version]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Hartley, R.C.; Cocheme, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Capeloa, T.; Krzystyniak, J.; Canas Rodriguez, A.; Payen, V.L.; Zampieri, L.X.; Pranzini, E.; Derouane, F.; Vazeille, T.; Bouzin, C.; Duhoux, F.P.; et al. MitoQ prevents human breast cancer recurrence and lung metastasis in mice. Cancers 2022, 14, 1488. [Google Scholar] [CrossRef]
- Liu, X.; Murphy, M.P.; Xing, W.; Wu, H.; Zhang, R.; Sun, H. Mitochondria-targeted antioxidant MitoQ reduced renal damage caused by ischemia-reperfusion injury in rodent kidneys: Longitudinal observations of T2 -weighted imaging and dynamic contrast-enhanced MRI. Magn. Reson. Med. 2018, 79, 1559–1567. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Cuenca, S.; Cocheme, H.M.; Logan, A.; Abakumova, I.; Prime, T.A.; Rose, C.; Vidal-Puig, A.; Smith, A.C.; Rubinsztein, D.C.; Fearnley, I.M.; et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic. Biol. Med. 2010, 48, 161–172. [Google Scholar] [CrossRef]
- Cailleau, R.; Young, R.; Olive, M.; Reeves, W.J., Jr. Breast tumor cell lines from pleural effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar] [CrossRef]
- Fogh, J.; Trempe, G. New human tumor cell lines. In Human Tumor Cells In Vitro; Fogh, J., Ed.; Plenum Publishing Corp: New York, NY, USA, 1975; pp. 115–159. [Google Scholar]
- Cailleau, R.; Olive, M.; Cruciger, Q.V. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. In Vitro 1978, 14, 911–915. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Scheinok, S.; Capeloa, T.; Porporato, P.E.; Sonveaux, P.; Gallez, B. An EPR study using cyclic hydroxylamines to assess the level of mitochondrial ROS in superinvasive cancer cells. Cell. Biochem. Biophys. 2020, 78, 249–254. [Google Scholar] [CrossRef]
- Piret, J.P.; Vankoningsloo, S.; Mejia, J.; Noel, F.; Boilan, E.; Lambinon, F.; Zouboulis, C.C.; Masereel, B.; Lucas, S.; Saout, C.; et al. Differential toxicity of copper (II) oxide nanoparticles of similar hydrodynamic diameter on human differentiated intestinal Caco-2 cell monolayers is correlated in part to copper release and shape. Nanotoxicology 2012, 6, 789–803. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef]
- Pulze, L.; Congiu, T.; Brevini, T.A.L.; Grimaldi, A.; Tettamanti, G.; D’Antona, P.; Baranzini, N.; Acquati, F.; Ferraro, F.; de Eguileor, M. MCF7 spheroid development: New insight about spatio/temporal arrangements of TNTs, amyloid fibrils, cell connections, and cellular bridges. Int. J. Mol. Sci. 2020, 21, 5400. [Google Scholar] [CrossRef]
- Porteous, C.M.; Logan, A.; Evans, C.; Ledgerwood, E.C.; Menon, D.K.; Aigbirhio, F.; Smith, R.A.; Murphy, M.P. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: Implications for mitochondria-specific therapies and probes. Biochim. Biophys. Acta 2010, 1800, 1009–1017. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [Green Version]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Kvokackova, B.; Remsik, J.; Jolly, M.K.; Soucek, K. Phenotypic heterogeneity of triple-negative breast cancer mediated by epithelial-mesenchymal plasticity. Cancers 2021, 13, 2188. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Steeg, P.S. Meaningful prevention of breast cancer metastasis: Candidate therapeutics, preclinical validation, and clinical trial concerns. J. Mol. Med. 2015, 93, 13–29. [Google Scholar] [CrossRef]
- Scully, O.J.; Bay, B.H.; Yip, G.; Yu, Y. Breast cancer metastasis. Cancer Genom. Proteom. 2012, 9, 311–320. [Google Scholar]
- Lim, B.; Hortobagyi, G.N. Current challenges of metastatic breast cancer. Cancer Metastasis Rev. 2016, 35, 495–514. [Google Scholar] [CrossRef]
- Beckman, J.S.; Minor, R.L., Jr.; White, C.W.; Repine, J.E.; Rosen, G.M.; Freeman, B.A. Superoxide dismutase and catalase conjugated to polyethylene glycol increases endothelial enzyme activity and oxidant resistance. J. Biol. Chem. 1988, 263, 6884–6892. [Google Scholar] [CrossRef]
- James, A.M.; Cocheme, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [Green Version]
- Urra, F.A.; Fuentes-Retamal, S.; Palominos, C.; Rodriguez-Lucart, Y.A.; Lopez-Torres, C.; Araya-Maturana, R. Extracellular matrix signals as drivers of mitochondrial bioenergetics and metabolic plasticity of cancer cells during metastasis. Front. Cell. Dev. Biol. 2021, 9, 751301. [Google Scholar] [CrossRef]
- Kemble, D.J.; Sun, G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc. Natl. Acad. Sci. USA 2009, 106, 5070–5075. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zou, L.; Huang, C.; Lei, Y. Redox regulation of cancer metastasis: Molecular signaling and therapeutic opportunities. Drug Dev. Res. 2014, 75, 331–341. [Google Scholar] [CrossRef]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS ONE 2016, 11, e0168283. [Google Scholar] [CrossRef] [Green Version]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-targeted drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Plecita-Hlavata, L.; Jezek, J.; Jezek, P. Pro-oxidant mitochondrial matrix-targeted ubiquinone MitoQ10 acts as anti-oxidant at retarded electron transport or proton pumping within Complex I. Int. J. Biochem. Cell. Biol. 2009, 41, 1697–1707. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Danes, J.M.; Hart, P.C.; Zhu, Y.; Huang, Y.; de Abreu, A.L.; O’Brien, J.; Mathison, A.J.; Tang, B.; Frasor, J.M.; et al. SOD2 acetylation on lysine 68 promotes stem cell reprogramming in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 23534–23541. [Google Scholar] [CrossRef]
- Shen, G.; Li, Y.; Hong, F.; Zhang, J.; Fang, Z.; Xiang, W.; Qi, W.; Yang, X.; Gao, G.; Zhou, T. A role for Snail-MnSOD axis in regulating epithelial-to-mesenchymal transition markers expression in RPE cells. Biochem. Biophys. Res. Commun. 2021, 585, 146–154. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Bill, R.; Christofori, G. The relevance of EMT in breast cancer metastasis: Correlation or causality? FEBS Lett. 2015, 589, 1577–1587. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Kang, Y. Cell lineage determinants as regulators of breast cancer metastasis. Cancer Metastasis Rev. 2016, 35, 631–644. [Google Scholar] [CrossRef]
- de Herreros, A.G.; Peiro, S.; Nassour, M.; Savagner, P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 135–147. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capeloa, T.; Krzystyniak, J.; d’Hose, D.; Canas Rodriguez, A.; Payen, V.L.; Zampieri, L.X.; Van de Velde, J.A.; Benyahia, Z.; Pranzini, E.; Vazeille, T.; et al. MitoQ Inhibits Human Breast Cancer Cell Migration, Invasion and Clonogenicity. Cancers 2022, 14, 1516. https://doi.org/10.3390/cancers14061516
Capeloa T, Krzystyniak J, d’Hose D, Canas Rodriguez A, Payen VL, Zampieri LX, Van de Velde JA, Benyahia Z, Pranzini E, Vazeille T, et al. MitoQ Inhibits Human Breast Cancer Cell Migration, Invasion and Clonogenicity. Cancers. 2022; 14(6):1516. https://doi.org/10.3390/cancers14061516
Chicago/Turabian StyleCapeloa, Tania, Joanna Krzystyniak, Donatienne d’Hose, Amanda Canas Rodriguez, Valery L. Payen, Luca X. Zampieri, Justine A. Van de Velde, Zohra Benyahia, Erica Pranzini, Thibaut Vazeille, and et al. 2022. "MitoQ Inhibits Human Breast Cancer Cell Migration, Invasion and Clonogenicity" Cancers 14, no. 6: 1516. https://doi.org/10.3390/cancers14061516
APA StyleCapeloa, T., Krzystyniak, J., d’Hose, D., Canas Rodriguez, A., Payen, V. L., Zampieri, L. X., Van de Velde, J. A., Benyahia, Z., Pranzini, E., Vazeille, T., Fransolet, M., Bouzin, C., Brusa, D., Michiels, C., Gallez, B., Murphy, M. P., Porporato, P. E., & Sonveaux, P. (2022). MitoQ Inhibits Human Breast Cancer Cell Migration, Invasion and Clonogenicity. Cancers, 14(6), 1516. https://doi.org/10.3390/cancers14061516