
CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML

 , , , and
, , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. RNA Expression Data from Human AML Patients
2.2. Cell Line Establishment and Cell Culture
2.3. RNA Isolation and RT-qPCR
2.4. RNA Sequencing
2.5. Immunoblot Analysis
2.6. CDK6 Degrader and CDK4/6 Inhibitor Treatments
2.7. Flow Cytometry
2.8. Immunoprecipitation (IP) and Mass Spectrometry (MS) Analysis
2.9. Statistical Analysis, Data Visualisation and Graphical Design
3. Results
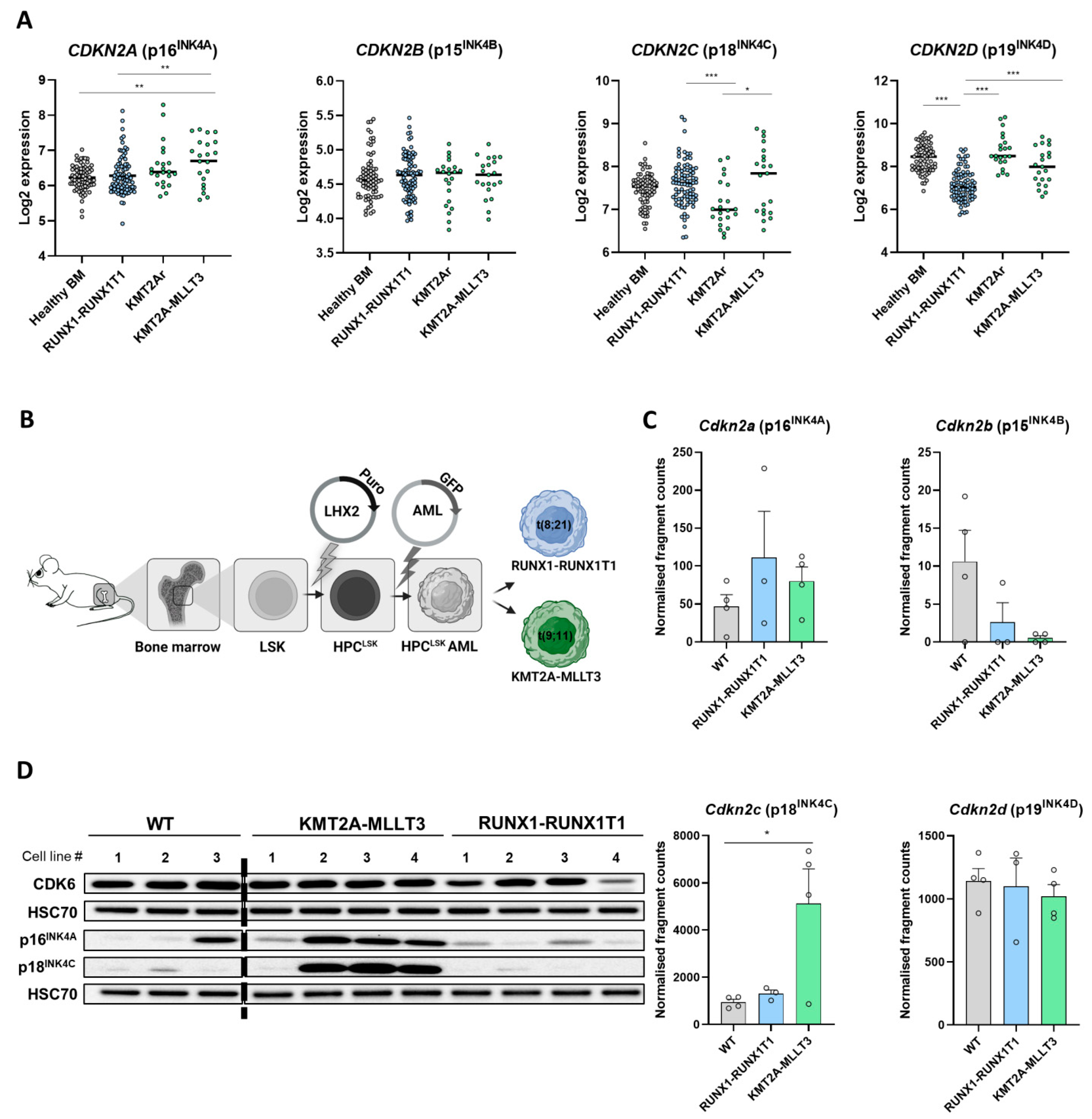
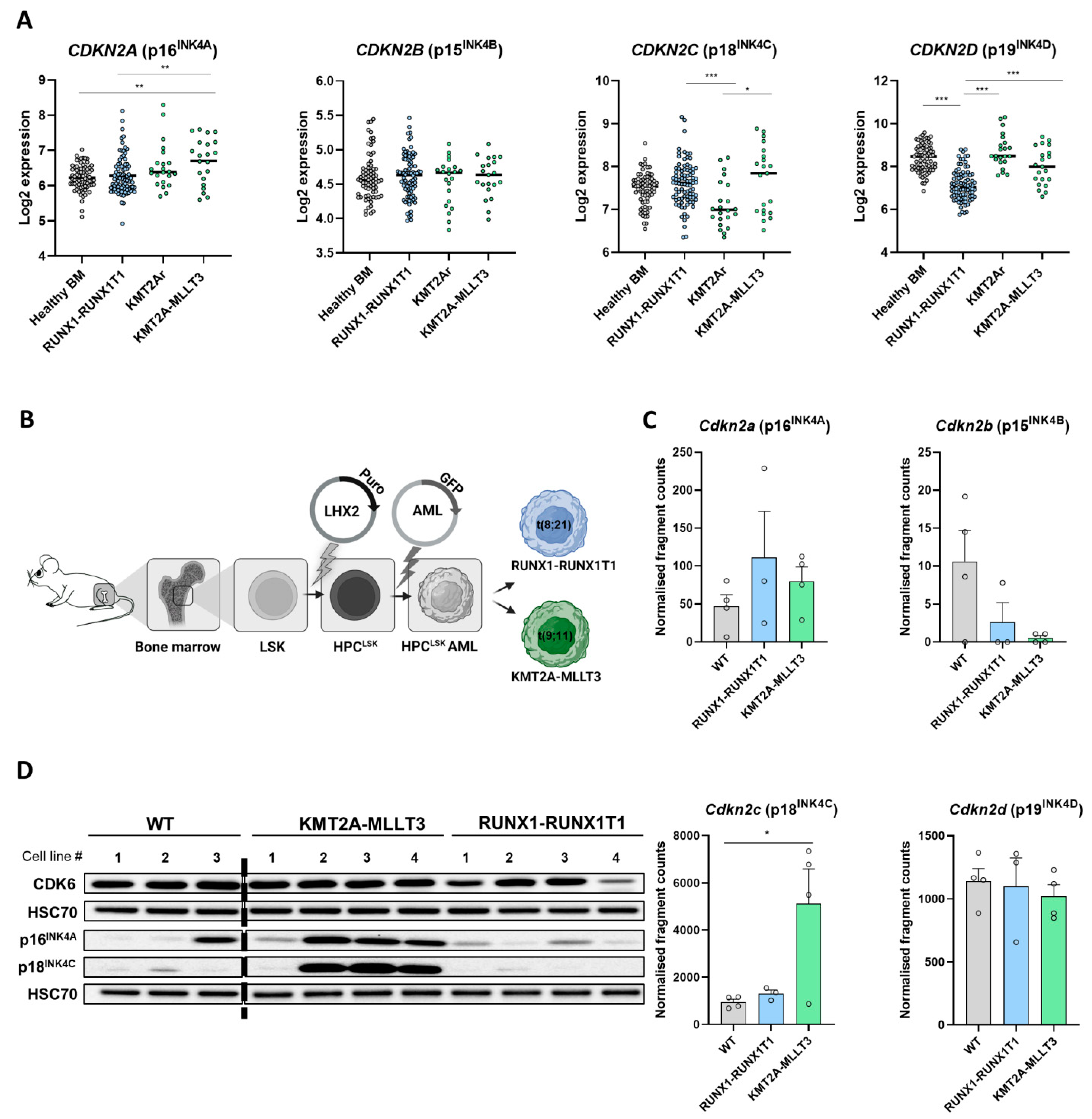
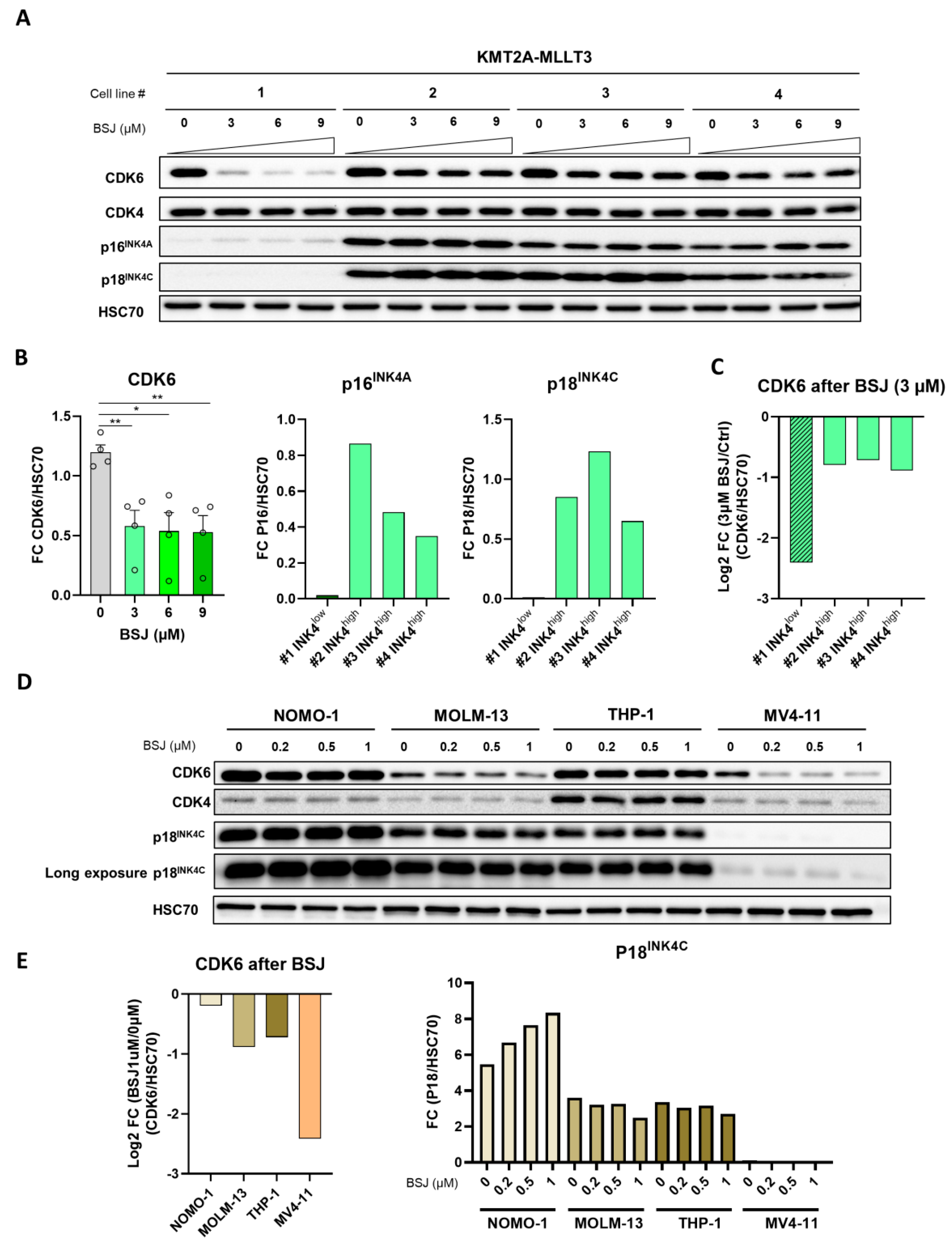
3.1. Differential INK4 Expression in KMT2A-MLLT3+ and RUNX1-RUNX1T1+ AML
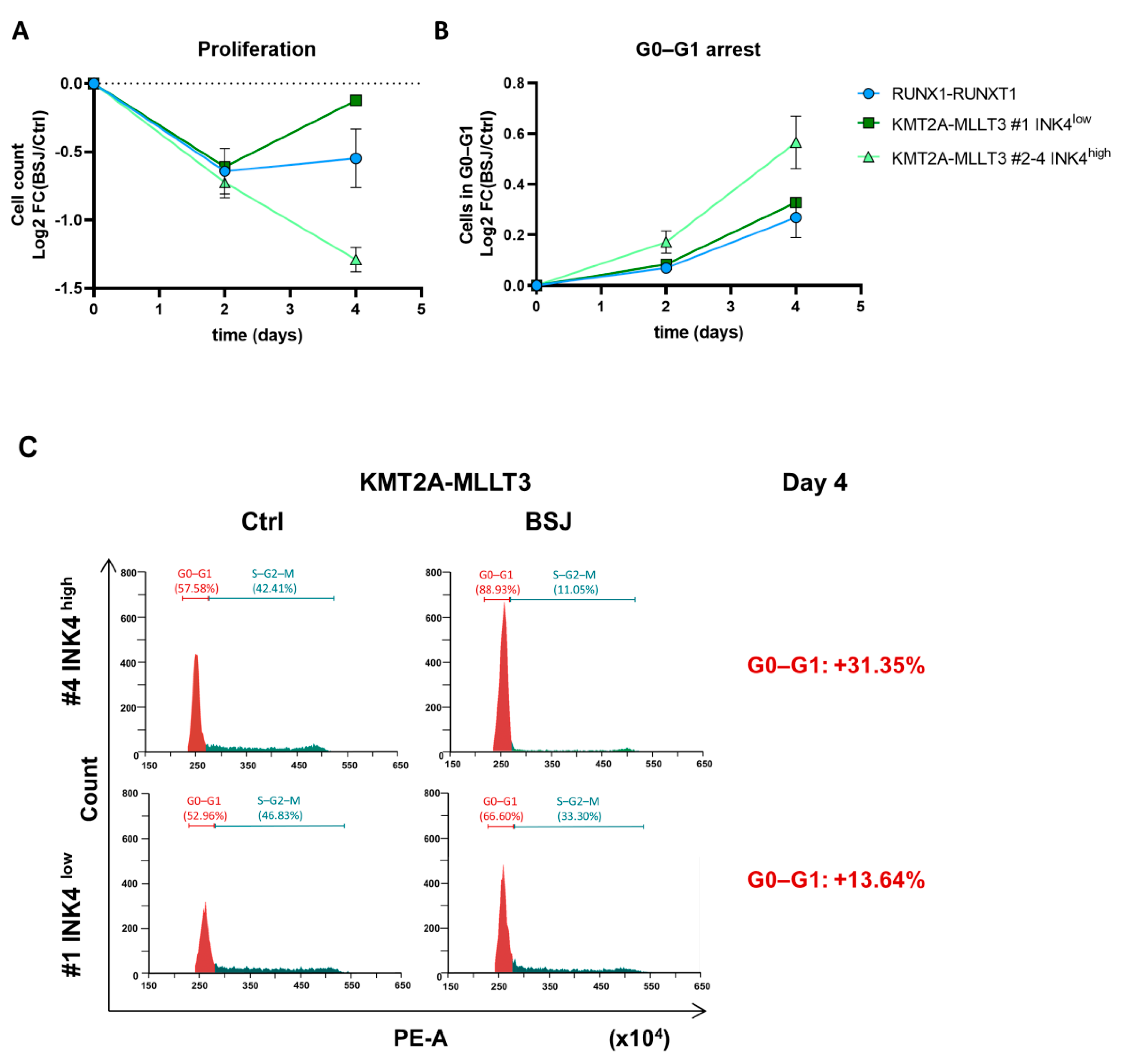
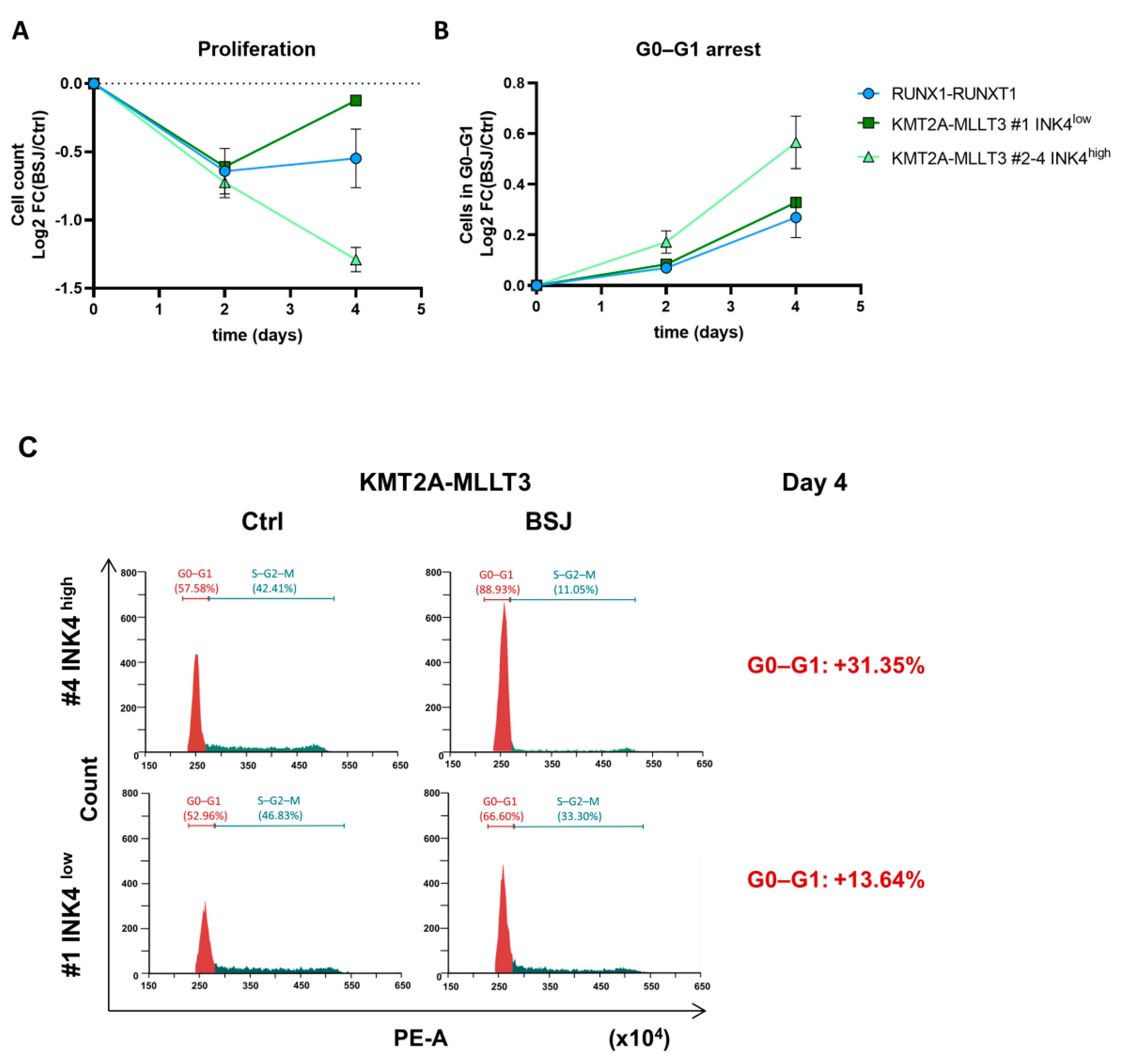
3.2. Cells with Low INK4 Levels Show Fast Cell Cycle Re-Entry after BSJ Treatment
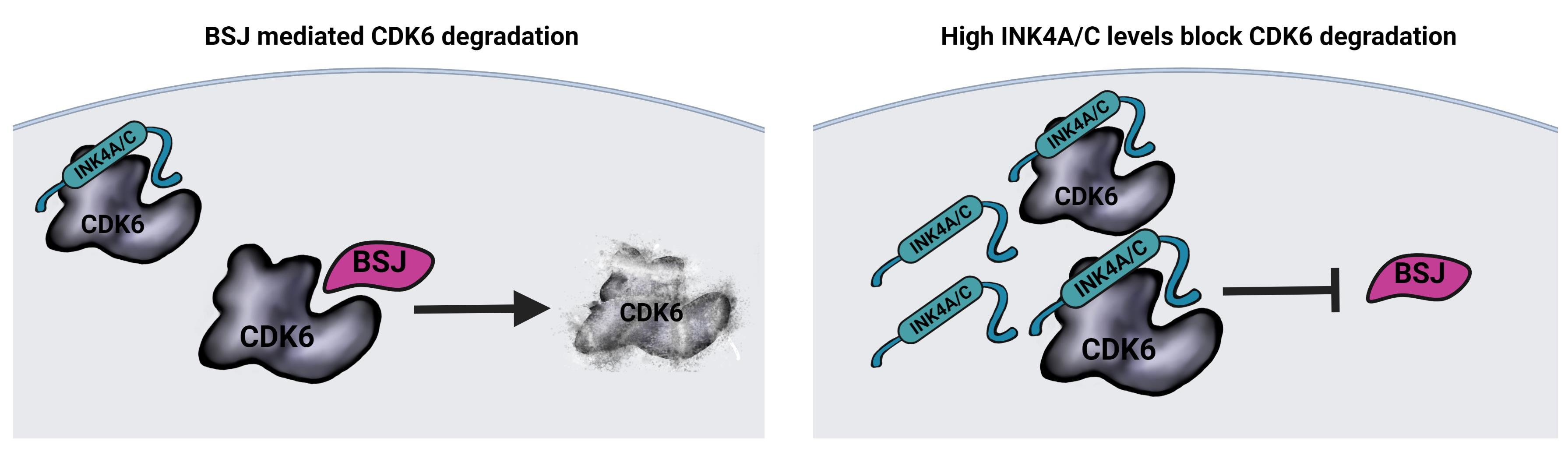
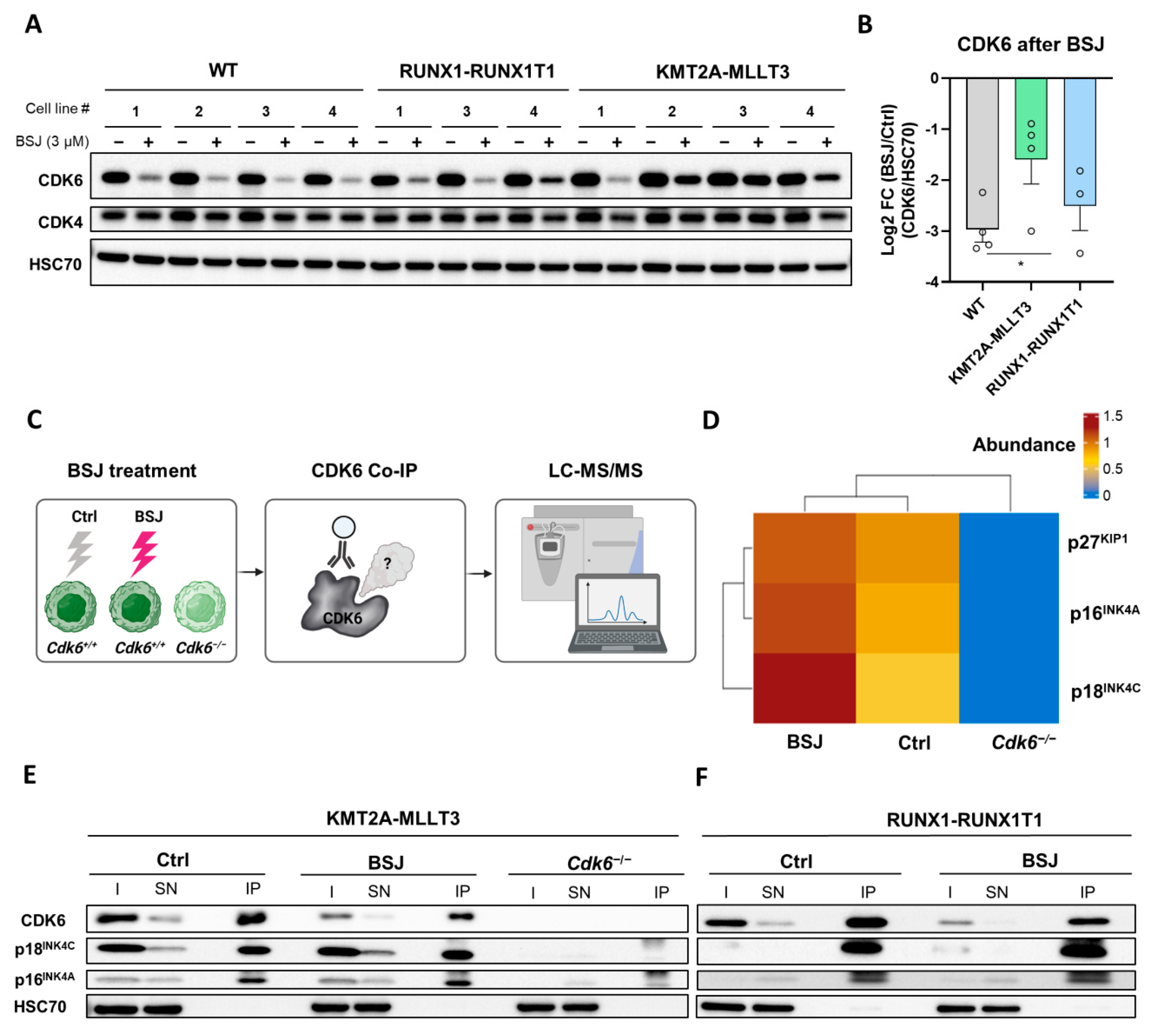
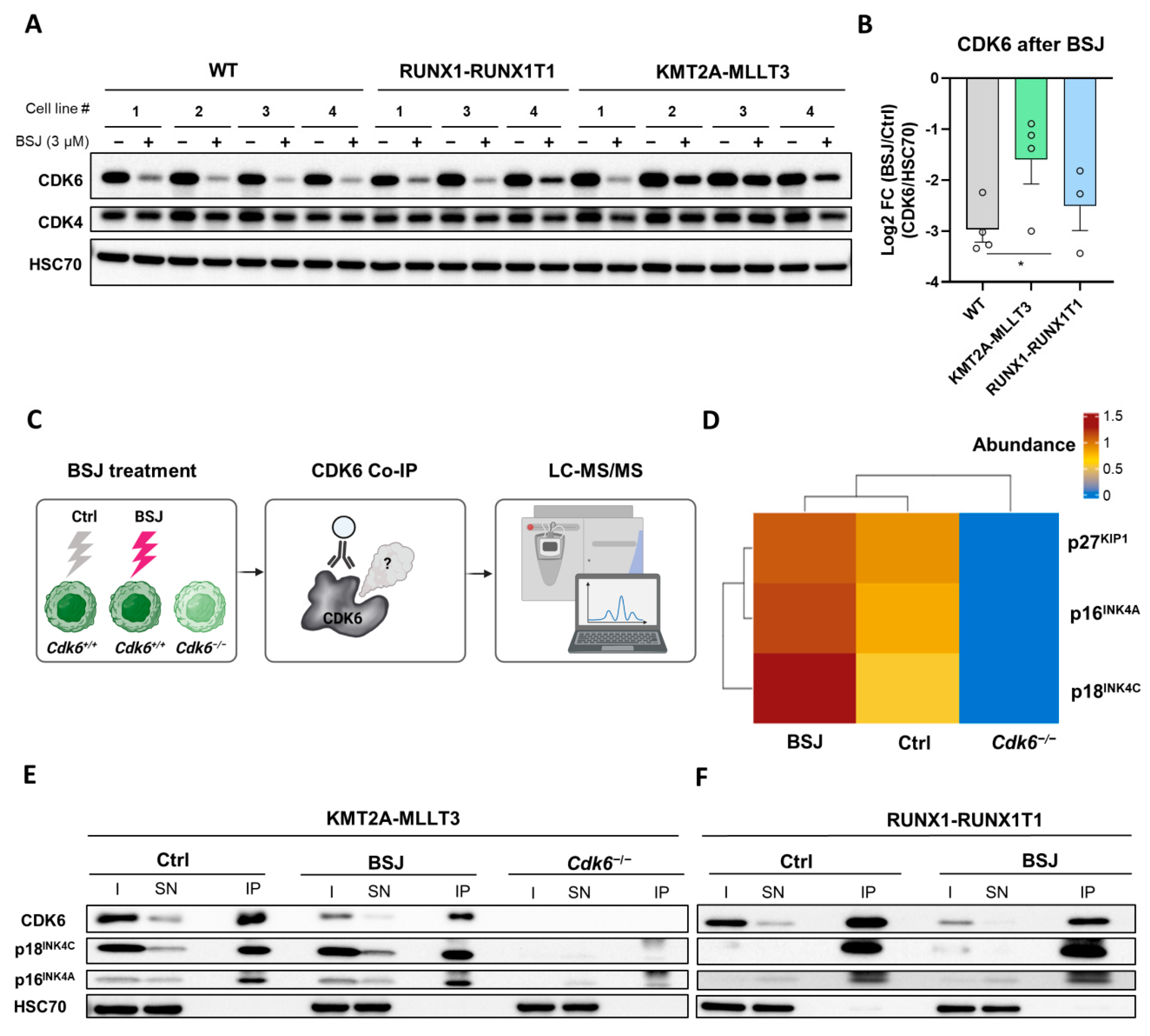
3.3. BSJ Treatment Enriches CDK6-INK4 Complexes
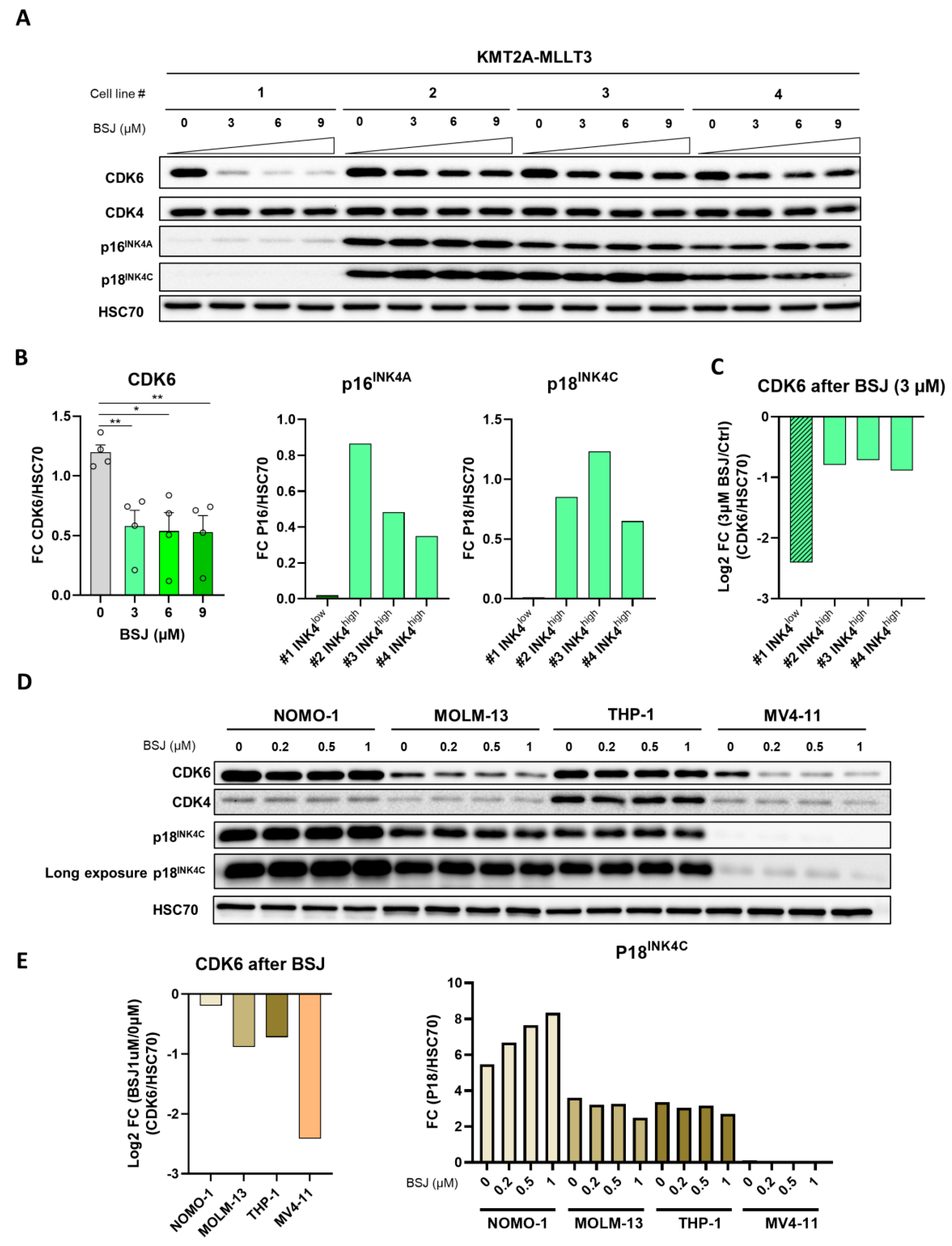
3.4. INK4 Levels Predict BSJ-Mediated CDK6 Degradation Efficacy in Murine and Human Leukaemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirtonia, A.; Pandya, G.; Sethi, G.; Pandey, A.K.; Das, B.C.; Garg, M. A comprehensive review of genetic alterations and molecular targeted therapies for the implementation of personalized medicine in acute myeloid leukemia. J. Mol. Med. 2020, 98, 1069–1091. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306. [Google Scholar] [CrossRef]
- Liu, W.; Yi, J.M.; Liu, Y.; Chen, C.; Zhang, K.X.; Zhou, C.; Zhan, H.E.; Zhao, L.; Morales, S.; Zhao, X.L.; et al. CDK6 Is a Potential Prognostic Biomarker in Acute Myeloid Leukemia. Front. Genet. 2020, 11, 600227. [Google Scholar] [CrossRef] [PubMed]
- Uras, I.Z.; Sexl, V.; Kollmann, K. CDK6 Inhibition: A Novel Approach in AML Management. Int. J. Mol. Sci. 2020, 21, 2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation. Cancer Cell 2018, 34, 626–642.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, J.; Blaser, B.W.; Duchemin, A.-M.; Kusewitt, D.F.; Liu, T.; Caligiuri, M.A.; Briesewitz, R. Pharmacologic inhibition of CDK4/6: Mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood 2007, 110, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheicher, R.; Hoelbl-Kovacic, A.; Bellutti, F.; Tigan, A.S.; Prchal-Murphy, M.; Heller, G.; Schneckenleithner, C.; Salazar-Roa, M.; Zöchbauer-Müller, S.; Zuber, J.; et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood 2015, 125, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, 147, 2988–2995. [Google Scholar] [CrossRef] [PubMed]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; de Luca, F.; Galardi, F.; Risi, E.; de Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef]
- Green, J.L.; Okerberg, E.S.; Sejd, J.; Palafox, M.; Monserrat, L.; Alemayehu, S.; Wu, J.; Sykes, M.; Aban, A.; Serra, V.; et al. Direct CDKN2 Modulation of CDK4 Alters Target Engagement of CDK4 Inhibitor Drugs. Mol. Cancer Ther. 2019, 18, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef] [PubMed]
- Ekholm, S.V.; Reed, S.I. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 2000, 12, 676–684. [Google Scholar] [CrossRef]
- Russo, A.A.; Tong, L.; Lee, J.O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Cechova, H.; Lassuthova, P.; Novakova, L.; Belickova, M.; Stemberkova, R.; Jencik, J.; Stankova, M.; Hrabakova, P.; Pegova, K.; Zizkova, H.; et al. Monitoring of methylation changes in 9p21 region in patients with myelodysplastic syndromes and acute myeloid leukemia. Neoplasma 2012, 59, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, H.J.M.; de Bont, E.S.J.M.; Valk, P.J.M.; Schuringa, J.J.; Kies, M.; Woolthuis, C.M.; Delwel, R.; Veeger, N.J.G.M.; Vellenga, E.; Löwenberg, B.; et al. AML at older age: Age-related gene expression profiles reveal a paradoxical down-regulation of p16INK4A mRNA with prognostic significance. Blood 2009, 114, 2869–2877. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, K.; Heller, G.; Schneckenleithner, C.; Warsch, W.; Scheicher, R.; Ott, R.G.; Schäfer, M.; Fajmann, S.; Schlederer, M.; Schiefer, A.I.; et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 2013, 24, 167–181. [Google Scholar] [CrossRef] [Green Version]
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573. [Google Scholar] [CrossRef]
- Doma, E.; Mayer, I.M.; Brandstoetter, T.; Maurer, B.; Grausenburger, R.; Menzl, I.; Zojer, M.; Hoelbl-Kovacic, A.; Carlsson, L.; Heller, G.; et al. A robust approach for the generation of functional hematopoietic progenitor cell lines to model leukemic transformation. Blood Adv. 2021, 5, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Nebenfuehr, S.; Bellutti, F.; Ünal, H.; Zojer, M.; Scheiblecker, L.; Sexl, V.; Kollmann, K. The Effect of CDK6 Expression on DNA Methylation and DNMT3B Regulation. iScience 2020, 23, 101602. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Version 4.0.3; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 18 November 2020).
- Zuber, J.; Radtke, I.; Pardee, T.S.; Zhao, Z.; Rappaport, A.R.; Luo, W.; McCurrach, M.E.; Yang, M.M.; Dolan, M.E.; Kogan, S.C.; et al. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009, 23, 877–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourrajab, F.; Zare-Khormizi, M.R.; Hashemi, A.S.; Hekmatimoghaddam, S. Genetic Characterization and Risk Stratification of Acute Myeloid Leukemia. Cancer Manag. Res. 2020, 12, 2231–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhang, X.; Gu, L.; Zhou, J.; Deng, D. P16 methylation increases the sensitivity of cancer cells to the CDK4/6 inhibitor palbociclib. PLoS ONE 2019, 14, e0223084. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Jiang, B.; Guo, J.; Shao, H.; Del Priore, I.S.; Chang, Q.; Kudo, R.; Li, Z.; Razavi, P.; Liu, B.; et al. INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors. Cancer Discov. 2021, 12, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Prange, K.H.M.; Mandoli, A.; Kuznetsova, T.; Wang, S.Y.; Sotoca, A.M.; Marneth, A.E.; van der Reijden, B.A.; Stunnenberg, H.G.; Martens, J.H.A. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene 2017, 36, 3346–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 2020, 11, 580251. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, P.; Shen, H.; Yu, H.; Song, X.; Zhang, L.; Zhang, P.; Cheng, H.; Xie, Z.; Hao, S.; et al. Small-molecule inhibitors targeting INK4 protein p18(INK4C) enhance ex vivo expansion of haematopoietic stem cells. Nat. Commun. 2015, 6, 6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, W.; Zhang, Y.; Ding, Y.; Yang, M.; He, M.; Liu, X.; Gu, J.; Xu, S.; Feng, Z.; et al. Enhanced self-renewal of human long-term hematopoietic stem cells by a sulfamoyl benzoate derivative targeting p18INK4C. Blood Adv. 2021, 5, 3362–3372. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.Q.; Yang, P.; Zhang, Y.; Zhang, P.; Wang, L.; Ding, Y.; Yang, M.; Tong, Q.; Cheng, H.; Ji, Q.; et al. Discovery of novel INK4C small-molecule inhibitors to promote human and murine hematopoietic stem cell ex vivo expansion. Sci. Rep. 2015, 5, 18115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; de Godoy, L.M.F.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmalzbauer, B.S.; Thondanpallil, T.; Heller, G.; Schirripa, A.; Sperl, C.-M.; Mayer, I.M.; Knab, V.M.; Nebenfuehr, S.; Zojer, M.; Mueller, A.C.; et al. CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML. Cancers 2022, 14, 1554. https://doi.org/10.3390/cancers14061554
Schmalzbauer BS, Thondanpallil T, Heller G, Schirripa A, Sperl C-M, Mayer IM, Knab VM, Nebenfuehr S, Zojer M, Mueller AC, et al. CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML. Cancers. 2022; 14(6):1554. https://doi.org/10.3390/cancers14061554
Chicago/Turabian StyleSchmalzbauer, Belinda S., Teresemary Thondanpallil, Gerwin Heller, Alessia Schirripa, Clio-Melina Sperl, Isabella M. Mayer, Vanessa M. Knab, Sofie Nebenfuehr, Markus Zojer, André C. Mueller, and et al. 2022. "CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML" Cancers 14, no. 6: 1554. https://doi.org/10.3390/cancers14061554
APA StyleSchmalzbauer, B. S., Thondanpallil, T., Heller, G., Schirripa, A., Sperl, C.-M., Mayer, I. M., Knab, V. M., Nebenfuehr, S., Zojer, M., Mueller, A. C., Fontaine, F., Klampfl, T., Sexl, V., & Kollmann, K. (2022). CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML. Cancers, 14(6), 1554. https://doi.org/10.3390/cancers14061554