
PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression

 ,
,  and
and
Abstract
Simple Summary
Abstract
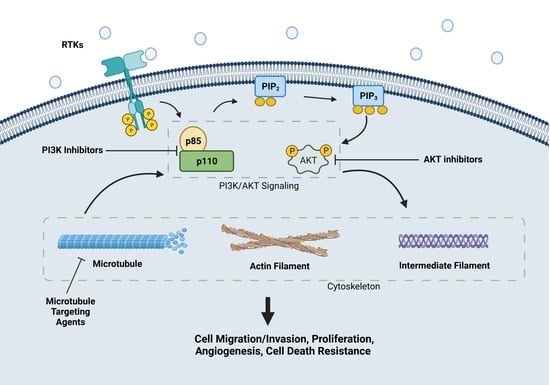
1. Introduction
1.1. PI3K/AKT Signaling Pathway in Cancer
1.2. Critical Roles of Cytoskeleton in Cancer
2. PI3K/AKT in Regulating Multiple Aspects of Cytoskeleton in Cancer Biology
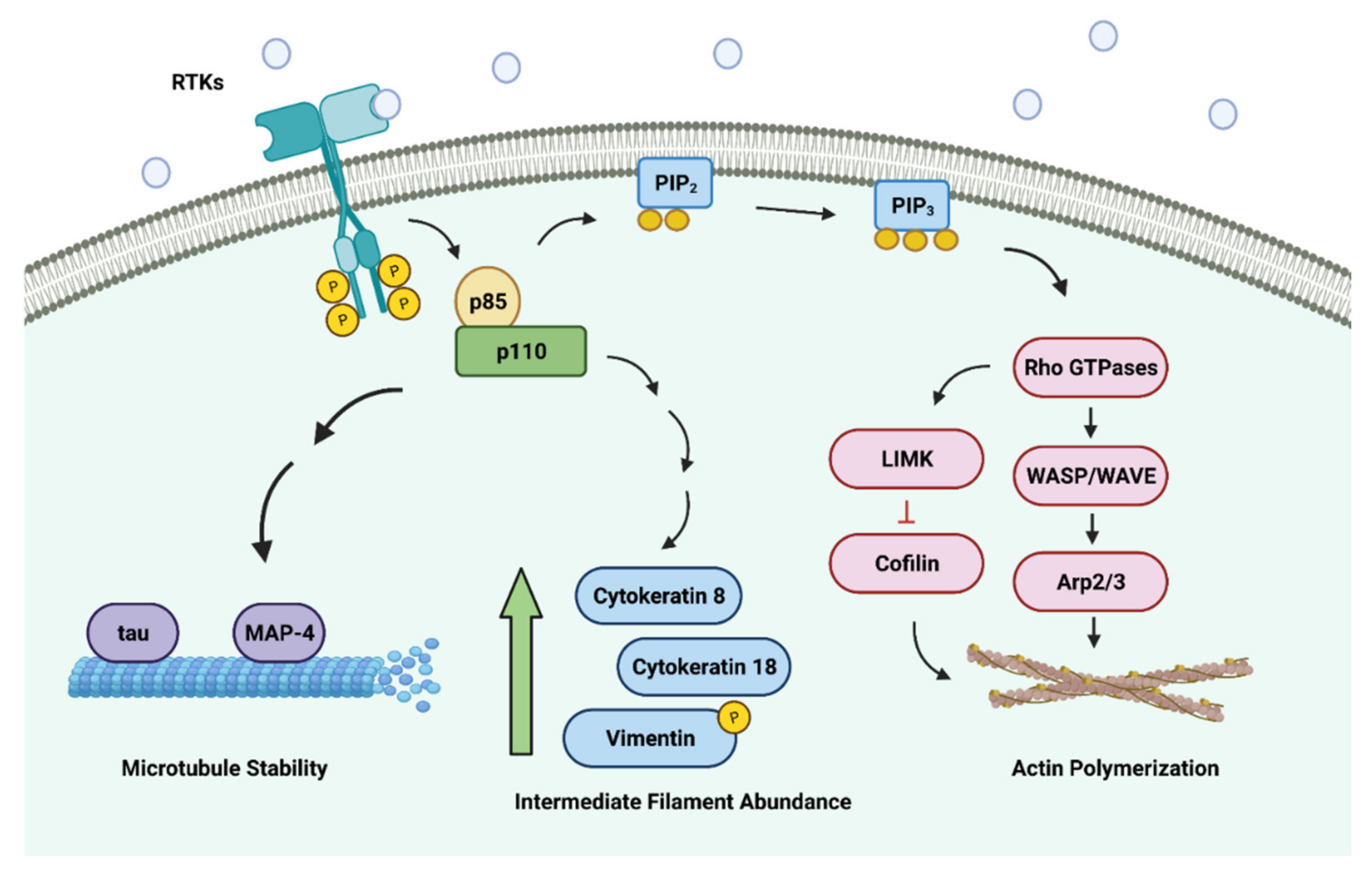
2.1. PI3K in Regulating the Actin Cytoskeleton
2.2. PI3K in Regulating Microtubules
2.3. PI3K in Regulating Intermediate Filaments
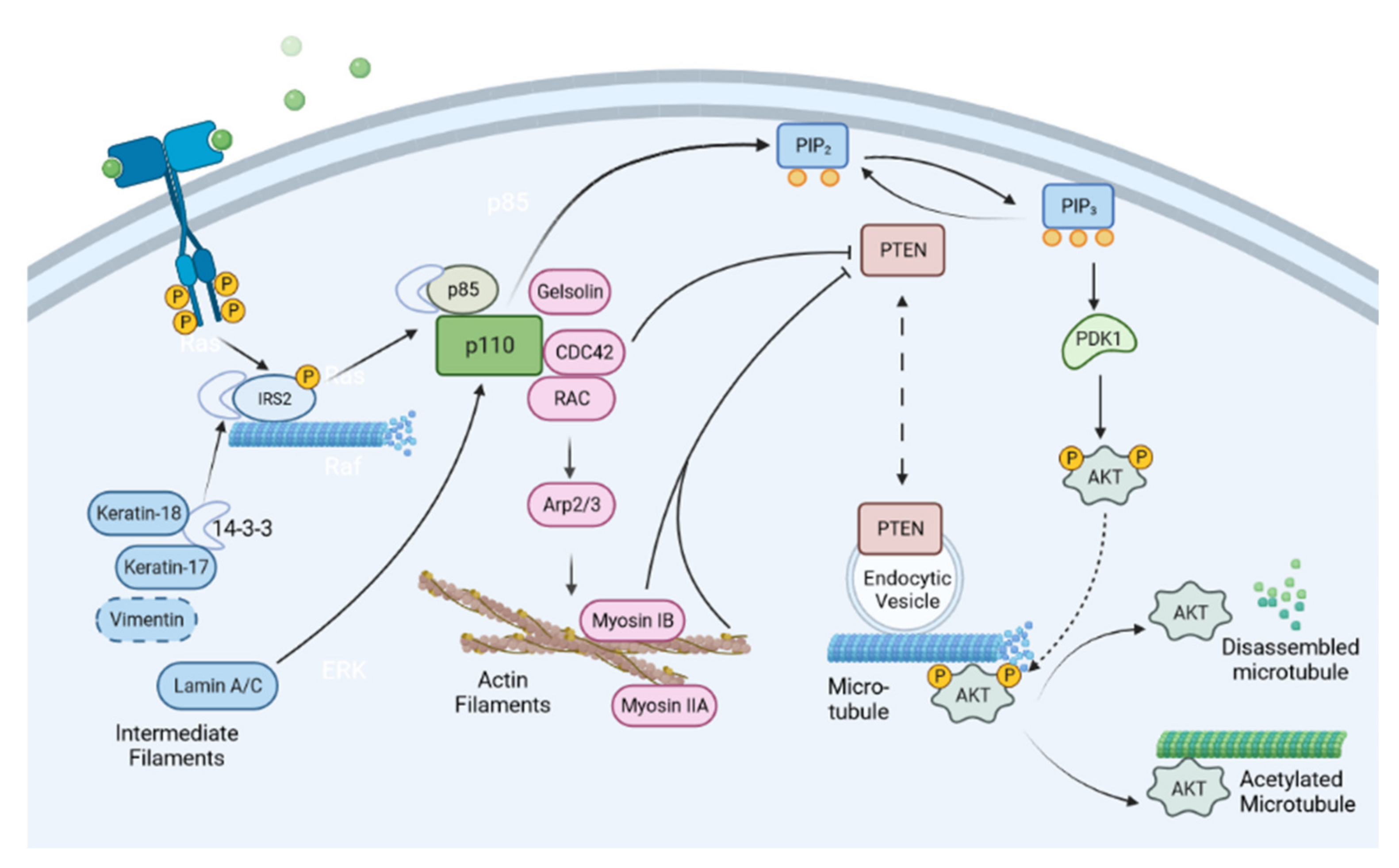
3. Cytoskeleton in Regulating PI3K Signaling
3.1. Actin Cytoskeleton and Its Regulators in Regulating PI3K Signaling
3.2. Microtubule Cytoskeleton in Regulating PI3K Signaling
3.3. Intermediate Filaments in Regulating PI3K Signaling
4. Clinical Relevance of PI3K/Akt-Cytoskeleton Crosstalk
4.1. Targeting PI3K/Akt in Cancer Treatment: PI3K Inhibitors in Clinical Trials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Phase | Treatment Composition | Disease Studied |
|---|---|---|---|
| Pan-PI3K inhibitors | |||
| Buparlisib | I/II | Buparlisib monotherapy [148,149] Buparlisib + mFOLFOX6 [150] Buparlisib + abiraterone acetate [151] Buparlisib + enzalutamide [152] | Advanced solid tumors [148,150] Patients with solid or hematologic malignancies with PI3K pathway activation [149] Castration-resistant prostate cancer [151] Metastatic castration-resistant prostate cancer [152] |
| Dual PI3K/mTOR inhibitors | |||
| Gedatolisib | I/II | Gedatolisib monotherapy [157,158] | Advanced solid tumors [157] Advanced solid tumors treated with palliative chemotherapy [158] |
| Isoform-selective PI3K inhibitors | |||
| Idelalisib | III/FDA approved (for treating SLL) | Idelalisib monotherapy [161,162] Idelalisib + rituximab [163] | Relapsed indolent lymphoma [161,162] Relapsed chronic lymphocytic leukemia [163] |
| ATP-competitive AKT inhibitors | |||
| Capivasertib | I/II | Capivasertib + fulvestrant [168,169] Capivasertib + paclitaxel [170] | PTEN-mutant ER + metastatic breast cancer [168] Estrogen receptor + HER2- metastatic/advanced breast cancer with aromatase inhibitor resistance [169] Metastatic triple-negative breast cancer [170] |
| Ipatasertib | II/III | Ipatasertib + mFOLFOX6 [171] Ipatasertib + abiraterone [172] Ipatasertib + abiraterone and prednisolone [173] | Locally advanced/metastatic gastric and gastroesophageal junction cancer [171] PTEN metastatic prostate cancer [172] Metastatic castration-resistant prostate cancer [173] |
| Allosteric AKT Inhibitors | |||
| BAY 1125976 | I | BAY 1125976 monotherapy [176] | Advanced solid cancer [176] |
| MK-2206 | II | MK-2206 + anastrozole [177] MK-2206 + standard neoadjuvant therapy [178] | Stage II/III ER+/HER2- breast cancer with PIK3CA mutation [177] HR-/HER2+ breast cancer [178] |
4.2. Targeting Cytoskeleton in Cancer Treatment
| Drug | Phase | Treatment Composition | Disease Studied |
|---|---|---|---|
| Vinca-site binders | |||
| Eribulin | II/ FDA approved (for metastatic breast cancer and liposarcoma) | Eribulin versus dacarbazine [186] Eribulin versus capecitabine [187] Eribulin + pembrolizumab [188,189] | Advanced liposarcoma or leiomyosarcoma [186] Advanced/metastatic breast cancer with prior anthracycline- and taxane-based treatment [187] HR+ HER2- metastatic breast cancer [188] Metastatic triple-negative breast cancer [189] |
| Glembatumumab vedotin (MMAE ADC) | II | Glembatumumab vedotin monotherapy [194,195,196,197] | Recurrent osteosarcoma [195] Advanced melanoma [196] Advanced glycoprotein NMB-expressing breast cancer [194] Metastatic glycoprotein NMB-expressing triple-negative breast cancer [197] |
| Brentuximab vedotin (MMAE ADC) | FDA approved | Brentuximab vedotin monotherapy [198,199,201] | Hodgkin’s lymphoma [199,201] Systemic anaplastic large cell lymphoma [198] |
| Colchicine-site binders | |||
| Fosbretabulin | II | Fosbretabulin + pazopanib [205] Fosbretabulin + bevacizumab [206] Fosbretabulin + paclitaxel/carboplatin [207] | Recurrent ovarian cancer [205,206] Anaplastic thyroid carcinoma [207] |
| Combretastatin A1 diphosphate | I | CA1P monotherapy [208] | Relapsed or refractory acute myeloid leukemia [208] |
| Plinabulin | III | Plinabulin + docetaxel | Metastatic non-small cell lung cancer (NCT02812667) |
| Lisavanbulin | I/II | Lisavanbulin monotherapy [216] | Advanced solid tumors [216] |
| Taxane-site binders | |||
| Cabazitaxel | III | Cabazitaxel versus docetaxel [220,221] | Metastatic castration-resistant prostate cancer [220,221] |
| Nab-paclitaxel | II/III | Nab-paclitaxel monotherapy [227] Nab-paclitaxel versus paclitaxel [226] Atezolizumab + nab-paclitaxel [228] | Advanced triple-negative breast cancer [228] Metastatic breast cancer patients with visceral metastases [227] Metastatic breast cancer [226] |
| Ixabepilone | III | Ixabepilone + capecitabine [238,240] | Metastatic breast cancer previously treated with anthracycline and taxanes [238,240] |
4.3. Potential Crosstalk of PI3K Inhibitors and Cytoskeletal Disruptors in Clinical Treatment of Cancer
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Najafi, M.; Ang, H.L.; Moghadam, E.R.; Mahabady, M.K.; Zabolian, A.; Jafaripour, L.; Bejandi, A.K.; Hushmandi, K.; Saleki, H.; et al. PTEN, a Barrier for Proliferation and Metastasis of Gastric Cancer Cells: From Molecular Pathways to Targeting and Regulation. Biomedicines 2020, 8, 264. [Google Scholar] [CrossRef]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Avvaru, P.; Furqan, M.; Song, Y.; Liu, D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J. Hematol. Oncol. 2013, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Hornsveld, M.; Dansen, T.B.; Derksen, P.W.; Burgering, B.M.T. Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol. 2018, 50, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Yart, A.; Chap, H.; Raynal, P. Phosphoinositide 3-kinases in lysophosphatidic acid signaling: Regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim. Biophys. Acta 2002, 1582, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, C.; Dance, M.; Montagner, A.; Edouard, T.; Malet, N.; Perret, B.; Yart, A.; Salles, J.P.; Raynal, P. Signal strength dictates phosphoinositide 3-kinase contribution to Ras/extracellular signal-regulated kinase 1 and 2 activation via differential Gab1/Shp2 recruitment: Consequences for resistance to epidermal growth factor receptor inhibition. Mol. Cell Biol. 2008, 28, 587–600. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.R.; Liao, L.; Yates, J.R.; Vogt, P.K. Essential role of Stat3 in PI3K-induced oncogenic transformation. Proc. Natl. Acad. Sci. USA 2011, 108, 13247–13252. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K.; Hart, J.R. PI3K and STAT3: A new alliance. Cancer Discov. 2011, 1, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Whale, A.D.; Colman, L.; Lensun, L.; Rogers, H.L.; Shuttleworth, S.J. Functional characterization of a novel somatic oncogenic mutation of. Signal. Transduct. Target Ther. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Waldman, T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top Microbiol. Immunol. 2010, 347, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Oda, K.; Stokoe, D.; Taketani, Y.; McCormick, F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005, 65, 10669–10673. [Google Scholar] [CrossRef]
- Levine, D.A.; Bogomolniy, F.; Yee, C.J.; Lash, A.; Barakat, R.R.; Borgen, P.I.; Boyd, J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef]
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 2005, 102, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Kang, S.; Vogt, P.K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Correia, N.C.; Gírio, A.; Antunes, I.; Martins, L.R.; Barata, J.T. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef]
- Ouderkirk, J.L.; Krendel, M. Non-muscle myosins in tumor progression, cancer cell invasion, and metastasis. Cytoskeleton 2014, 71, 447–463. [Google Scholar] [CrossRef]
- Dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect Biol. 2018, 10, a022608. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Bar, H.; Kreplak, L.; Strelkov, S.V.; Aebi, U. Intermediate filaments: From cell architecture to nanomechanics. Nat. Rev. Mol. Cell Biol. 2007, 8, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Leduc, C.; Etienne-Manneville, S. Intermediate filaments in cell migration and invasion: The unusual suspects. Curr. Opin. Cell Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Flitney, E.W.; Kuczmarski, E.R.; Adam, S.A.; Goldman, R.D. Insights into the mechanical properties of epithelial cells: The effects of shear stress on the assembly and remodeling of keratin intermediate filaments. FASEB J. 2009, 23, 2110–2119. [Google Scholar] [PubMed]
- Hall, A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Tochhawng, L.; Deng, S.; Pervaiz, S.; Yap, C.T. Redox regulation of cancer cell migration and invasion. Mitochondrion 2013, 13, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Deng, S.; Gopal, V.; Yap, K.C.; Halim, C.E.; Lye, M.L.; Ong, M.S.; Tan, T.Z.; Sethi, G.; Hooi, S.C.; et al. Cytoskeletal Dynamics in Epithelial-Mesenchymal Transition: Insights into Therapeutic Targets for Cancer Metastasis. Cancers 2021, 13, 1882. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Yan, B.; Yap, C.T.; Wang, S.; Lee, C.K.; Koh, S.; Omar, M.F.; Salto-Tellez, M.; Kumarasinghe, M.P. Cofilin immunolabelling correlates with depth of invasion in gastrointestinal endocrine cell tumors. Acta Histochem. 2010, 112, 101–106. [Google Scholar] [CrossRef]
- Zhuo, J.; Tan, E.H.; Yan, B.; Tochhawng, L.; Jayapal, M.; Koh, S.; Tay, H.K.; Maciver, S.K.; Hooi, S.C.; Salto-Tellez, M.; et al. Gelsolin induces colorectal tumor cell invasion via modulation of the urokinase-type plasminogen activator cascade. PLoS ONE 2012, 7, e43594. [Google Scholar] [CrossRef]
- Tochhawng, L.; Deng, S.; Pugalenthi, G.; Kumar, A.P.; Lim, K.H.; Tan, T.Z.; Yang, H.; Hooi, S.C.; Goh, Y.C.; Maciver, S.K.; et al. Gelsolin-Cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget 2016, 7, 52832–52848. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Invest. 2009, 119, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Desouza, M.; Gunning, P.W.; Stehn, J.R. The actin cytoskeleton as a sensor and mediator of apoptosis. Bioarchitecture 2012, 2, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.P.; Veltman, D.; Machesky, L.M. Actin-bundling proteins in cancer progression at a glance. J. Cell Sci. 2012, 125, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Zankov, D.P.; Ogita, H. Actin-tethered junctional complexes in angiogenesis and lymphangiogenesis in association with vascular endothelial growth factor. Biomed. Res. Int. 2015, 2015, 314178. [Google Scholar] [CrossRef]
- Ong, M.S.; Deng, S.; Halim, C.E.; Cai, W.; Tan, T.Z.; Huang, R.Y.; Sethi, G.; Hooi, S.C.; Kumar, A.P.; Yap, C.T. Cytoskeletal Proteins in Cancer and Intracellular Stress: A Therapeutic Perspective. Cancers 2020, 12, 238. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielinska, W.; Halas-Wisniewska, M.; Grzanka, A. Involvement of Actin and Actin-Binding Proteins in Carcinogenesis. Cells 2020, 9, 2245. [Google Scholar] [CrossRef]
- Abedini, M.R.; Wang, P.W.; Huang, Y.F.; Cao, M.; Chou, C.Y.; Shieh, D.B.; Tsang, B.K. Cell fate regulation by gelsolin in human gynecologic cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 14442–14447. [Google Scholar] [CrossRef]
- Sun, M.Y.; Xu, B.; Wu, Q.X.; Chen, W.L.; Cai, S.; Zhang, H.; Tang, Q.F. Cisplatin-Resistant Gastric Cancer Cells Promote the Chemoresistance of Cisplatin-Sensitive Cells via the Exosomal RPS3-Mediated PI3K-Akt-Cofilin-1 Signaling Axis. Front. Cell Dev. Biol. 2021, 9, 618899. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, Y.; Bryce, N.S.; Tang, K.; Meagher, N.S.; Kang, E.Y.; Kelemen, L.E.; Köbel, M.; Ramus, S.J.; Friedlander, M.; et al. Targeting the actin/tropomyosin cytoskeleton in epithelial ovarian cancer reveals multiple mechanisms of synergy with anti-microtubule agents. Br. J. Cancer 2021, 125, 265–276. [Google Scholar] [CrossRef]
- Roque, D.M.; Bellone, S.; English, D.P.; Buza, N.; Cocco, E.; Gasparrini, S.; Bortolomai, I.; Ratner, E.; Silasi, D.A.; Azodi, M.; et al. Tubulin-β-III overexpression by uterine serous carcinomas is a marker for poor overall survival after platinum/taxane chemotherapy and sensitivity to epothilones. Cancer 2013, 119, 2582–2592. [Google Scholar] [CrossRef] [PubMed]
- Hari, M.; Loganzo, F.; Annable, T.; Tan, X.; Musto, S.; Morilla, D.B.; Nettles, J.H.; Snyder, J.P.; Greenberger, L.M. Paclitaxel-resistant cells have a mutation in the paclitaxel-binding region of beta-tubulin (Asp26Glu) and less stable microtubules. Mol. Cancer Ther. 2006, 5, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Martello, L.A.; Verdier-Pinard, P.; Shen, H.J.; He, L.; Torres, K.; Orr, G.A.; Horwitz, S.B. Elevated levels of microtubule destabilizing factors in a Taxol-resistant/dependent A549 cell line with an alpha-tubulin mutation. Cancer Res. 2003, 63, 1207–1213. [Google Scholar] [PubMed]
- Verdier-Pinard, P.; Wang, F.; Martello, L.; Burd, B.; Orr, G.A.; Horwitz, S.B. Analysis of tubulin isotypes and mutations from taxol-resistant cells by combined isoelectrofocusing and mass spectrometry. Biochemistry 2003, 42, 5349–5357. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Z.; Wang, L.; Lv, W.; Liu, J.; Ding, C.; Yuan, Y.; Lei, G.; Xu, C. Overexpression of stathmin is resistant to paclitaxel treatment in patients with non-small cell lung cancer. Tumour. Biol. 2015, 36, 7195–7204. [Google Scholar] [CrossRef]
- Jiménez, C.; Portela, R.A.; Mellado, M.; Rodríguez-Frade, J.M.; Collard, J.; Serrano, A.; Martínez, A.C.; Avila, J.; Carrera, A.C. Role of the PI3K regulatory subunit in the control of actin organization and cell migration. J. Cell Biol. 2000, 151, 249–262. [Google Scholar] [CrossRef]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef]
- Ebi, H.; Costa, C.; Faber, A.C.; Nishtala, M.; Kotani, H.; Juric, D.; Della Pelle, P.; Song, Y.; Yano, S.; Mino-Kenudson, M.; et al. PI3K regulates MEK/ERK signaling in breast cancer via the Rac-GEF, P-Rex1. Proc. Nat. Acad. Sci. USA 2013, 110, 21124–21129. [Google Scholar] [CrossRef]
- McCormick, B.; Chu, J.Y.; Vermeren, S. Cross-talk between Rho GTPases and PI3K in the neutrophil. Small GTPases 2019, 10, 187–195. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Bompard, G.; Caron, E. Regulation of WASP/WAVE proteins: Making a long story short. J. Cell Biol. 2004, 166, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac Take Center Stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 642–652. [Google Scholar] [CrossRef]
- Machesky, L.M. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 2008, 582, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Carmona, G.; Perera, U.; Gillett, C.; Naba, A.; Law, A.L.; Sharma, V.P.; Wang, J.; Wyckoff, J.; Balsamo, M.; Mosis, F.; et al. Lamellipodin promotes invasive 3D cancer cell migration via regulated interactions with Ena/VASP and SCAR/WAVE. Oncogene 2016, 35, 5155–5169. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Xu, R.; Du, J.; Hu, Z.; Yang, L.; Chen, Y.; Zhu, Y.; Gu, L. PI3K/Akt-dependent phosphorylation of GSK3β and activation of RhoA regulate Wnt5a-induced gastric cancer cell migration. Cell Signal 2013, 25, 447–456. [Google Scholar] [CrossRef]
- Garcin, C.; Straube, A. Microtubules in cell migration. Essays Biochem. 2019, 63, 509–520. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Microtubules in cell migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.; Yang, H.; Sharma, R.; Patel, K.D.; Cabral, F. The Role of Microtubules and Their Dynamics in Cell Migration. J. Biol. Chem. 2012, 287, 43359–43369. [Google Scholar] [CrossRef]
- Onishi, K.; Higuchi, M.; Asakura, T.; Masuyama, N.; Gotoh, Y. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes Cells 2007, 12, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Thapa, N.; Chen, M.; Horn, H.T.; Choi, S.; Wen, T.; Anderson, R.A. Phosphatidylinositol-3-OH kinase signalling is spatially organized at endosomal compartments by microtubule-associated protein 4. Nat. Cell Biol. 2020, 22, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Batrouni, A.G.; Baskin, J.M. A MAP for PI3K activation on endosomes. Nat. Cell Biol. 2020, 22, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C. Microtubule Formation in vitro in Solutions Containing Low Calcium Concentrations. Science 1972, 177, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Chaaban, S.; Brouhard, G.J. A microtubule bestiary: Structural diversity in tubulin polymers. Mol. Biol. Cell 2017, 28, 2924–2931. [Google Scholar] [CrossRef]
- Meiring, J.C.M.; Shneyer, B.I.; Akhmanova, A. Generation and regulation of microtubule network asymmetry to drive cell polarity. Curr. Opin. Cell Biol. 2020, 62, 86–95. [Google Scholar] [CrossRef]
- Mukherjee, A.; Brooks, P.S.; Bernard, F.; Guichet, A.; Conduit, P.T. Microtubules originate asymmetrically at the somatic golgi and are guided via Kinesin2 to maintain polarity within neurons. eLife 2020, 9, e58943. [Google Scholar] [CrossRef]
- Higuchi, M.; Masuyama, N.; Fukui, Y.; Suzuki, A.; Gotoh, Y. Akt mediates Rac/Cdc42-regulated cell motility in growth factor-stimulated cells and in invasive PTEN knockout cells. Curr. Biol. 2001, 11, 1958–1962. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Chun, C.; Takeda, K.; Firtel, R.A. Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J. Cell Biol. 2004, 167, 505–518. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Hosokawa, Y.; Watanabe, K.; Tanimura, S.; Ozaki, K.; Kohno, M. Blockade of the phosphatidylinositol-3-kinase-Akt signaling pathway enhances the induction of apoptosis by microtubule-destabilizing agents in tumor cells in which the pathway is constitutively activated. Mol. Cancer Ther. 2007, 6, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef]
- Kavallaris, M.; Tait, A.S.; Walsh, B.J.; He, L.; Horwitz, S.B.; Norris, M.D.; Haber, M. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001, 61, 5803–5809. [Google Scholar] [PubMed]
- Mozzetti, S.; Ferlini, C.; Concolino, P.; Filippetti, F.; Raspaglio, G.; Prislei, S.; Gallo, D.; Martinelli, E.; Ranelletti, F.O.; Ferrandina, G.; et al. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin. Cancer Res. 2005, 11, 298–305. [Google Scholar] [PubMed]
- Nishimura, Y.; Kasahara, K.; Inagaki, M. Intermediate filaments and IF-associated proteins: From cell architecture to cell proliferation. Proc. Jpn. Acad. Ser. B 2019, 95, 479–493. [Google Scholar] [CrossRef]
- Cheng, F.; Eriksson, J.E. Intermediate Filaments and the Regulation of Cell Motility during Regeneration and Wound Healing. Cold Spring Harb. Perspect Biol. 2017, 9, a022046. [Google Scholar] [CrossRef] [PubMed]
- Fortier, A.-M.; Van Themsche, C.; Asselin, É.; Cadrin, M. Akt isoforms regulate intermediate filament protein levels in epithelial carcinoma cells. FEBS Lett. 2010, 584, 984–988. [Google Scholar] [CrossRef]
- Weng, Y.-R.; Cui, Y.; Fang, J.-Y. Biological Functions of Cytokeratin 18 in Cancer. Mol. Cancer Res. 2012, 10, 485. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Matthias, C.; Mack, B.; Berghaus, A.; Gires, O. Keratin 8 expression in head and neck epithelia. BMC Cancer 2008, 8, 267. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, S.; Li, Y. KRT18 is correlated with the malignant status and acts as an oncogene in colorectal cancer. Biosci. Rep. 2019, 39, BSR20190884. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Trevor, K.T. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am. J. Pathol. 1997, 150, 483–495. [Google Scholar] [PubMed]
- Chu, Y.W.; Runyan, R.B.; Oshima, R.G.; Hendrix, M.J. Expression of complete keratin filaments in mouse L cells augments cell migration and invasion. Proc. Natl. Acad. Sci. USA 1993, 90, 4261–4265. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef]
- Cizmecioglu, O.; Ni, J.; Xie, S.; Zhao, J.J.; Roberts, T.M. Rac1-mediated membrane raft localization of PI3K/p110β is required for its activation by GPCRs or PTEN loss. eLife 2016, 5, e17635. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, J.; Wen, J.; Wang, Y.; Niu, W.; Teng, Z.; Zhao, T.; Dai, Y.; Zhang, Y.; Wang, C.; et al. CDC42 controls the activation of primordial follicles by regulating PI3K signaling in mouse oocytes. BMC Biol. 2018, 16, 73. [Google Scholar] [CrossRef]
- Li, Z.; Hannigan, M.; Mo, Z.; Liu, B.; Lu, W.; Wu, Y.; Smrcka, A.V.; Wu, G.; Li, L.; Liu, M.; et al. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell 2003, 114, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Wang, Z.; Liu, W.; Deng, N.; Ding, Y.; Tang, L.; Hla, T.; Zeng, R.; Li, L.; et al. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005, 7, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Meyer, T. Synthetic activation of endogenous PI3K and Rac identifies an AND-gate switch for cell polarization and migration. PLoS ONE 2008, 3, e3068. [Google Scholar] [CrossRef]
- Wang, F.; Herzmark, P.; Weiner, O.D.; Srinivasan, S.; Servant, G.; Bourne, H.R. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 2002, 4, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Weiner, O.D.; Neilsen, P.O.; Prestwich, G.D.; Kirschner, M.W.; Cantley, L.C.; Bourne, H.R. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 2002, 4, 509–513. [Google Scholar] [CrossRef]
- Srinivasan, S.; Wang, F.; Glavas, S.; Ott, A.; Hofmann, F.; Aktories, K.; Kalman, D.; Bourne, H.R. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 2003, 160, 375–385. [Google Scholar] [CrossRef]
- Huang, B.; Deng, S.; Loo, S.Y.; Datta, A.; Yap, Y.L.; Yan, B.; Ooi, C.H.; Dinh, T.D.; Zhuo, J.; Tochhawng, L.; et al. Gelsolin-mediated activation of PI3K/Akt pathway is crucial for hepatocyte growth factor-induced cell scattering in gastric carcinoma. Oncotarget 2016, 7, 25391–25407. [Google Scholar] [CrossRef]
- Yu, Y.; Xiong, Y.; Ladeiras, D.; Yang, Z.; Ming, X.F. Myosin 1b Regulates Nuclear AKT Activation by Preventing Localization of PTEN in the Nucleus. iScience 2019, 19, 39–53. [Google Scholar] [CrossRef]
- Choi, C.; Kwon, J.; Lim, S.; Helfman, D.M. Integrin β1, myosin light chain kinase and myosin IIA are required for activation of PI3K-AKT signaling following MEK inhibition in metastatic triple negative breast cancer. Oncotarget 2016, 7, 63466–63487. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, J.; Huang, Y.; Li, W.; Cheng, H. MYO18B promotes hepatocellular carcinoma progression by activating PI3K/AKT/mTOR signaling pathway. Diagn. Pathol. 2018, 13, 85. [Google Scholar] [CrossRef]
- Naguib, A.; Bencze, G.; Cho, H.; Zheng, W.; Tocilj, A.; Elkayam, E.; Faehnle, C.R.; Jaber, N.; Pratt, C.P.; Chen, M.; et al. PTEN functions by recruitment to cytoplasmic vesicles. Mol. Cell 2015, 58, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Matos, J.; Clark, J.L.; Piper, A.J.; Janusis, J.; Shaw, L.M. Differential involvement of the microtubule cytoskeleton in insulin receptor substrate 1 (IRS-1) and IRS-2 signaling to AKT determines the response to microtubule disruption in breast carcinoma cells. J. Biol. Chem. 2017, 292, 7806–7816. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Cheung, S.C.; Lan, L.L.; Ho, S.K.; Chan, J.C.; Tong, P.C. Microtubule network is required for insulin-induced signal transduction and actin remodeling. Mol. Cell Endocrinol. 2013, 365, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Loison, F.; Luo, H.R. Microtubule dynamics regulates Akt signaling via dynactin p150. Cell Signal 2014, 26, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Kumar, S.; Zaman, N.; Pan, C.C.; Bloodworth, J.C.; Lei, W.; Streicher, J.M.; Hempel, N.; Mythreye, K.; Lee, N.Y. TAK1 activation of alpha-TAT1 and microtubule hyperacetylation control AKT signaling and cell growth. Nat. Commun. 2018, 9, 1696. [Google Scholar] [CrossRef]
- Yan, X.; Yang, C.; Hu, W.; Chen, T.; Wang, Q.; Pan, F.; Qiu, B.; Tang, B. Knockdown of KRT17 decreases osteosarcoma cell proliferation and the Warburg effect via the AKT/mTOR/HIF1α pathway. Oncol. Rep. 2020, 44, 103–114. [Google Scholar] [CrossRef]
- Li, C.; Su, H.; Ruan, C.; Li, X. Keratin 17 knockdown suppressed malignancy and cisplatin tolerance of bladder cancer cells, as well as the activation of AKT and ERK pathway. Folia Histochem. Cytobiol. 2021, 59, 40–48. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, S.; Ye, S.; Shen, Z.; Gao, L.; Han, Z.; Zhang, P.; Luo, F.; Chen, S.; Kang, M. Keratin 17 activates AKT signalling and induces epithelial-mesenchymal transition in oesophageal squamous cell carcinoma. J. Proteom. 2020, 211, 103557. [Google Scholar] [CrossRef]
- Sankar, S.; Tanner, J.M.; Bell, R.; Chaturvedi, A.; Randall, R.L.; Beckerle, M.C.; Lessnick, S.L. A novel role for keratin 17 in coordinating oncogenic transformation and cellular adhesion in Ewing sarcoma. Mol. Cell Biol. 2013, 33, 4448–4460. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Liu, Y.; Wang, R.; Liao, J.; Wu, S.; Fan, J.; Peng, Z.; Li, B.; Wang, Z. Keratin 80 promotes migration and invasion of colorectal carcinoma by interacting with PRKDC via activating the AKT pathway. Cell Death Dis. 2018, 9, 1009. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.H.; Yang, W.; Lee, K.M.; Oh, S.; Nam, K.; Shim, S.; Shin, S.Y.; Gye, M.C.; Chu, I.S.; Shin, I. Regulation of cell proliferation and migration by keratin19-induced nuclear import of early growth response-1 in breast cancer cells. Clin. Cancer Res. 2013, 19, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.; Loranger, A.; Lavoie, J.N.; Marceau, N. Keratin 8/18 regulation of insulin receptor signaling and trafficking in hepatocytes through a concerted phosphoinositide-dependent Akt and Rab5 modulation. FASEB J. 2017, 31, 3555–3573. [Google Scholar] [CrossRef] [PubMed]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef]
- Deng, M.; Zhang, W.; Tang, H.; Ye, Q.; Liao, Q.; Zhou, Y.; Wu, M.; Xiong, W.; Zheng, Y.; Guo, X.; et al. Lactotransferrin acts as a tumor suppressor in nasopharyngeal carcinoma by repressing AKT through multiple mechanisms. Oncogene 2013, 32, 4273–4283. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Kim, S.; Yoon, H.N.; Ku, N.O. Keratin 8/18 Regulate the Akt Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 9227. [Google Scholar] [CrossRef]
- Kim, S.; Wong, P.; Coulombe, P.A. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006, 441, 362–365. [Google Scholar] [CrossRef]
- Tzivion, G.; Luo, Z.J.; Avruch, J. Calyculin A-induced vimentin phosphorylation sequesters 14-3-3 and displaces other 14-3-3 partners in vivo. J. Biol. Chem. 2000, 275, 29772–29778. [Google Scholar] [CrossRef]
- Kong, L.; Schäfer, G.; Bu, H.; Zhang, Y.; Klocker, H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 2012, 33, 751–759. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Tarantelli, C.; Lupia, A.; Stathis, A.; Bertoni, F. Is There a Role for Dual PI3K/mTOR Inhibitors for Patients Affected with Lymphoma? Int. J. Mol. Sci. 2020, 21, 1060. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, K.; Jokar, M.H.; Sedighi, S.; Moradzadeh, M. Therapeutic Potency of PI3K Pharmacological Inhibitors of Gastrointestinal Cancer. Middle East J. Dig. Dis. 2019, 11, 5–16. [Google Scholar] [CrossRef]
- Cleary, J.M.; Shapiro, G.I. Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr. Oncol. Rep. 2010, 12, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Speranza, M.-C.; Nowicki, M.O.; Behera, P.; Cho, C.-F.; Chiocca, E.A.; Lawler, S.E. BKM-120 (Buparlisib): A Phosphatidyl-Inositol-3 Kinase Inhibitor with Anti-Invasive Properties in Glioblastoma. Sci. Rep. 2016, 6, 20189. [Google Scholar] [CrossRef]
- Criscitiello, C.; Viale, G.; Curigliano, G.; Goldhirsch, A. Profile of buparlisib and its potential in the treatment of breast cancer: Evidence to date. Breast Cancer 2018, 10, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Yang, J.; Gu, Y.; Yi, J. Research update on the anticancer effects of buparlisib (Review). Oncol. Lett. 2021, 21, 266. [Google Scholar] [CrossRef]
- Rodon, J.; Braña, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Taylor, M.H.; Spitz, D.; Schwartzberg, L.; Beck, J.T.; Bauer, T.M.; Meric-Bernstam, F.; Purkayastha, D.; Karpiak, L.; Szpakowski, S.; et al. Efficacy and safety of buparlisib, a PI3K inhibitor, in patients with malignancies harboring a PI3K pathway activation: A phase 2, open-label, single-arm study. Oncotarget 2019, 10, 6526–6535. [Google Scholar] [CrossRef]
- McRee, A.J.; Sanoff, H.K.; Carlson, C.; Ivanova, A.; O’Neil, B.H. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Investig. New Drugs 2015, 33, 1225–1231. [Google Scholar] [CrossRef]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef] [PubMed]
- Markman, B.; Dienstmann, R.; Tabernero, J. Targeting the PI3K/Akt/mTOR pathway—Beyond rapalogs. Oncotarget 2010, 1, 530–543. [Google Scholar] [CrossRef]
- Vargaftig, J.; Farhat, H.; Ades, L.; Briaux, A.; Benoist, C.; Turbiez, I.; Vey, N.; Glaisner, S.; Callens, C. Phase 2 Trial of Single Agent Gedatolisib (PF-05212384), a Dual PI3K/mTOR Inhibitor, for Adverse Prognosis and Relapse/Refractory AML: Clinical and Transcriptomic Results. Blood 2018, 132, 5233. [Google Scholar] [CrossRef]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef]
- Colombo, I.; Genta, S.; Martorana, F.; Guidi, M.; Samartzis, E.S.P.; Brandt, S.; Gaggetta, S.; Moser, L.; Pascale, M.R.; Terrot, T.; et al. 568P Phase I dose-escalation study of the dual PI3K/mTORC1/2 inhibitor Gedatolisib (PF-05212384) in combination with paclitaxel (P) and carboplatin (C) in patients (pts) with advanced solid tumours. Ann. Oncol. 2020, 31, S487. [Google Scholar] [CrossRef]
- Wang, X.; Ding, J.; Meng, L.-h. PI3K isoform-selective inhibitors: Next-generation targeted cancer therapies. Acta Pharmacol. Sin. 2015, 36, 1170–1176. [Google Scholar] [CrossRef]
- Miller, B.W.; Przepiorka, D.; de Claro, R.A.; Lee, K.; Nie, L.; Simpson, N.; Gudi, R.; Saber, H.; Shord, S.; Bullock, J.; et al. FDA Approval: Idelalisib Monotherapy for the Treatment of Patients with Follicular Lymphoma and Small Lymphocytic Lymphoma. Clin. Cancer Res. 2015, 21, 1525. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Kahl, B.S.; Leonard, J.P.; Furman, R.R.; Brown, J.R.; Byrd, J.C.; Wagner-Johnston, N.D.; Coutre, S.E.; Benson, D.M.; Peterman, S.; et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood 2014, 123, 3406–3413. [Google Scholar] [CrossRef] [PubMed]
- Sharman, J.P.; Coutre, S.E.; Furman, R.R.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.W.; et al. Final Results of a Randomized, Phase III Study of Rituximab with or without Idelalisib Followed by Open-Label Idelalisib in Patients with Relapsed Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2019, 37, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Landel, I.; Quambusch, L.; Depta, L.; Rauh, D. Spotlight on AKT: Current Therapeutic Challenges. ACS Med. Chem. Lett. 2020, 11, 225–227. [Google Scholar] [CrossRef]
- Kumar, C.C.; Madison, V. AKT crystal structure and AKT-specific inhibitors. Oncogene 2005, 24, 7493–7501. [Google Scholar] [CrossRef]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Chen, J.; Wang, J.; Liu, J.; Jiang, Y. Targeting Akt in cancer for precision therapy. J. Hematol. Oncol. 2021, 14, 128. [Google Scholar] [CrossRef]
- Smyth, L.M.; Batist, G.; Meric-Bernstam, F.; Kabos, P.; Spanggaard, I.; Lluch, A.; Jhaveri, K.; Varga, A.; Wong, A.; Schram, A.M.; et al. Selective AKT kinase inhibitor capivasertib in combination with fulvestrant in PTEN-mutant ER-positive metastatic breast cancer. NPJ Breast Cancer 2021, 7, 44. [Google Scholar] [CrossRef]
- Jones, R.H.; Casbard, A.; Carucci, M.; Cox, C.; Butler, R.; Alchami, F.; Madden, T.A.; Bale, C.; Bezecny, P.; Joffe, J.; et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): A multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020, 21, 345–357. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef]
- Bang, Y.J.; Kang, Y.K.; Ng, M.; Chung, H.C.; Wainberg, Z.A.; Gendreau, S.; Chan, W.Y.; Xu, N.; Maslyar, D.; Meng, R.; et al. A phase II, randomised study of mFOLFOX6 with or without the Akt inhibitor ipatasertib in patients with locally advanced or metastatic gastric or gastroesophageal junction cancer. Eur. J. Cancer 2019, 108, 17–24. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): A multicentre, randomised, double-blind, phase 3 trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Zhao, Z.; Leister, W.H.; Robinson, R.G.; Barnett, S.F.; Defeo-Jones, D.; Jones, R.E.; Hartman, G.D.; Huff, J.R.; Huber, H.E.; et al. Allosteric Akt (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 761–764. [Google Scholar] [CrossRef]
- Schneeweiss, A.; Hess, D.; Joerger, M.; Varga, A.; Moulder, S.; Tsimberidou, A.M.; Ma, C.; Hurvitz, S.A.; Rentzsch, C.; Rudolph, M.; et al. Phase 1 Dose Escalation Study of the Allosteric AKT Inhibitor BAY 1125976 in Advanced Solid Cancer-Lack of Association between Activating AKT Mutation and AKT Inhibition-Derived Efficacy. Cancers 2019, 11, 1987. [Google Scholar] [CrossRef]
- Ma, C.X.; Suman, V.; Goetz, M.P.; Northfelt, D.; Burkard, M.E.; Ademuyiwa, F.; Naughton, M.; Margenthaler, J.; Aft, R.; Gray, R.; et al. A Phase II Trial of Neoadjuvant MK-2206, an AKT Inhibitor, with Anastrozole in Clinical Stage II or III. Clin. Cancer Res. 2017, 23, 6823–6832. [Google Scholar] [CrossRef]
- Chien, A.J.; Tripathy, D.; Albain, K.S.; Symmans, W.F.; Rugo, H.S.; Melisko, M.E.; Wallace, A.M.; Schwab, R.; Helsten, T.; Forero-Torres, A.; et al. MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients with Human Epidermal Growth Factor Receptor 2-Positive and/or Hormone Receptor-Negative Breast Cancers in the I-SPY 2 Trial. J. Clin. Oncol. 2020, 38, 1059–1069. [Google Scholar] [CrossRef]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Smith, J.A.; Slusher, B.S.; Wozniak, K.M.; Farah, M.H.; Smiyun, G.; Wilson, L.; Feinstein, S.; Jordan, M.A. Structural Basis for Induction of Peripheral Neuropathy by Microtubule-Targeting Cancer Drugs. Cancer Res. 2016, 76, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [Google Scholar] [PubMed]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Shetty, N.; Gupta, S. Eribulin drug review. South Asian J. Cancer 2014, 3, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open-label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Kaufman, P.A.; Awada, A.; Twelves, C.; Yelle, L.; Perez, E.A.; Velikova, G.; Olivo, M.S.; He, Y.; Dutcus, C.E.; Cortes, J. Phase III open-label randomized study of eribulin mesylate versus capecitabine in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 2015, 33, 594–601. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Barroso-Sousa, R.; Keenan, T.; Li, T.; Trippa, L.; Vaz-Luis, I.; Wulf, G.; Spring, L.; Sinclair, N.F.; Andrews, C.; et al. Effect of Eribulin with or without Pembrolizumab on Progression-Free Survival for Patients with Hormone Receptor-Positive, ERBB2-Negative Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1598–1605. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Kalinsky, K.; Kaklamani, V.G.; D’Adamo, D.R.; Aktan, G.; Tsai, M.L.; O’Regan, R.M.; Kaufman, P.A.; Wilks, S.T.; Andreopoulou, E.; et al. Eribulin Plus Pembrolizumab in Patients with Metastatic Triple-Negative Breast Cancer (ENHANCE 1): A Phase Ib/II Study. Clin. Cancer Res. 2021, 27, 3061–3068. [Google Scholar] [CrossRef]
- Akaiwa, M.; Martin, T.; Mendelsohn, B.A. Synthesis and Evaluation of Linear and Macrocyclic Dolastatin 10 Analogues Containing Pyrrolidine Ring Modifications. ACS Omega 2018, 3, 5212–5221. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Y.; Hua, H.; Li, D.; Tang, C. Marine Antitumor Peptide Dolastatin 10: Biological Activity, Structural Modification and Synthetic Chemistry. Mar Drugs 2021, 19. [Google Scholar] [CrossRef]
- Kindler, H.L.; Tothy, P.K.; Wolff, R.; McCormack, R.A.; Abbruzzese, J.L.; Mani, S.; Wade-Oliver, K.T.; Vokes, E.E. Phase II trials of dolastatin-10 in advanced pancreaticobiliary cancers. Investig. New Drugs 2005, 23, 489–493. [Google Scholar] [CrossRef]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investig. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef]
- Yardley, D.A.; Weaver, R.; Melisko, M.E.; Saleh, M.N.; Arena, F.P.; Forero, A.; Cigler, T.; Stopeck, A.; Citrin, D.; Oliff, I.; et al. EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB-Expressing Breast Cancer. J. Clin. Oncol. 2015, 33, 1609–1619. [Google Scholar] [CrossRef]
- Kopp, L.M.; Malempati, S.; Krailo, M.; Gao, Y.; Buxton, A.; Weigel, B.J.; Hawthorne, T.; Crowley, E.; Moscow, J.A.; Reid, J.M.; et al. Phase II trial of the glycoprotein non-metastatic B-targeted antibody-drug conjugate, glembatumumab vedotin (CDX-011), in recurrent osteosarcoma AOST1521: A report from the Children’s Oncology Group. Eur. J. Cancer 2019, 121, 177–183. [Google Scholar] [CrossRef]
- Ott, P.A.; Pavlick, A.C.; Johnson, D.B.; Hart, L.L.; Infante, J.R.; Luke, J.J.; Lutzky, J.; Rothschild, N.E.; Spitler, L.E.; Cowey, C.L.; et al. A phase 2 study of glembatumumab vedotin, an antibody-drug conjugate targeting glycoprotein NMB, in patients with advanced melanoma. Cancer 2019, 125, 1113–1123. [Google Scholar] [CrossRef]
- Vahdat, L.T.; Schmid, P.; Forero-Torres, A.; Blackwell, K.; Telli, M.L.; Melisko, M.; Möbus, V.; Cortes, J.; Montero, A.J.; Ma, C.; et al. Glembatumumab vedotin for patients with metastatic, gpNMB overexpressing, triple-negative breast cancer (“METRIC”): A randomized multicenter study. NPJ Breast Cancer 2021, 7, 57. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Noort, S.; Wander, P.; Alonzo, T.A.; Smith, J.; Ries, R.E.; Gerbing, R.B.; Dolman, M.E.M.; Locatelli, F.; Reinhardt, D.; Baruchel, A.; et al. The clinical and biological characteristics of NUP98-KDM5A in pediatric acute myeloid leukemia. Haematologica 2021, 106, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Granata, R.; Locati, L.D.; Licitra, L. Fosbretabulin for the treatment of anaplastic thyroid cancer. Future Oncol. 2014, 10, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef]
- Morgan, R.D.; Banerjee, S.; Hall, M.; Clamp, A.R.; Zhou, C.; Hasan, J.; Orbegoso, C.; Taylor, S.; Tugwood, J.; Lyon, A.R.; et al. Pazopanib and Fosbretabulin in recurrent ovarian cancer (PAZOFOS): A multi-centre, phase 1b and open-label, randomised phase 2 trial. Gynecol. Oncol. 2020, 156, 545–551. [Google Scholar] [CrossRef]
- Monk, B.J.; Sill, M.W.; Walker, J.L.; Darus, C.J.; Sutton, G.; Tewari, K.S.; Martin, L.P.; Schilder, J.M.; Coleman, R.L.; Balkissoon, J.; et al. Randomized Phase II Evaluation of Bevacizumab Versus Bevacizumab Plus Fosbretabulin in Recurrent Ovarian, Tubal, or Peritoneal Carcinoma: An NRG Oncology/Gynecologic Oncology Group Study. J. Clin. Oncol. 2016, 34, 2279–2286. [Google Scholar] [CrossRef]
- Sosa, J.A.; Elisei, R.; Jarzab, B.; Balkissoon, J.; Lu, S.P.; Bal, C.; Marur, S.; Gramza, A.; Yosef, R.B.; Gitlitz, B.; et al. Randomized safety and efficacy study of fosbretabulin with paclitaxel/carboplatin against anaplastic thyroid carcinoma. Thyroid 2014, 24, 232–240. [Google Scholar] [CrossRef]
- Cogle, C.R.; Collins, B.; Turner, D.; Pettiford, L.C.; Bossé, R.; Hawkins, K.E.; Beachamp, Z.; Wise, E.; Cline, C.; May, W.S.; et al. Safety, feasibility and preliminary efficacy of single agent combretastatin A1 diphosphate (OXi4503) in patients with relapsed or refractory acute myeloid leukemia or myelodysplastic syndromes. Br. J. Haematol. 2020, 189, e211–e213. [Google Scholar] [CrossRef]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T. NPI-2358 is a tubulin-depolymerizing agent: In-vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer. Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef]
- La Sala, G.; Olieric, N.; Sharma, A.; Viti, F.; de Asis Balaguer Perez, F.; Huang, L.; Tonra, J.R.; Lloyd, G.K.; Decherchi, S.; Díaz, J.F.; et al. Structure, Thermodynamics, and Kinetics of Plinabulin Binding to Two Tubulin Isotypes. Chem 2019, 5, 2969–2986. [Google Scholar] [CrossRef]
- Mita, M.M.; Spear, M.A.; Yee, L.K.; Mita, A.C.; Heath, E.I.; Papadopoulos, K.P.; Federico, K.C.; Reich, S.D.; Romero, O.; Malburg, L.; et al. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin(NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. 2010, 16, 5892–5899. [Google Scholar] [CrossRef] [PubMed]
- Blayney, D.W.; Zhang, Q.; Feng, J.; Zhao, Y.; Bondarenko, I.; Vynnychenko, I.; Kovalenko, N.; Nair, S.; Ibrahim, E.; Udovista, D.P.; et al. Efficacy of Plinabulin vs Pegfilgrastim for Prevention of Chemotherapy-Induced Neutropenia in Adults with Non-Small Cell Lung Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204429. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Broggini-Tenzer, A.; Vuong, V.; Messikommer, A.; Nytko, K.J.; Guckenberger, M.; Bachmann, F.; Lane, H.A.; Pruschy, M. The novel microtubule targeting agent BAL101553 in combination with radiotherapy in treatment-refractory tumor models. Radiother Oncol. 2017, 124, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Duran, G.E.; Lane, H.; Bachmann, F.; Sikic, B.I. Abstract 4412: In vitro activity of the novel tubulin active agent BAL27862 in MDR1(+) and MDR1(−) human breast and ovarian cancer variants selected for resistance to taxanes. Cancer Res. 2010, 70, 4412. [Google Scholar] [CrossRef]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Kristeleit, R.; Evans, J.; Molife, L.R.; Tunariu, N.; Shaw, H.; Slater, S.; Haris, N.R.M.; Brown, N.F.; Forster, M.D.; Diamantis, N.; et al. Phase 1/2a trial of intravenous BAL101553, a novel controller of the spindle assembly checkpoint, in advanced solid tumours. Br. J. Cancer 2020, 123, 1360–1369. [Google Scholar] [CrossRef]
- Alushin, G.M.; Lander, G.C.; Kellogg, E.H.; Zhang, R.; Baker, D.; Nogales, E. High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell 2014, 157, 1117–1129. [Google Scholar] [CrossRef]
- Cortes, J.; Roché, H. Docetaxel combined with targeted therapies in metastatic breast cancer. Cancer Treat Rev. 2012, 38, 387–396. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Mirzaei, S.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Saleki, H.; Sharifzadeh, S.O.; Soleymani, L.; Daneshi, S.; Hushmandi, K.; et al. New insight towards development of paclitaxel and docetaxel resistance in cancer cells: EMT as a novel molecular mechanism and therapeutic possibilities. Biomed. Pharm. 2021, 141, 111824. [Google Scholar] [CrossRef]
- Oudard, S.; Fizazi, K.; Sengeløv, L.; Daugaard, G.; Saad, F.; Hansen, S.; Hjälm-Eriksson, M.; Jassem, J.; Thiery-Vuillemin, A.; Caffo, O.; et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients with Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial-FIRSTANA. J. Clin. Oncol. 2017, 35, 3189–3197. [Google Scholar] [CrossRef]
- Baciarello, G.; Delva, R.; Gravis, G.; Tazi, Y.; Beuzeboc, P.; Gross-Goupil, M.; Bompas, E.; Joly, F.; Greilsamer, C.; Hon, T.N.T.; et al. Patient Preference Between Cabazitaxel and Docetaxel for First-line Chemotherapy in Metastatic Castration-resistant Prostate Cancer: The CABADOC Trial. Eur. Urol. 2022, 81, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ling, Y.; Guo, W.; Pang, J.; Liu, W.; Fang, Y.; Wen, X.; Wei, K.; Gao, X. Docetaxel loaded oleic acid-coated hydroxyapatite nanoparticles enhance the docetaxel-induced apoptosis through activation of caspase-2 in androgen independent prostate cancer cells. J. Control. Release 2010, 147, 278–288. [Google Scholar] [CrossRef]
- Miele, E.; Spinelli, G.P.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int. J. Nanomed. 2009, 4, 99–105. [Google Scholar] [CrossRef]
- Singer, J.W. Paclitaxel poliglumex (XYOTAX, CT-2103): A macromolecular taxane. J. Control. Release 2005, 109, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.L.; Savin, M.A.; Edelman, G.; Pippen, J.E.; Robert, N.J.; Geister, B.V.; Kirby, R.L.; Clawson, A.; O’Shaughnessy, J.A. Phase II study of weekly albumin-bound paclitaxel for patients with metastatic breast cancer heavily pretreated with taxanes. Clin. Breast Cancer 2007, 7, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [Google Scholar] [CrossRef]
- Xie, Y.; Gong, C.; Zhang, J.; Wang, L.; Cao, J.; Tao, Z.; Li, T.; Zhao, Y.; Li, Y.; Hu, S.; et al. Phase II trail of nab-paclitaxel in metastatic breast cancer patients with visceral metastases. BMC Cancer 2021, 21, 1174. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar]
- Giannakakou, P.; Gussio, R.; Nogales, E.; Downing, K.H.; Zaharevitz, D.; Bollbuck, B.; Poy, G.; Sackett, D.; Nicolaou, K.C.; Fojo, T. A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2904–2909. [Google Scholar] [CrossRef]
- Forli, S. Epothilones: From discovery to clinical trials. Curr. Top Med. Chem. 2014, 14, 2312–2321. [Google Scholar] [CrossRef]
- Lee, F.Y.; Borzilleri, R.; Fairchild, C.R.; Kim, S.H.; Long, B.H.; Reventos-Suarez, C.; Vite, G.D.; Rose, W.C.; Kramer, R.A. BMS-247550: A novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin. Cancer Res. 2001, 7, 1429–1437. [Google Scholar]
- Chou, T.C.; Zhang, X.G.; Balog, A.; Su, D.S.; Meng, D.; Savin, K.; Bertino, J.R.; Danishefsky, S.J. Desoxyepothilone B: An efficacious microtubule-targeted antitumor agent with a promising in vivo profile relative to epothilone B. Proc. Natl. Acad. Sci. USA 1998, 95, 9642–9647. [Google Scholar] [CrossRef]
- Newman, R.A.; Yang, J.; Raymond, M.; Finlay, V.; Cabral, F.; Vourloumis, D.; Stephens, L.C.; Troncoso, P.; Wu, X.; Logothetis, C.J.; et al. Antitumor efficacy of 26-fluoroepothilone B against human prostate cancer xenografts. Cancer Chemother. Pharmacol. 2001, 48, 319–326. [Google Scholar] [CrossRef]
- Ibrahim, N.K. Ixabepilone: Overview of Effectiveness, Safety, and Tolerability in Metastatic Breast Cancer. Front. Oncol. 2021, 11, 617874. [Google Scholar] [CrossRef]
- Lam, E.T.; Goel, S.; Schaaf, L.J.; Cropp, G.F.; Hannah, A.L.; Zhou, Y.; McCracken, B.; Haley, B.I.; Johnson, R.G.; Mani, S.; et al. Phase I dose escalation study of KOS-1584, a novel epothilone, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 69, 523–531. [Google Scholar] [CrossRef][Green Version]
- Campone, M.; Berton-Rigaud, D.; Joly-Lobbedez, F.; Baurain, J.F.; Rolland, F.; Stenzl, A.; Fabbro, M.; van Dijk, M.; Pinkert, J.; Schmelter, T.; et al. A double-blind, randomized phase II study to evaluate the safety and efficacy of acetyl-L-carnitine in the prevention of sagopilone-induced peripheral neuropathy. Oncologist 2013, 18, 1190–1191. [Google Scholar] [CrossRef]
- Thomas, E.S.; Gomez, H.L.; Li, R.K.; Chung, H.C.; Fein, L.E.; Chan, V.F.; Jassem, J.; Pivot, X.B.; Klimovsky, J.V.; de Mendoza, F.H.; et al. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J. Clin. Oncol. 2007, 25, 5210–5217. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Gomez, H.L.; Li, R.K.; Chung, H.C.; Fein, L.E.; Chan, V.F.; Jassem, J.; Lerzo, G.L.; Pivot, X.B.; Hurtado de Mendoza, F.; et al. Analysis of overall survival from a phase III study of ixabepilone plus capecitabine versus capecitabine in patients with MBC resistant to anthracyclines and taxanes. Breast Cancer Res. Treat 2010, 122, 409–418. [Google Scholar] [CrossRef]
- Sparano, J.A.; Vrdoljak, E.; Rixe, O.; Xu, B.; Manikhas, A.; Medina, C.; Da Costa, S.C.; Ro, J.; Rubio, G.; Rondinon, M.; et al. Randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 2010, 28, 3256–3263. [Google Scholar] [CrossRef]
- Maffei, R.; Fiorcari, S.; Martinelli, S.; Potenza, L.; Luppi, M.; Marasca, R. Targeting neoplastic B cells and harnessing microenvironment: The “double face” of ibrutinib and idelalisib. J. Hematol. Oncol. 2015, 8, 60. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Meadows, S.A.; Sivina, M.; Wierda, W.G.; Kantarjian, H.; Keating, M.J.; Giese, N.; O’Brien, S.; Yu, A.; Miller, L.L.; et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 2011, 118, 3603–3612. [Google Scholar] [CrossRef]
- Fiorcari, S.; Brown, W.S.; McIntyre, B.W.; Estrov, Z.; Maffei, R.; O’Brien, S.; Sivina, M.; Hoellenriegel, J.; Wierda, W.G.; Keating, M.J.; et al. The PI3-Kinase Delta Inhibitor Idelalisib (GS-1101) Targets Integrin-Mediated Adhesion of Chronic Lymphocytic Leukemia (CLL) Cell to Endothelial and Marrow Stromal Cells. PLoS ONE 2013, 8, e83830. [Google Scholar] [CrossRef]
- Song, J. Targeting epithelial-mesenchymal transition pathway in hepatocellular carcinoma. Clin. Mol. Hepatol. 2020, 26, 484–486. [Google Scholar] [CrossRef]
- Bohnacker, T.; Prota, A.E.; Beaufils, F.; Burke, J.E.; Melone, A.; Inglis, A.J.; Rageot, D.; Sele, A.M.; Cmiljanovic, V.; Cmiljanovic, N.; et al. Deconvolution of Buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nat. Commun. 2017, 8, 14683. [Google Scholar] [CrossRef]
- Henriques, A.C.; Ribeiro, D.; Pedrosa, J.; Sarmento, B.; Silva, P.M.A.; Bousbaa, H. Mitosis inhibitors in anticancer therapy: When blocking the exit becomes a solution. Cancer Lett. 2019, 440–441, 64–81. [Google Scholar] [CrossRef]
- Toyoshima, F.; Matsumura, S.; Morimoto, H.; Mitsushima, M.; Nishida, E. PtdIns(3,4,5)P3 regulates spindle orientation in adherent cells. Dev. Cell. 2007, 13, 796–811. [Google Scholar] [CrossRef]
- Silió, V.; Redondo-Muñoz, J.; Carrera, A.C. Phosphoinositide 3-kinase β regulates chromosome segregation in mitosis. Mol. Biol. Cell 2012, 23, 4526–4542. [Google Scholar] [CrossRef]
- Liu, X.; Shi, Y.; Woods, K.W.; Hessler, P.; Kroeger, P.; Wilsbacher, J.; Wang, J.; Wang, J.Y.; Li, C.; Li, Q.; et al. Akt inhibitor a-443654 interferes with mitotic progression by regulating aurora a kinase expression. Neoplasia 2008, 10, 828–837. [Google Scholar] [CrossRef]
- Leonard, M.K.; Hill, N.T.; Bubulya, P.A.; Kadakia, M.P. The PTEN-Akt pathway impacts the integrity and composition of mitotic centrosomes. Cell Cycle 2013, 12, 1406–1415. [Google Scholar] [CrossRef]
- Gulluni, F.; Martini, M.; De Santis, M.C.; Campa, C.C.; Ghigo, A.; Margaria, J.P.; Ciraolo, E.; Franco, I.; Ala, U.; Annaratone, L.; et al. Mitotic Spindle Assembly and Genomic Stability in Breast Cancer Require PI3K-C2α Scaffolding Function. Cancer Cell 2017, 32, 444–459.e447. [Google Scholar] [CrossRef]
- Loibl, S.; de la Pena, L.; Nekljudova, V.; Zardavas, D.; Michiels, S.; Denkert, C.; Rezai, M.; Bermejo, B.; Untch, M.; Lee, S.C.; et al. Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: A randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur. J. Cancer 2017, 85, 133–145. [Google Scholar] [CrossRef]
- Soulières, D.; Licitra, L.; Mesía, R.; Remenár, É.; Li, S.H.; Karpenko, A.; Chol, M.; Wang, Y.A.; Solovieff, N.; Bourdeau, L.; et al. Molecular Alterations and Buparlisib Efficacy in Patients with Squamous Cell Carcinoma of the Head and Neck: Biomarker Analysis from BERIL-1. Clin. Cancer Res. 2018, 24, 2505–2516. [Google Scholar] [CrossRef]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, S.; Leong, H.C.; Datta, A.; Gopal, V.; Kumar, A.P.; Yap, C.T. PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression. Cancers 2022, 14, 1652. https://doi.org/10.3390/cancers14071652
Deng S, Leong HC, Datta A, Gopal V, Kumar AP, Yap CT. PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression. Cancers. 2022; 14(7):1652. https://doi.org/10.3390/cancers14071652
Chicago/Turabian StyleDeng, Shuo, Hin Chong Leong, Arpita Datta, Vennila Gopal, Alan Prem Kumar, and Celestial T. Yap. 2022. "PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression" Cancers 14, no. 7: 1652. https://doi.org/10.3390/cancers14071652
APA StyleDeng, S., Leong, H. C., Datta, A., Gopal, V., Kumar, A. P., & Yap, C. T. (2022). PI3K/AKT Signaling Tips the Balance of Cytoskeletal Forces for Cancer Progression. Cancers, 14(7), 1652. https://doi.org/10.3390/cancers14071652