
CSA Antisense Targeting Enhances Anticancer Drug Sensitivity in Breast Cancer Cells, including the Triple-Negative Subtype

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Oligonucleotides Transfection
2.3. OXA, and PTX Treatment in Presence or Not of Oligonucleotide Antisense
2.4. Protein Expression Analysis by Western Blot
2.5. Retrotranscription and Real-Time Quantitative PCR
2.6. Cell Viability MTT Assay
2.7. Proliferation Tripan Blue Exclusion (TBE) Assay
2.8. Apoptosis Assay
2.9. Statistical Analysis
3. Results
3.1. CSA Gene Is Overexpressed in Breast Cancer Cells
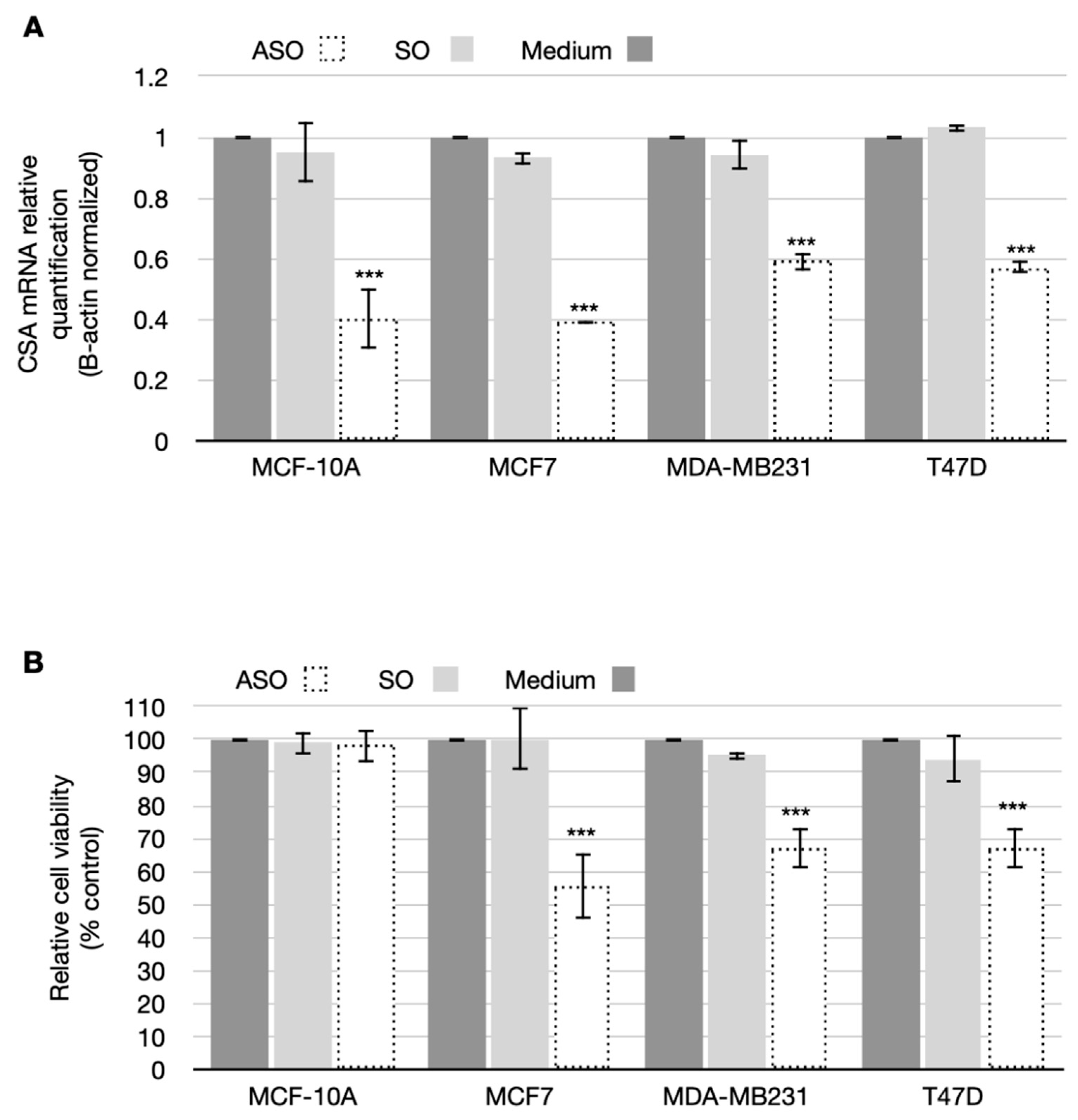
3.2. CSA Silencing Reduced Viability and Proliferation in BC Cell Lines
3.3. CSA Antisense Targeting Enhances Anticancer Drug Sensitivity in Breast Cancer Cell Lines
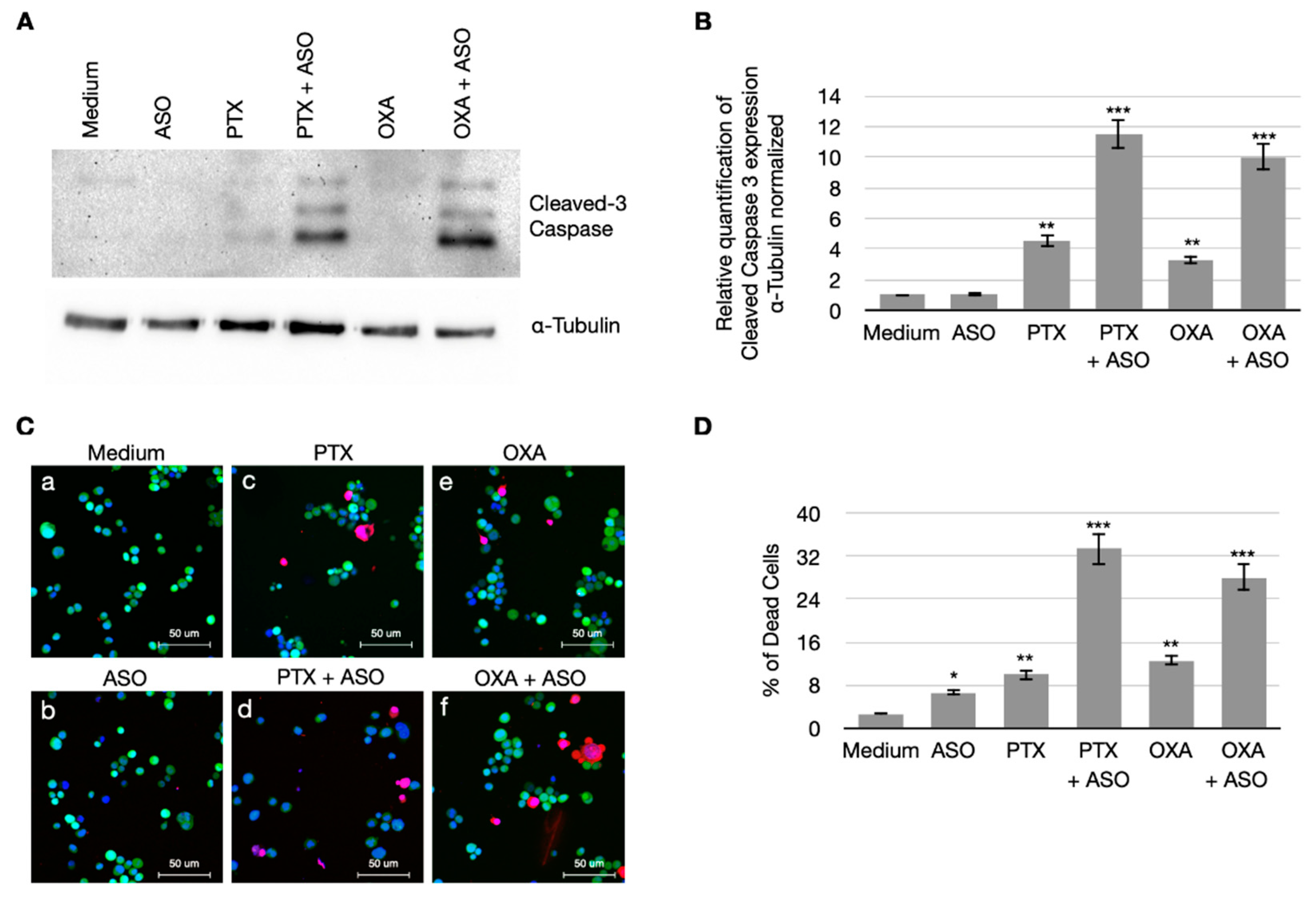
3.4. CSA Suppression Massively Induces Apoptosis in BC Cells When Treated with PTX and OXA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riaz, M.; Van Jaarsveld, M.T.; Hollestelle, A.; Prager-van Der Smissen, W.J.; Heine, A.A.; Boersma, A.W.; Liu, J.; Helmijr, J.; Ozturk, B.; Smid, M.; et al. miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation-specific miRNAs. Breast Cancer Res. 2013, 15, R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer (Unabridged Version). J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular Responses to Cisplatin-Induced DNA Damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef] [Green Version]
- Foland, T.B.; Dentler, W.L.; Suprenant, K.A.; Gupta, M.L., Jr.; Himes, R.H. Paclitaxel-induced microtubule stabilization causes mitotic block and apoptotic-like cell death in a paclitaxel-sensitive strain ofSaccharomyces cerevisiae. Yeast 2005, 22, 971–978. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of Taxane Resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F. The Multifunctions of WD40 Proteins in Genome Integrity and Cell Cycle Progression. J. Genom. 2015, 3, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.-I.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The Ubiquitin Ligase Activity in the DDB2 and CSA Complexes Is Differentially Regulated by the COP9 Signalosome in Response to DNA Damage. Cell 2003, 113, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.S.; Scrima, A.; Böhm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The Molecular Basis of CRL4DDB2/CSA Ubiquitin Ligase Architecture, Targeting, and Activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef]
- Beerens, N.; Hoeijmakers, J.H.J.; Kanaar, R.; Vermeulen, W.; Wyman, C. The CSB Protein Actively Wraps DNA. J. Biol. Chem. 2005, 280, 4722–4729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bregman, D.B.; Halaban, R.; van Gool, A.J.; Henning, K.A.; Friedberg, E.C.; Warren, S.L. UV-induced ubiquitination of RNA polymerase II: A novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11586–11590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svejstrup, J.Q. Rescue of arrested RNA polymerase II complexes. J. Cell Sci. 2003, 116, 447–451. [Google Scholar] [CrossRef] [Green Version]
- Van Der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.; Heuvel, D.V.D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Epanchintsev, A.; Costanzo, F.; Rauschendorf, M.-A.; Caputo, M.; Ye, T.; Donnio, L.-M.; Proietti-De-Santis, L.; Coin, F.; Laugel, V.; Egly, J.-M. Cockayne’s Syndrome A and B Proteins Regulate Transcription Arrest after Genotoxic Stress by Promoting ATF3 Degradation. Mol. Cell 2017, 68, 1054–1066.e6. [Google Scholar] [CrossRef] [Green Version]
- Paccosi, E.; Costanzo, F.; Costantino, M.; Balzerano, A.; Monteonofrio, L.; Soddu, S.; Prantera, G.; Brancorsini, S.; Egly, J.-M.; Proietti-De-Santis, L. The Cockayne syndrome group A and B proteins are part of a ubiquitin–proteasome degradation complex regulating cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30498–30508. [Google Scholar] [CrossRef]
- Latini, P.; Frontini, M.; Caputo, M.; Gregan, J.; Cipak, L.; Filippi, S.; Kumar, V.; Velez-Cruz, R.; Stefanini, M.; Proietti-De-Santis, L. CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle 2011, 10, 3719–3730. [Google Scholar] [CrossRef] [Green Version]
- Paccosi, E.; Costantino, M.; Balzerano, A.; Filippi, S.; Brancorsini, S.; Proietti-De-Santis, L. Neuroblastoma Cells Depend on CSB for Faithful Execution of Cytokinesis and Survival. Int. J. Mol. Sci. 2021, 22, 10070. [Google Scholar] [CrossRef] [PubMed]
- Filippi, S.; Latini, P.; Frontini, M.; Palitti, F.; Egly, J.-M.; Proietti-De-Santis, L. CSB protein is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. EMBO J. 2008, 27, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, S.; Filippi, S.; Caputo, M.; Cipak, L.; Gregan, J.; Ammerer, G.; Frontini, M.; Willems, D.; Prantera, G.; Balajee, A.S.; et al. Identification of Novel Proteins Co-Purifying with Cockayne Syndrome Group B (CSB) Reveals Potential Roles for CSB in RNA Metabolism and Chromatin Dynamics. PLoS ONE 2015, 10, e0128558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, M.; Balzerano, A.; Arisi, I.; D’Onofrio, M.; Brandi, R.; Bongiorni, S.; Brancorsini, S.; Frontini, M.; Proietti-De-Santis, L. CSB ablation induced apoptosis is mediated by increased endoplasmic reticulum stress response. PLoS ONE 2017, 12, e0172399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; O’Donovan, N.; Stordal, B.; O’Donovan, N. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botta, L.; Filippi, S.; Bizzarri, B.M.; Meschini, R.; Caputo, M.; Proietti-De-Santis, L.; Iside, C.; Nebbioso, A.; Gualandi, G.; Saladino, R. Oxidative nucleophilic substitution selectively produces cambinol derivatives with antiproliferative activity on bladder cancer cell lines. Bioorganic Med. Chem. Lett. 2018, 29, 78–82. [Google Scholar] [CrossRef]
- Caputo, M.; Frontini, M.; Velez-Cruz, R.; Nicolai, S.; Prantera, G.; Proietti-De-Santis, L. The CSB repair factor is overexpressed in cancer cells, increases apoptotic resistance, and promotes tumor growth. DNA Repair 2013, 12, 293–299. [Google Scholar] [CrossRef] [Green Version]
- De Santis, L.P.; Garcia, C.L.; Balajee, A.S.; Calvo, G.T.B.; Bassi, L.; Palitti, F. Transcription coupled repair deficiency results in increased chromosomal aberrations and apoptotic death in the UV61 cell line, the Chinese hamster homologue of Cockayne’s syndrome B. Mutat. Res. Repair 2001, 485, 121–132. [Google Scholar] [CrossRef]
- Weerapreeyakul, N.; Junhom, C.; Barusrux, S.; Thitimetharoch, T. Induction of apoptosis in human hepatocellular carcinoma cells by extracts of Lannea coromandelica (Houtt.) Merr. and Diospyros castanea (Craib) Fletcher. Chin. Med. 2016, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. NMR illuminates intrinsic disorder. Curr. Opin. Struct. Biol. 2021, 70, 44–52. [Google Scholar] [CrossRef]
- Vavouri, T.; Semple, J.I.; Garcia-Verdugo, R.; Lehner, B. Intrinsic Protein Disorder and Interaction Promiscuity Are Widely Associated with Dosage Sensitivity. Cell 2009, 138, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Versatility from Protein Disorder. Science 2012, 337, 1460–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) OXA | ||
| Cell Line | IC50 (µM) | SI |
| MCF-10A | 42.10 ± 0.66 | |
| MCF-10A + ASO | 19.99 ± 1.0 | |
| MCF-7 | 56.85 ± 1.4 | 0.74 |
| MCF7 + ASO | 4 ± 0.04 | 4.99 |
| MDA-MB231 | 21.91 ± 0.46 | 1.92 |
| MDA-MB231 + ASO | 3.7 ± 0.29 | 5.4 |
| T47D | 67.35 ± 1.04 | 0.62 |
| T47D + ASO | 3.6 ± 0.02 | 5.5 |
| (B) PTX | ||
| Cell Line | IC50 (µM) | SI |
| MCF-10A | 0.51 ± 0.04 | |
| MCF-10A + ASO | 0.16 ± 0.05 | |
| MCF-7 | 0.08 ± 0.02 | 6.4 |
| MCF7 + ASO | 0.02 ± 0.001 | 8 |
| MDA-MB231 | 0.20 ± 0.04 | 2.55 |
| MDA-MB231 + ASO | 0.02 ± 0.002 | 8 |
| T47D | 0.15 ± 0.02 | 3.4 |
| T47D + ASO | 0.02 ± 0.003 | 8 |
| (C) OXA | ||
| Cell Line | IC50 (µM) | |
| MDA-MB321 | 21.91 ± 0.46 | |
| MDA-MB231resistant | 54.9 ± 0.56 | |
| MDA-MB231resistant + ASO | 22.90 ± 0.90 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippi, S.; Paccosi, E.; Balzerano, A.; Ferretti, M.; Poli, G.; Taborri, J.; Brancorsini, S.; Proietti-De-Santis, L. CSA Antisense Targeting Enhances Anticancer Drug Sensitivity in Breast Cancer Cells, including the Triple-Negative Subtype. Cancers 2022, 14, 1687. https://doi.org/10.3390/cancers14071687
Filippi S, Paccosi E, Balzerano A, Ferretti M, Poli G, Taborri J, Brancorsini S, Proietti-De-Santis L. CSA Antisense Targeting Enhances Anticancer Drug Sensitivity in Breast Cancer Cells, including the Triple-Negative Subtype. Cancers. 2022; 14(7):1687. https://doi.org/10.3390/cancers14071687
Chicago/Turabian StyleFilippi, Silvia, Elena Paccosi, Alessio Balzerano, Margherita Ferretti, Giulia Poli, Juri Taborri, Stefano Brancorsini, and Luca Proietti-De-Santis. 2022. "CSA Antisense Targeting Enhances Anticancer Drug Sensitivity in Breast Cancer Cells, including the Triple-Negative Subtype" Cancers 14, no. 7: 1687. https://doi.org/10.3390/cancers14071687