
Refining AML Treatment: The Role of Genetics in Response and Resistance Evaluation to New Agents

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
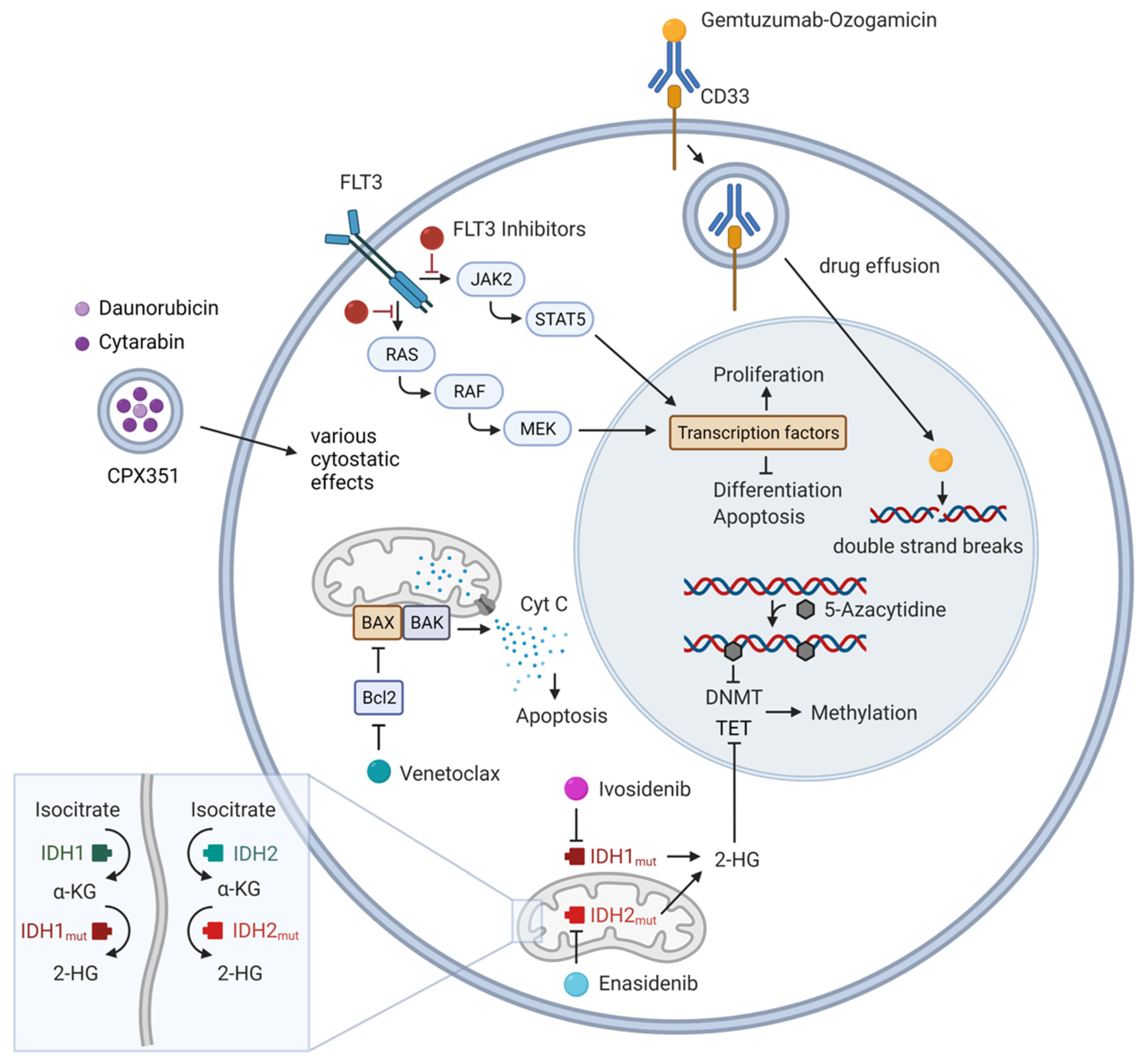
2. Targetable Gene Mutations
2.1. FLT3 Inhibitors
2.1.1. Midostaurin
2.1.2. Gilteritinib
2.1.3. Sorafenib
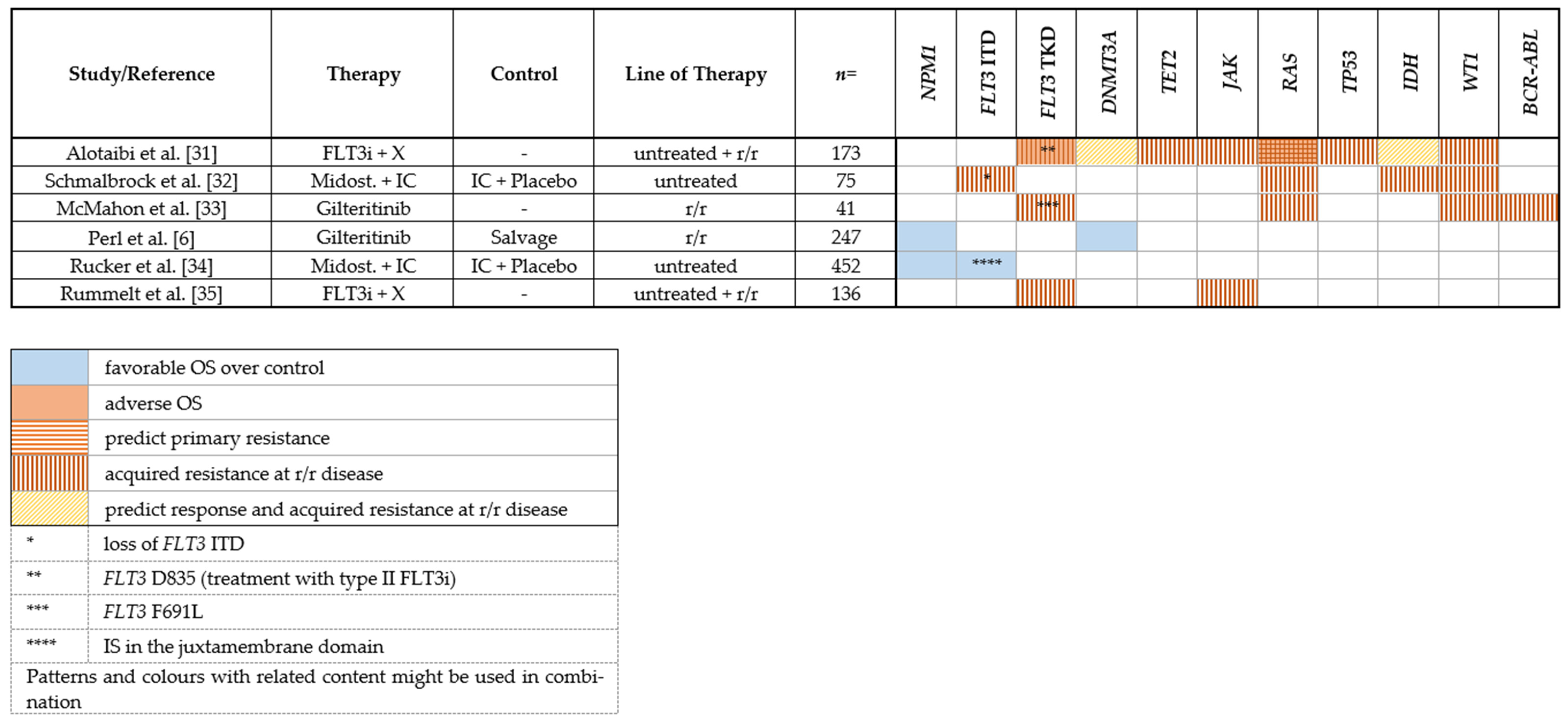
2.1.4. Mechanisms of Resistance to FLT3 Inhibitors
2.1.5. Clonal Evolution
2.1.6. On-Target Mutations
2.1.7. Off-Target Mutations
2.2. IDH Inhibitors
2.2.1. IDH1
2.2.2. IDH2
3. Hypomethylating Agents (HMAs)
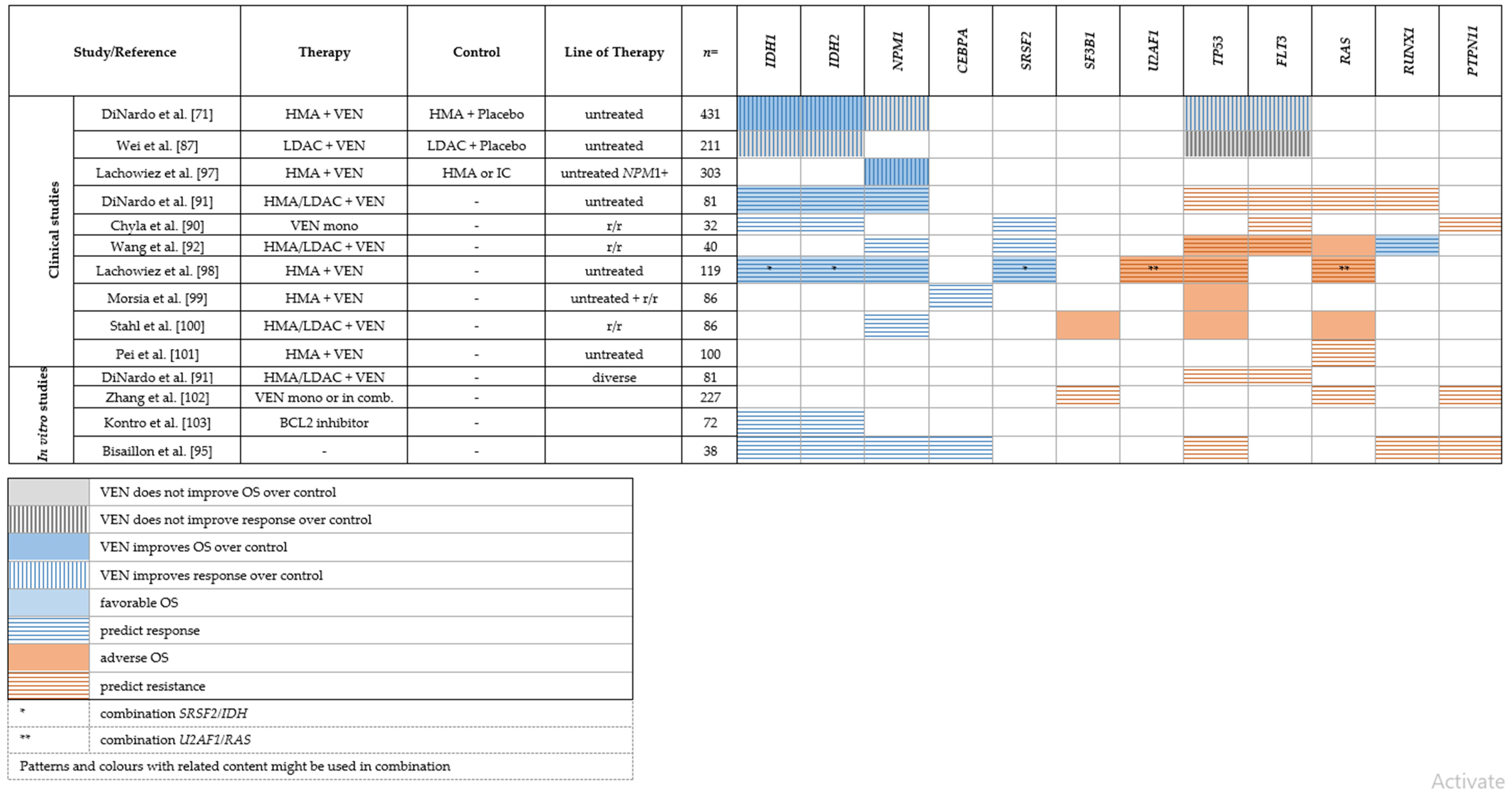
Genetic Predictors of Response or Resistance to HMA as Monotherapy
4. Other Molecular Targets
4.1. BCL-2/Venetoclax
Genetic Predictors of Response or Resistance to Venetoclax
4.2. Glasdegib/Inhibition of the Hedgehog Signaling Pathway
5. Antigen-Targeting
CD33/Gemtuzumab Ozogamicin
6. Novel Chemotherapeutic Agents
CPX-351
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nat. Cell Biol. 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Stein, E.M.; DE Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib inIDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2018, 130, 722–731. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients With Acute Myeloblastic Leukemia Who Benefit From the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2018, 33, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosnet, O.; Buhring, H.J.; Marchetto, S.; Rappold, I.; Lavagna, C.; Sainty, D.; Arnoulet, C.; Chabannon, C.; Kanz, L.; Hannum, C.; et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 1996, 10, 238–248. [Google Scholar] [PubMed]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef]
- Nagel, G.; German-Austrian AML Study Group (AMLSG); Weber, D.; Fromm, E.; Erhardt, S.; Lübbert, M.; Fiedler, W.; Kindler, T.; Krauter, J.; Brossart, P.; et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann. Hematol. 2017, 96, 1993–2003. [Google Scholar] [CrossRef] [Green Version]
- Herold, T.; Rothenberg-Thurley, M.; Grunwald, V.V.; Janke, H.; Goerlich, D.; Sauerland, M.C.; Konstandin, N.P.; Dufour, A.; Schneider, S.; Neusser, M.; et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia 2020, 34, 3161–3172. [Google Scholar] [CrossRef] [Green Version]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.-S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Propper, D.J.; McDonald, A.C.; Man, A.; Thavasu, P.; Balkwill, F.; Braybrooke, J.P.; Caponigro, F.; Graf, P.; Dutreix, C.; Blackie, R.; et al. Phase I and Pharmacokinetic Study of PKC412, an Inhibitor of Protein Kinase C. J. Clin. Oncol. 2001, 19, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Buchdunger, E.; Wood, J.; Mestan, J.; Hofmann, F.; Ferrari, S.; Mett, H.; O’Reilly, T.; Meyer, T. Inhibitors of Protein KinasesCGP 41251, a Protein Kinase Inhibitor with Potential as an Anticancer Agent. Pharmacol. Ther. 1999, 82, 293–301. [Google Scholar] [CrossRef]
- Ke, Y.-Y.; Singh, V.K.; Coumar, M.; Hsu, Y.C.; Wang, W.-C.; Song, J.-S.; Chen, C.-H.; Lin, W.-H.; Wu, S.-H.; Hsu, J.T.A.; et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci. Rep. 2015, 5, srep11702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef]
- Mori, M.; Kaneko, N.; Ueno, Y.; Yamada, M.; Tanaka, R.; Saito, R.; Shimada, I.; Mori, K.; Kuromitsu, S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. New Drugs 2017, 35, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Auclair, D.; Miller, D.; Yatsula, V.; Pickett, W.; Carter, C.; Chang, Y.; Zhang, X.; Wilkie, D.; Burd, A.; Shi, H.; et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia 2007, 21, 439–445. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3–Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, JCO1903345. [Google Scholar] [CrossRef]
- Xuan, L.; Wang, Y.; Huang, F.; Fan, Z.; Xu, Y.; Sun, J.; Xu, N.; Deng, L.; Li, X.; Liang, X.; et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: An open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020, 21, 1201–1212. [Google Scholar] [CrossRef]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2020, 2, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Du, L.; Luck, T.J.; Benner, A.; Krzykalla, J.; Gathmann, I.; Voso, M.T.; Amadori, S.; Prior, T.W.; Brandwein, J.M.; et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia 2021, 36, 90–99. [Google Scholar] [CrossRef]
- Rummelt, C.; Gorantla, S.P.; Meggendorfer, M.; Charlet, A.; Endres, C.; Döhner, K.; Heidel, F.H.; Fischer, T.; Haferlach, T.; Duyster, J.; et al. Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia 2020, 35, 2017–2029. [Google Scholar] [CrossRef]
- Chen, F.; Ishikawa, Y.; Akashi, A.; Naoe, T.; Kiyoi, H. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget 2016, 7, 47018–47032. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.-S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.H.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2005, 107, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.D.; Zimmerman, E.I.; Wang, Y.-D.; Orwick, S.; Zatechka, D.S.; Buaboonnam, J.; Neale, G.A.; Olsen, S.R.; Enemark, E.J.; Shurtleff, S.; et al. Emergence of Polyclonal FLT3 Tyrosine Kinase Domain Mutations during Sequential Therapy with Sorafenib and Sunitinib in FLT3-ITD–Positive Acute Myeloid Leukemia. Clin. Cancer Res. 2013, 19, 5758–5768. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, E.; Yoshida, K.; Frick, M.; Hoyer, K.; Christen, F.; Kaeda, J.; Obenaus, M.; Noerenberg, D.; Hennch, C.; Chan, W.; et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; A Eide, C.; Harlow, A.; Corbin, A.S.; Mauro, M.J.; Druker, B.; Corless, C.L.; Heinrich, M.; Deininger, M.W. An activating KRAS mutation in imatinib-resistant chronic myeloid leukemia. Leukemia 2008, 22, 2269–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauth, M.-T.; Alpermann, T.; Bacher, U.; Eder, C.; Dicker, F.; Ulke, M.; Kuznia, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; et al. WT1 mutations are secondary events in AML, show varying frequencies and impact on prognosis between genetic subgroups. Leukemia 2014, 29, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Whitman, S.P.; Mrózek, K.; Maharry, K.; Langer, C.; Baldus, C.D.; Zhao, W.; Powell, B.L.; et al. Wilms’ Tumor 1 Gene Mutations Independently Predict Poor Outcome in Adults With Cytogenetically Normal Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2008, 26, 4595–4602. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Schlenk, R.F.; Moschny, S.; Becker, A.; Bullinger, L.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; Döhner, H.; et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: A study of the German-Austrian AML Study Group. Blood 2009, 113, 4505–4511. [Google Scholar] [CrossRef] [Green Version]
- Damm, F.; Heuser, M.; Morgan, M.; Yun, H.; Großhennig, A.; Göhring, G.; Schlegelberger, B.; Döhner, K.; Ottmann, O.; Lübbert, M.; et al. Single Nucleotide Polymorphism in the Mutational Hotspot of WT1 Predicts a Favorable Outcome in Patients With Cytogenetically Normal Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 578–585. [Google Scholar] [CrossRef]
- Alotaibi, A.S.; Yilmaz, M.; Loghavi, S.; Dinardo, C.; Borthakur, G.; Kadia, T.M.; Thakral, B.; Pemmaraju, N.; Issa, G.C.; Konopleva, M.; et al. Emergence of BCR–ABL1 Fusion in AML Post–FLT3 Inhibitor-Based Therapy: A Potentially Targetable Mechanism of Resistance – A Case Series. Front. Oncol. 2020, 10, 588876. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744, Erratum in Nature 2010, 465, 966. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F.; et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2019, 37, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DelaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [Green Version]
- Roboz, G.J.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Choe, S.; Wang, H.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef] [Green Version]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.; Marteyn, B.; Farnoud, N.R.; DE Botton, S.; Bernard, O.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Fathi, A.T.; Pollyea, D.A.; Stone, R.M.; Altman, J.K.; Roboz, G.J.; Patel, M.R.; Collins, R.; Flinn, I.W.; et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 2019, 133, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Morita, K.; DiNardo, C.D.; Furudate, K.; Tanaka, T.; Yan, Y.; Patel, K.P.; MacBeth, K.J.; Wu, B.; Liu, G.; et al. Leukemia stemness and co-occurring mutations drive resistance to IDH inhibitors in acute myeloid leukemia. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Tallman, M.S.; De Botton, S.; Kantarjian, H.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, V.; Tiacci, E.; Holmes, A.B.; Kohlmann, A.; Martelli, M.P.; Kern, W.; Spanhol-Rosseto, A.; Klein, H.-U.; Dugas, M.; Schindela, S.; et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood 2011, 118, 6153–6163. [Google Scholar] [CrossRef] [Green Version]
- Damm, F.; Chesnais, V.; Nagata, Y.; Yoshida, K.; Scourzic, L.; Okuno, Y.; Itzykson, R.; Sanada, M.; Shiraishi, Y.; Gelsi-Boyer, V.; et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood 2013, 122, 3169–3177. [Google Scholar] [CrossRef] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Jasielec, J.; Saloura, V.; A Godley, L. The mechanistic role of DNA methylation in myeloid leukemogenesis. Leukemia 2014, 28, 1765–1773. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA Methylation Signatures Identify Biologically Distinct Subtypes in Acute Myeloid Leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.-H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Faramand, R.; Carter-Cooper, B.; Tolu, S.; Ford, L.A.; Lapidus, R.G.; Wetzler, M.; Wang, E.S.; Etemadi, A.; Griffiths, E.A. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am. J. Hematol. 2015, 90, E77–E79. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.L.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.J.; et al. DNMT3AMutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Damm, F.; Lüdeking, A.; Winschel, C.; Wagner, K.; Morgan, M.; Yun, H.; Göhring, G.; Schlegelberger, B.; Hoelzer, D.; et al. Incidence and Prognostic Influence of DNMT3A Mutations in Acute Myeloid Leukemia. J. Clin. Oncol. 2011, 29, 2889–2896. [Google Scholar] [CrossRef]
- Metzeler, K.; Walker, A.; Geyer, S.; Garzon, R.; Klisovic, R.B.; Bloomfield, C.D.; Blum, W.; Marcucci, G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia 2011, 26, 1106–1107. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Döhner, H.; Dolnik, A.; Tang, L.; Seymour, J.F.; Minden, M.D.; Stone, R.M.; Del Castillo, T.B.; Al-Ali, H.K.; Santini, V.; Vyas, P.; et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia 2018, 32, 2546–2557. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensi, L.; Longo, R.; Vecchi, A.; Messora, C.; Garagnani, L.; Bernardi, S.; Tamassia, M.G.; Sacchi, S. Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica 1995, 80, 98–102. [Google Scholar] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; DeBose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes On-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.-Z.; Fiedler, W.; Lin, T.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; Dinardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.C.; Roberts, A.W.; Reynolds, J.; Fong, C.Y.; Ting, S.B.; Salmon, J.M.; MacRaild, S.; Ivey, A.; Tiong, I.S.; Fleming, S.; et al. Chemotherapy and Venetoclax in Elderly Acute Myeloid Leukemia Trial (CAVEAT): A Phase Ib Dose-Escalation Study of Venetoclax Combined With Modified Intensive Chemotherapy. J. Clin. Oncol. 2020, 38, 3506–3517. [Google Scholar] [CrossRef]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am. J. Hematol. 2018, 93, E202–E205. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Tsai, C.; Lin, C.-C.; Tien, F.-M.; Chen, Y.-W.; Lin, H.-Y.; Yao, M.; Lin, Y.-C.; Cheng, C.-L.; Tang, J.-L.; et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann. Hematol. 2020, 99, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollyea, D.A.; DiNardo, C.D.; Arellano, M.L.; Pigneux, A.; Fiedler, W.; Konopleva, M.; Rizzieri, D.A.; Smith, B.D.; Shinagawa, A.; Lemoli, R.M.; et al. Impact of Venetoclax and Azacitidine in Treatment-Naïve Patients with Acute Myeloid Leukemia and IDH1/2 Mutations. Clin. Cancer Res. 2022. [Google Scholar] [CrossRef]
- Bisaillon, R.; Moison, C.; Thiollier, C.; Krosl, J.; Bordeleau, M.-E.; Lehnertz, B.; Lavallée, V.-P.; MacRae, T.; Mayotte, N.; Labelle, C.; et al. Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 2019, 34, 63–74. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Lachowiez, C.A.; Loghavi, S.; Kadia, T.M.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Naqvi, K.; Alvarado, Y.; Yilmaz, M.; Short, N.; et al. Outcomes of older patients with NPM1-mutated AML: Current treatments and the promise of venetoclax-based regimens. Blood Adv. 2020, 4, 1311–1320. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Furudate, K.; Montalban-Bravo, G.; Maiti, A.; Kadia, T.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Sasaki, K.; et al. Impact of splicing mutations in acute myeloid leukemia treated with hypomethylating agents combined with venetoclax. Blood Adv. 2021, 5, 2173–2183. [Google Scholar] [CrossRef]
- Morsia, E.; McCullough, K.; Joshi, M.; Cook, J.; Alkhateeb, H.B.; Al-Kali, A.; Begna, K.; Elliott, M.; Hogan, W.; Litzow, M.; et al. Venetoclax and hypomethylating agents in acute myeloid leukemia: Mayo Clinic series on 86 patients. Am. J. Hematol. 2020, 95, 1511–1521. [Google Scholar] [CrossRef]
- Stahl, M.; Menghrajani, K.; Derkach, A.; Chan, A.; Xiao, W.; Glass, J.; King, A.C.; Daniyan, A.F.; Famulare, C.; Cuello, B.M.; et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021, 5, 1552–1564. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Kontro, M.; Kumar, A.; Majumder, M.M.; Eldfors, S.; Parsons, A.; Pemovska, T.; Saarela, J.; Yadav, B.; Malani, D.; Fløisand, Y.; et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 2016, 31, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53 -mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Venugopal, S.; Shoukier, M.; Konopleva, M.; Dinardo, C.D.; Ravandi, F.; Short, N.J.; Andreeff, M.; Borthakur, G.; Daver, N.; Pemmaraju, N.; et al. Outcomes in patients with newly diagnosed TP53 -mutated acute myeloid leukemia with or without venetoclax-based therapy. Cancer 2021, 127, 3541–3551. [Google Scholar] [CrossRef]
- Brinton, L.T.; Zhang, P.; Williams, K.; Canfield, D.; Orwick, S.; Sher, S.; Wasmuth, R.; Beaver, L.; Cempre, C.; Skinner, J.; et al. Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Mali, R.S.; Zhang, Q.; DeFilippis, R.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2020, 106, 1034–1046. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Maiti, A.; Rausch, C.R.; Pemmaraju, N.; Naqvi, K.; Daver, N.G.; Kadia, T.M.; Borthakur, G.; Ohanian, M.; Alvarado, Y.; et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: A single-centre, phase 2 trial. Lancet Haematol. 2020, 7, e724–e736. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moujalled, D.M.; Pomilio, G.; Ghiurau, C.; Ivey, A.; Salmon, J.; Rijal, S.; MacRaild, S.; Zhang, L.; Teh, T.-C.; Tiong, I.-S.; et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 2018, 33, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Hormi, M.; Birsen, R.; Belhadj, M.; Huynh, T.; Aguilar, L.C.; Grignano, E.; Haddaoui, L.; Guillonneau, F.; Mayeux, P.; Hunault, M.; et al. Pairing MCL-1 inhibition with venetoclax improves therapeutic efficiency of BH3-mimetics in AML. Eur. J. Haematol. 2020, 105, 588–596. [Google Scholar] [CrossRef]
- Wang, Q.; Wan, J.; Zhang, W.; Hao, S. MCL-1 or BCL-xL-dependent resistance to the BCL-2 antagonist (ABT-199) can be overcome by specific inhibitor as single agents and in combination with ABT-199 in acute myeloid leukemia cells. Leuk. Lymphoma 2019, 60, 2170–2180. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- Han, L.; Zhang, Q.; Dail, M.; Shi, C.; Cavazos, A.; Ruvolo, V.R.; Zhao, Y.; Kim, E.; Rahmani, M.; Mak, D.H.; et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica 2019, 105, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2017, 32, 575–587. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Schuurhuis, G.J.; Kessler, F.L.; Piersma, S.R.; Knol, J.C.; Pham, T.V.; Jansen, G.; Musters, R.J.P.; van Meerloo, J.; Assaraf, Y.G.; et al. Exosomes Secreted by Apoptosis-Resistant Acute Myeloid Leukemia (AML) Blasts Harbor Regulatory Network Proteins Potentially Involved in Antagonism of Apoptosis. Mol. Cell. Proteom. 2016, 15, 1281–1298. [Google Scholar] [CrossRef] [Green Version]
- Mirfakhraie, R.; Noorazar, L.; Mohammadian, M.; Hajifathali, A.; Gholizadeh, M.; Salimi, M.; Sankanian, G.; Roshandel, E.; Mehdizadeh, M. Treatment Failure in Acute Myeloid Leukemia: Focus on the Role of Extracellular Vesicles. Leuk. Res. 2021, 112, 106751. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Matsui, W. Hedgehog Signaling in Myeloid Malignancies. Cancers 2021, 13, 4888. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Copland, M. Targeting hedgehog in hematologic malignancy. Blood 2012, 119, 2196–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, K.C.S.; Ruela-De-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.E.; Heidel, F.H.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Candoni, A.; Leber, B.; Sekeres, M.A.; Pollyea, D.A.; et al. Survival outcomes and clinical benefit in patients with acute myeloid leukemia treated with glasdegib and low-dose cytarabine according to response to therapy. J. Hematol. Oncol. 2020, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Smith, B.D.; Fiedler, W.; Sekeres, M.A.; Montesinos, P.; Leber, B.; Merchant, A.; Papayannidis, C.; Pérez-Simón, J.A.; Hoang, C.J.; et al. Clinical benefit of glasdegib plus low-dose cytarabine in patients with de novo and secondary acute myeloid leukemia: Long-term analysis of a phase II randomized trial. Ann. Hematol. 2021, 100, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Linenberger, M.L. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: Progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia 2004, 19, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Choice of Linker. Bioconjugate Chem. 2001, 13, 40–46. [Google Scholar] [CrossRef]
- Taksin, A.-L.; Legrand, O.; Raffoux, E.; De Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: A prospective study of the alfa group. Leukemia 2006, 21, 66–71. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2018, 104, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; on behalf of the UK NCRI-AML Study Group; Hills, R.K.; Virgo, P.; Couzens, S.; Clark, N.; Gilkes, A.; Richardson, P.; Knapper, S.; Grimwade, D.; et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia 2017, 31, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, E.; Duployez, N.; Ducourneau, B.; Raffoux, E.; Turlure, P.; Caillot, D.; Thomas, X.; Marceau-Renaut, A.; Chantepie, S.P.; Malfuson, J.-V.; et al. Mutational profile and benefit of gemtuzumab ozogamicin in acute myeloid leukemia. Blood 2020, 135, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.J.; Lancet, J.E.; Kolitz, J.E.; Ritchie, E.K.; Roboz, G.J.; List, A.; Allen, S.; Asatiani, E.; Mayer, L.D.; Swenson, C.; et al. First-In-Man Study of CPX-351: A Liposomal Carrier Containing Cytarabine and Daunorubicin in a Fixed 5:1 Molar Ratio for the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2011, 29, 979–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, E.; Kolitz, J.; Trang, J.; Liboiron, B.; Swenson, C.; Chiarella, M.; Mayer, L.; Louie, A.; Lancet, J. Pharmacokinetics of CPX-351; a nano-scale liposomal fixed molar ratio formulation of cytarabine:daunorubicin, in patients with advanced leukemia. Leuk. Res. 2012, 36, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Administration, U.S.F.a.D. Drug Approval Package: VYXEOS (daunorubicin and cytarabine). FDA webpage. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209401Orig1s000TOC.cfm (accessed on 11 February 2022).
- Agency, E.M. Vyxeos liposomal (previously known as Vyxeos) EMA 2018. Available online: https://www.ema.europa.eu/en/medicines/hu-1102man/EPAR/vyxeos-liposomal (accessed on 11 February 2022).
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Chiche, E.; Rahmé, R.; Bertoli, S.; Dumas, P.-Y.; Micol, J.-B.; Hicheri, Y.; Pasquier, F.; Peterlin, P.; Chevallier, P.; Thomas, X.; et al. Real-life experience with CPX-351 and impact on the outcome of high-risk AML patients: A multicentric French cohort. Blood Adv. 2021, 5, 176–184. [Google Scholar] [CrossRef]
- Rautenberg, C.; Stölzel, F.; Röllig, C.; Stelljes, M.; Gaidzik, V.; Lauseker, M.; Kriege, O.; Verbeek, M.; Unglaub, J.M.; Thol, F.; et al. Real-world experience of CPX-351 as first-line treatment for patients with acute myeloid leukemia. Blood Cancer J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Guolo, F.; Fianchi, L.; Minetto, P.; Clavio, M.; Gottardi, M.; Galimberti, S.; Rizzuto, G.; Rondoni, M.; Bertani, G.; Dargenio, M.; et al. CPX-351 treatment in secondary acute myeloblastic leukemia is effective and improves the feasibility of allogeneic stem cell transplantation: Results of the Italian compassionate use program. Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Cluzeau, T.; Loschi, M.; Fenaux, P.; Komrokji, R.; Sallman, D.A. Personalized Medicine for TP53 Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 10105. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Degtjarik, O.; Golovenko, D.; Diskin-Posner, Y.; Abrahmsén, L.; Rozenberg, H.; Shakked, Z. Structural basis of reactivation of oncogenic p53 mutants by a small molecule: Methylene quinuclidinone (MQ). Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Tashakori, M.; Yang, H.; Loghavi, S.; Wang, Y.; Wang, J.; Piya, S.; Borthakur, G. Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation. Cancers 2020, 12, 3511. [Google Scholar] [CrossRef]
- Tambe, M.; Karjalainen, E.; Vähä-Koskela, M.; Bulanova, D.; Gjertsen, B.T.; Kontro, M.; Porkka, K.; Heckman, C.A.; Wennerberg, K. Pan-RAF inhibition induces apoptosis in acute myeloid leukemia cells and synergizes with BCL2 inhibition. Leukemia 2020, 34, 3186–3196. [Google Scholar] [CrossRef]
- Morales, M.L.; Arenas, A.; Ortiz-Ruiz, A.; Leivas, A.; Rapado, I.; Rodríguez-García, A.; Castro, N.; Zagorac, I.; Quintela-Fandino, M.; López, G.G.; et al. MEK inhibition enhances the response to tyrosine kinase inhibitors in acute myeloid leukemia. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Li, Z.-Y.; Ding, M.; Liang, C.; Weng, X.-Q.; Sheng, Y.; Wu, J.; Cai, X. Trametinib enhances ATRA-induced differentiation in AML cells. Leuk. Lymphoma 2021, 1–12. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II Study of the Oral MEK Inhibitor Selumetinib in Advanced Acute Myelogenous Leukemia: A University of Chicago Phase II Consortium Trial. Clin. Cancer Res. 2013, 20, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 Inhibitor Trametinib in Combination With AKT Inhibitor GSK2141795 in Patients With Acute Myeloid Leukemia With RAS Mutations: A Phase II Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440. [Google Scholar] [CrossRef]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef]
- Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2019, 134, 570. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Lareau, C.A.; Chen, K.Y.; Zorzetto-Fernandes, A.L.; Hao, Y.; Takeshima, Y.; Luo, W.; Huang, T.-S.; Yeung, B.Z.; Papalexi, E.; et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 2021, 39, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Ludwig, L.S.; Muus, C.; Gohil, S.H.; Zhao, T.; Chiang, Z.; Pelka, K.; Verboon, J.M.; Luo, W.; Christian, E.; et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 2020, 39, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Story, B.A.; Hernández-Malmierca, P.; Raffel, S.; Leonce, D.R.; Milbank, J.; Paulsen, M.; Demir, A.; Szu-Tu, C.; Frömel, R.; et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stetson, L.C.; Balasubramanian, D.; Ribeiro, S.P.; Stefan, T.; Gupta, K.; Xu, X.; Fourati, S.; Roe, A.; Jackson, Z.; Schauner, R.; et al. Single cell RNA sequencing of AML initiating cells reveals RNA-based evolution during disease progression. Leukemia 2021, 35, 2799–2812. [Google Scholar] [CrossRef]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
 |
 |
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halik, A.; Arends, C.M.; Bullinger, L.; Damm, F.; Frick, M. Refining AML Treatment: The Role of Genetics in Response and Resistance Evaluation to New Agents. Cancers 2022, 14, 1689. https://doi.org/10.3390/cancers14071689
Halik A, Arends CM, Bullinger L, Damm F, Frick M. Refining AML Treatment: The Role of Genetics in Response and Resistance Evaluation to New Agents. Cancers. 2022; 14(7):1689. https://doi.org/10.3390/cancers14071689
Chicago/Turabian StyleHalik, Adriane, Christopher Maximilian Arends, Lars Bullinger, Frederik Damm, and Mareike Frick. 2022. "Refining AML Treatment: The Role of Genetics in Response and Resistance Evaluation to New Agents" Cancers 14, no. 7: 1689. https://doi.org/10.3390/cancers14071689