
Transcriptional CDK Inhibitors as Potential Treatment Option for Testicular Germ Cell Tumors

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Protein Extraction, SDS-PAGE and Western Blot
2.4. 7AAD/AnnexinV (Apoptosis) and Hoechst-FACS Analysis (Cell Cycle)
2.5. RNA Sequencing Analysis
2.6. Peptide Chip Array
3. Results
3.1. Cell Cycle and Transcriptional CDKs Are Expressed in TGCT Cell Lines
3.2. CDK Inhibitors Display Cytotoxic Effect on TGCT Cell Lines
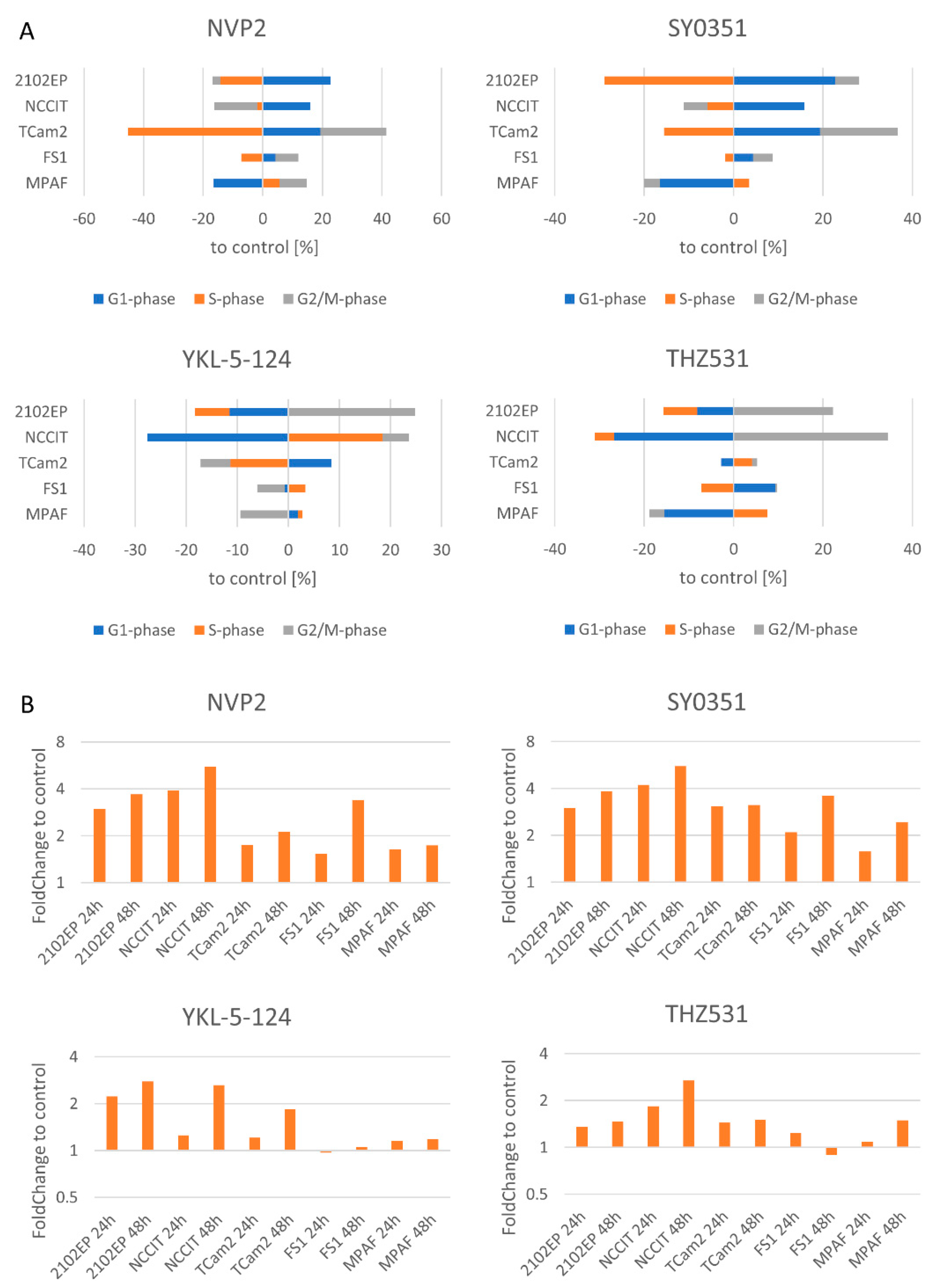
3.3. NVP2, SY0351, YKL-5-124 and THZ531 Affect Cell Cycle Progression and Induce Apoptosis in TGCT Cells
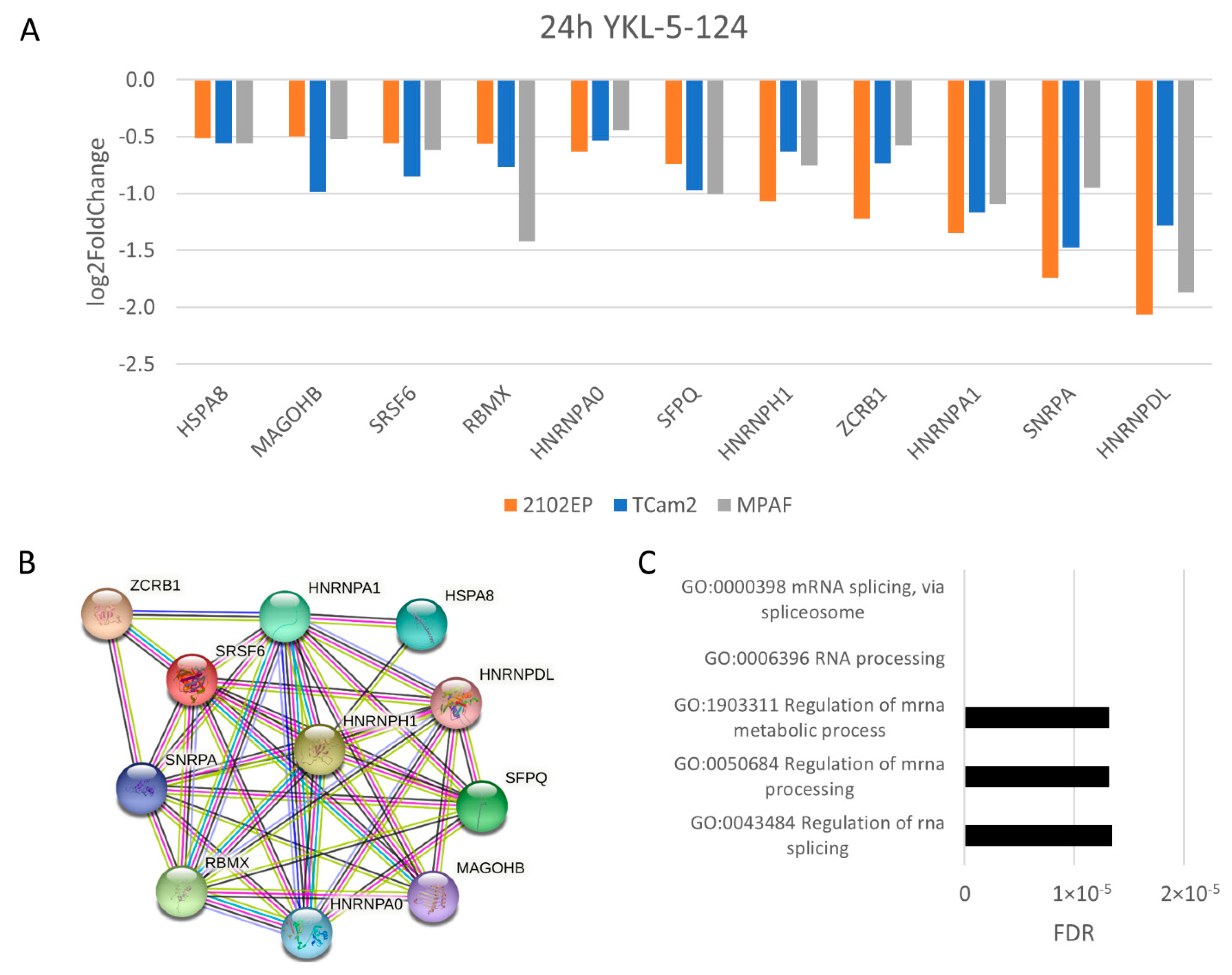
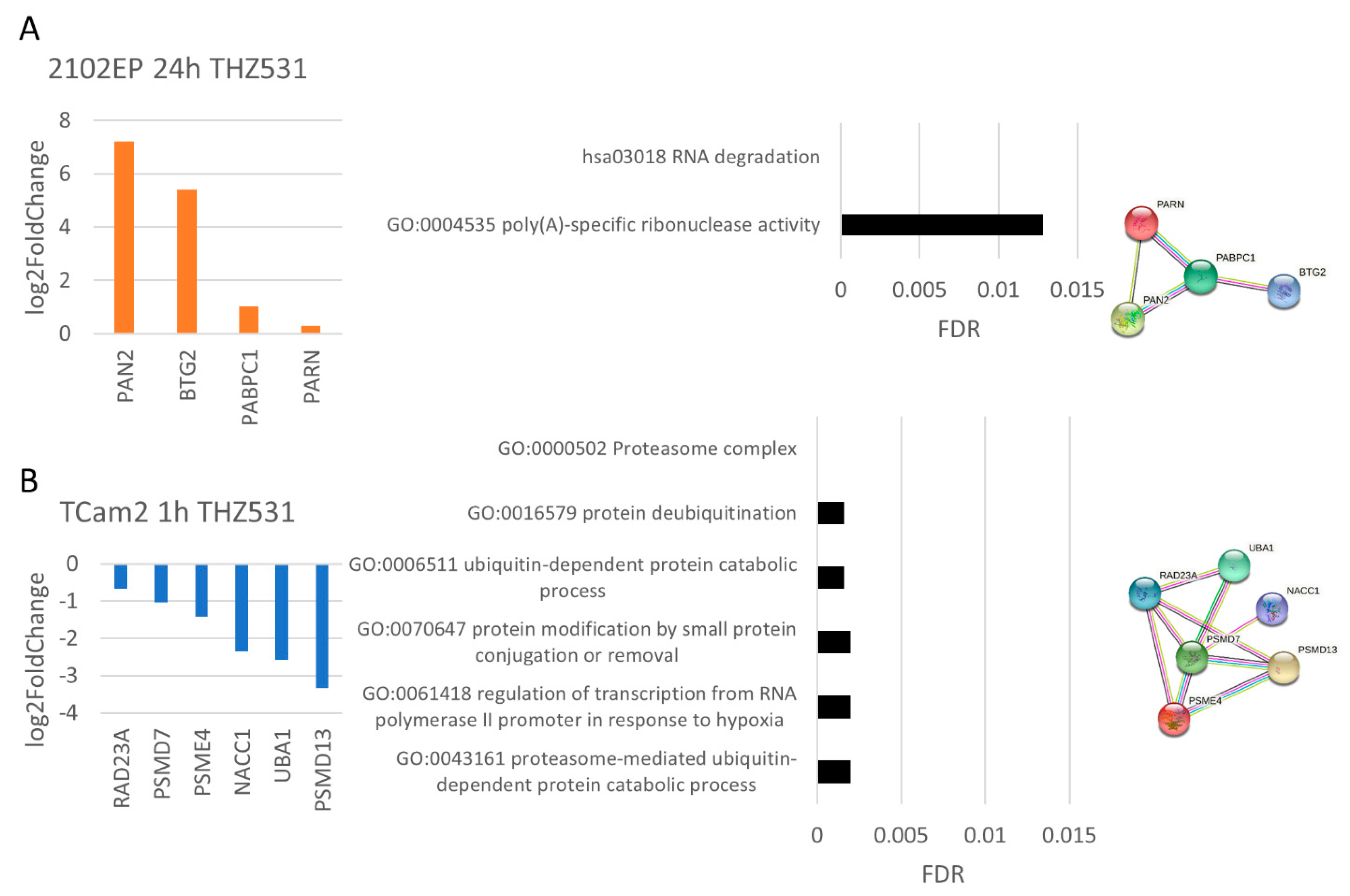
3.4. The Molecular Response Is Cell-Line-Specific for NVP2, SY0351 and THZ531 but Not for YKL-5-124 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes-Lattin, B.; Nichols, C.R. Testicular Cancer: A Prototypic Tumor of Young Adults. Semin. Oncol. 2009, 36, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oing, C.; Seidel, C.; Bokemeyer, C. Expert review of anticancer therapy therapeutic approaches for refractory germ cell cancer therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer Ther 2018, 18, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Albers, P.; Berney, D.M.; Feldman, D.R.; Daugaard, G.; Gilligan, T.; Looijenga, L. Testicular cancer. Nat. Rev. Dis. Prim. 2018, 4, 29. [Google Scholar] [CrossRef]
- Meyts, E.R.-D.; McGlynn, K.A.; Okamoto, K.; Jewett, M.A.S.; Bokemeyer, C. Testicular germ cell tumours. Lancet 2015, 387, 1762–1774. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L. Testicular germ-cell tumours in a broader perspective. Nat. Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Loehr, A.; Pierpont, T.; Gelsleichter, E.; Galang, A.; Fernandez, I.; Moore, E.; Guo, M.; Miller, A.; Weiss, R. Targeting Cancer Stem Cells with Differentiation Agents as an Alternative to Genotoxic Chemotherapy for the Treatment of Malignant Testicular Germ Cell Tumors. Cancers 2021, 13, 2045. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Buck Louis, G.M.; Toppari, J.; Andersson, A.-M.; Eisenberg, M.L.; Jensen, T.K.; Jørgensen, N.; Swan, S.H.; Sapra, K.J.; et al. Male Reproductive Disorders and Fertility Trends: Influences of Environment and Genetic Susceptibility. Physiol. Rev. 2016, 96, 55–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berney, D.M.; Looijenga, L.H.J.; Idrees, M.; Oosterhuis, J.W.; Rajpert-De Meyts, E.; Ulbright, T.M. Germ cell neoplasia in situ (GCNIS): Evolution of the current nomenclature for testicular pre-invasive germ cell malignancy. Histopathology 2016, 69, 7–10. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Schorle, H. Epigenetic drugs and their molecular targets in testicular germ cell tumours. Nat. Rev. Urol. 2019, 16, 245–259. [Google Scholar] [CrossRef]
- Oing, C.; Giannatempo, P.; Honecker, F.; Oechsle, K.; Bokemeyer, C.; Beyer, J. Palliative treatment of germ cell cancer. Cancer Treat. Rev. 2018, 71, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Oing, C.; Lorch, A. The Role of Salvage High-Dose Chemotherapy in Relapsed Male Germ Cell Tumors. Oncol. Res. Treat. 2018, 41, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schönberger, S.; Calaminus, G.; Köhrer, K.; Albers, P.; et al. CDK4/6 inhibition presents as a therapeutic option for paediatric and adult germ cell tumours and induces cell cycle arrest and apoptosis via canonical and non-canonical mechanisms. Br. J. Cancer 2020, 123, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Spangler, L.; Wang, X.; Conaway, J.W.; Conaway, R.C.; Dvir, A. TFIIH action in transcription initiation and promoter escape requires distinct regions of downstream promoter DNA. Proc. Natl. Acad. Sci. USA 2001, 98, 5544–5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serizawa, H.; Mäkelä, T.P.; Conaway, J.W.; Conaway, R.C.; Weinberg, R.A.; Young, R.A. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature 1995, 374, 280–282. [Google Scholar] [CrossRef]
- Wang, S.; Fischer, P. Cyclin-dependent kinase 9: A key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol. Sci. 2008, 29, 302–313. [Google Scholar] [CrossRef]
- Vervoort, S.J.; Devlin, J.R.; Kwiatkowski, N.; Teng, M.; Gray, N.S.; Johnstone, R.W. Targeting transcription cycles in cancer. Nat. Cancer 2021, 22, 5–24. [Google Scholar] [CrossRef]
- Dubbury, S.; Boutz, P.L.; Sharp, P.A. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018, 564, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.; Mueller, S.; Malenke, E.; Kuczyk, M.; Hartmann, J.T.; Bokemeyer, C. Induction of apoptosis by flavopiridol unrelated to cell cycle arrest in germ cell tumour derived cell lines. Investig. New Drugs 2005, 23, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.; Dickson, M.A.; Feldman, D.R.; Carvajal, R.D.; Shah, M.A.; Wu, N.; Lefkowitz, R.; Gonen, M.; Cane, L.M.; Dials, H.J.; et al. Phase I Study of Flavopiridol with Oxaliplatin and Fluorouracil/Leucovorin in Advanced Solid Tumors. Clin. Cancer Res. 2009, 15, 7405–7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-X.; Xie, F.-F.; Zhu, X.-J.; Lin, F.; Pan, S.-S.; Gong, L.-H.; Qiu, J.-G.; Zhang, W.-J.; Jiang, Q.-W.; Mei, X.-L.; et al. Cyclin-dependent kinase inhibitor dinaciclib potently synergizes with cisplatin in preclinical models of ovarian cancer. Oncotarget 2015, 6, 14926–14939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.-M.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.-L.; Small, K.; et al. Randomized Phase II Trial of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Capecitabine in Patients With Advanced Breast Cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef]
- Hu, S.; Marineau, J.J.; Rajagopal, N.; Hamman, K.B.; Choi, Y.J.; Schmidt, D.R.; Ke, N.; Johannessen, L.; Bradley, M.J.; Orlando, D.A.; et al. Discovery and Characterization of SY-1365, a Selective, Covalent Inhibitor of CDK7. Cancer Res. 2019, 79, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Rimel, J.K.; Poss, Z.C.; Erickson, B.; Maas, Z.L.; Ebmeier, C.C.; Johnson, J.L.; Decker, T.-M.; Yaron, T.M.; Bradley, M.J.; Hamman, K.B.; et al. Selective inhibition of CDK7 reveals high-confidence targets and new models for TFIIH function in transcription. Genes Dev. 2020, 34, 1452–1473. [Google Scholar] [CrossRef]
- Olson, C.M.; Liang, Y.; Leggett, A.; Park, W.D.; Li, L.; Mills, C.E.; Elsarrag, S.Z.; Ficarro, S.B.; Zhang, T.; Düster, R.; et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem. Biol. 2019, 26, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Christensen, C.L.; Dries, R.; Oser, M.G.; Deng, J.; Diskin, B.; Li, F.; Pan, Y.; Zhang, X.; Yin, Y.; et al. CDK7 Inhibition Potentiates Genome Instability Triggering Anti-tumor Immunity in Small Cell Lung Cancer. Cancer Cell 2019, 37, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019, 33, 5990–6000. [Google Scholar] [CrossRef]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocrine-Related Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [Green Version]
- Storch, K.; Cordes, N. The impact of CDK9 on radiosensitivity, DNA damage repair and cell cycling of HNSCC cancer cells. Int. J. Oncol. 2015, 48, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Tang, L.; Zhang, Q.; Zhang, Z.; Wei, W. MicroRNA-613 inhibits the progression of gastric cancer by targeting CDK9. Artif. Cells, Nanomedicine, Biotechnol. 2017, 46, 980–984. [Google Scholar] [CrossRef]
- Nettersheim, D.; Heukamp, L.; Fronhoffs, F.; Grewe, M.J.; Haas, N.; Waha, A.; Honecker, F.; Waha, A.; Kristiansen, G.; Schorle, H. Analysis of TET Expression/Activity and 5mC Oxidation during Normal and Malignant Germ Cell Development. PLoS ONE 2013, 8, e82881. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumoursin vitroandin vivo. J. Cell. Mol. Med. 2016, 21, 1300–1314. [Google Scholar] [CrossRef]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 24 March 2022).
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R.; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Hartline, E.; et al. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Research 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Research 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. (2007–2015) Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 24 March 2022).
- Schwill, M.; Tamaskovic, R.; Gajadhar, A.S.; Kast, F.; White, F.M.; Plückthun, A. Systemic analysis of tyrosine kinase signaling reveals a common adaptive response program in a HER2-positive breast cancer. Sci. Signal. 2019, 12, eaau2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, D.; Nettersheim, D.; Heukamp, L.; Kitazawa, S.; Biermann, K.; Schorle, H. TCam-2 but not JKT-1 cells resemble seminoma in cell culture. Cell Tissue Res. 2007, 331, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, Y.; Dai, H.; Zhang, H.; Wan, D.; Zhou, X.; Situ, C.; Zhu, H. Cyclin-dependent kinase 7 is essential for spermatogenesis by regulating retinoic acid signaling pathways and the STAT3 molecular pathway. IUBMB Life 2021, 73, 1446–1459. [Google Scholar] [CrossRef]
- Liang, S.; Hu, L.; Wu, Z.; Chen, Z.; Liu, S.; Xu, X.; Qian, A. CDK12: A Potent Target and Biomarker for Human Cancer Therapy. Cells 2020, 9, 1483. [Google Scholar] [CrossRef]
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline genome protection: Implications for gamete quality and germ cell tumorigenesis. Andrology 2019, 7, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Bird, G.; Zorio, D.A.R.; Bentley, D.L. RNA Polymerase II Carboxy-Terminal Domain Phosphorylation Is Required for Cotranscriptional Pre-mRNA Splicing and 3′-End Formation. Mol. Cell. Biol. 2004, 24, 8963–8969. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Poulose, N.; Girmay, S.; Helevä, A.; Doultsinos, D.; Gondane, A.; Steele, R.E.; Liu, X.; Loda, M.; Liu, S.; et al. Inhibition of CDK9 activity compromises global splicing in prostate cancer cells. RNA Biol. 2021, 18, 722–729. [Google Scholar] [CrossRef]
- Tellier, M.; Zaborowska, J.; Neve, J.; Nojima, T.; Hester, S.; Furger, A.; Murphy, S. CDK9 and PP2A regulate RNA polymerase II transcription termination and coupled RNA maturation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kalan, S.; Amat, R.; Schachter, M.M.; Kwiatkowski, N.; Abraham, B.; Liang, Y.; Zhang, T.; Olson, C.M.; Larochelle, S.; Young, R.A.; et al. Activation of the p53 Transcriptional Program Sensitizes Cancer Cells to Cdk7 Inhibitors. Cell Rep. 2017, 21, 467–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Kwiatkowski, N.P.; Zhang, T.; Nabet, B.; Xu, M.; Liang, Y.; Quan, C.; Wang, J.; Hao, M.; Palakurthi, S.; et al. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. eLife 2018, 7, e39030. [Google Scholar] [CrossRef] [PubMed]
- Marineau, J.J.; Hamman, K.B.; Hu, S.; Alnemy, S.; Mihalich, J.; Kabro, A.; Whitmore, K.M.; Winter, D.K.; Roy, S.; Ciblat, S.; et al. Discovery of SY-5609: A Selective, Noncovalent Inhibitor of CDK7. J. Med. Chem. 2021, 65, 1458–1480. [Google Scholar] [CrossRef]
- Patel, H.; Periyasamy, M.; Sava, G.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; et al. ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol. Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sava, G.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Bunch, H.; Choe, H.; Kim, J.; Jo, D.S.; Jeon, S.; Lee, S.; Cho, D.-H.; Kang, K. P-TEFb Regulates Transcriptional Activation in Non-coding RNA Genes. Front. Genet. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2019, 121, 109679. [Google Scholar] [CrossRef]
- Park, M.H.; Kim, S.Y.; Kim, Y.J.; Chung, Y.-H. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem. Biophys. Res. Commun. 2014, 450, 129–134. [Google Scholar] [CrossRef]
- Arrouchi, H.; Lakhlili, W.; Ibrahimi, A. A review on PIM kinases in tumors. Bioinformation 2019, 15, 40–45. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Company | Species | Dilution | Order No. |
|---|---|---|---|---|
| CDK7 | Invitrogen, Waltham, MA, USA | Rabbit | 1:1000 | PA5-34791 |
| CDK9 | Cell signaling, Danvers, MA, USA | Rabbit | 1:1000 | 2316T |
| CDK10 | Cell signaling, Danvers, MA, USA | Rabbit | 1:500 | 36106S |
| CDK12 | Merck, Darmstadt, Germany | Rabbit | 1:500 | ABE1861 |
| CDK13 | Antibodies-online.com (01/2022) | Rabbit | 1:1000 | ABIN6130965 |
| β-Actin | Merck, Darmstadt Germany | Mouse | 1:35,000 | a5441 |
| Anti-mouse HRP | Agilent Technologies (Dako), Santa Clara, CA, USA | Rabbit | 1:750 | P0260 |
| Anti-rabbit HRP | Agilent Technologies (Dako), Santa Clara, CA, USA | Goat | 1:2000 | P0447 |
| IC50 [nM] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cell Line and Treatment Time | NVP2 | SY0351 | YKL-5-124 | THZ531 | THZ1 | Dinaciclib | Flavo-Piridol | THAL-SNS-032 |
| 2102EP_24 h | 502.7 | 38.7 | 317.6 | >1000 | 95.6 | 76.8 | >1000 | 164.15 |
| 2102EP_48 h | 10.5 | 7.5 | 43.6 | 179.6 | 26.3 | 3.4 | 39.94 | 39.51 |
| 2102EP_72 h | 6.1 | 6.7 | 17.2 | 74.5 | 6.7 | 0.8 | 18.01 | 34.82 |
| 2102EP-R_24 h | 884.1 | 15.9 | >1000 | >1000 | 235.3 | 102.2 | >1000 | |
| 2102EP-R_48 h | 11.2 | 3.4 | 30.5 | 227.5 | 16.3 | 2.0 | 21.85 | |
| 2102EP-R_72 h | 8.6 | 6.4 | 23.6 | 163.9 | 9.9 | 1.1 | 7.56 | |
| NCCIT_24 h | 90.6 | 104.5 | >1000 | >1000 | >1000 | 593.5 | 710.47 | 149.39 |
| NCCIT_48 h | 40.7 | 19.1 | >1000 | >1000 | 92.9 | 21.8 | 21.85 | 50.62 |
| NCCIT_72 h | 6.5 | 9.0 | 121.1 | 96.6 | 9.3 | 1.5 | 34.07 | 30.09 |
| NCCIT-R_24 h | 103.7 | 24.9 | >1000 | >1000 | 929.4 | 314.8 | 638.83 | |
| NCCIT-R_48 h | 17.1 | 14.0 | >1000 | >1000 | 145.0 | 19.6 | 106.83 | |
| NCCIT-R_72 h | 12.2 | 11.6 | 178.1 | 137.2 | 16.2 | 4.3 | 43.28 | |
| TCam2_24 h | 215.7 | 21.6 | >1000 | >1000 | >1000 | 35.1 | >1000 | 139.83 |
| TCam2_48 h | 60.0 | 8.9 | >1000 | >1000 | >1000 | 11.5 | 581.81 | 76.70 |
| TCam2_72 h | 16.1 | 8.6 | 263.4 | >1000 | 847.0 | 1.6 | 272.74 | 39.57 |
| FS1_24 h | 98.2 | 85.4 | >1000 | >1000 | >1000 | 392.6 | >1000 | >1000 |
| FS1_48 h | 33.5 | 30.1 | >1000 | >1000 | >1000 | 85.7 | 695.75 | >1000 |
| FS1_72 h | 8.9 | 7.7 | 95.2 | >1000 | >1000 | 14.3 | 91.47 | >1000 |
| MPAF_24 h | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| MPAF_48 h | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| MPAF_72 h | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funke, K.; Düster, R.; Wilson, P.D.-G.; Arévalo, L.; Geyer, M.; Schorle, H. Transcriptional CDK Inhibitors as Potential Treatment Option for Testicular Germ Cell Tumors. Cancers 2022, 14, 1690. https://doi.org/10.3390/cancers14071690
Funke K, Düster R, Wilson PD-G, Arévalo L, Geyer M, Schorle H. Transcriptional CDK Inhibitors as Potential Treatment Option for Testicular Germ Cell Tumors. Cancers. 2022; 14(7):1690. https://doi.org/10.3390/cancers14071690
Chicago/Turabian StyleFunke, Kai, Robert Düster, Prince De-Graft Wilson, Lena Arévalo, Matthias Geyer, and Hubert Schorle. 2022. "Transcriptional CDK Inhibitors as Potential Treatment Option for Testicular Germ Cell Tumors" Cancers 14, no. 7: 1690. https://doi.org/10.3390/cancers14071690