
Evaluation of the TCR Repertoire as a Predictive and Prognostic Biomarker in Cancer: Diversity or Clonality?

 ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract

1. Introduction
2. T Cell Receptor
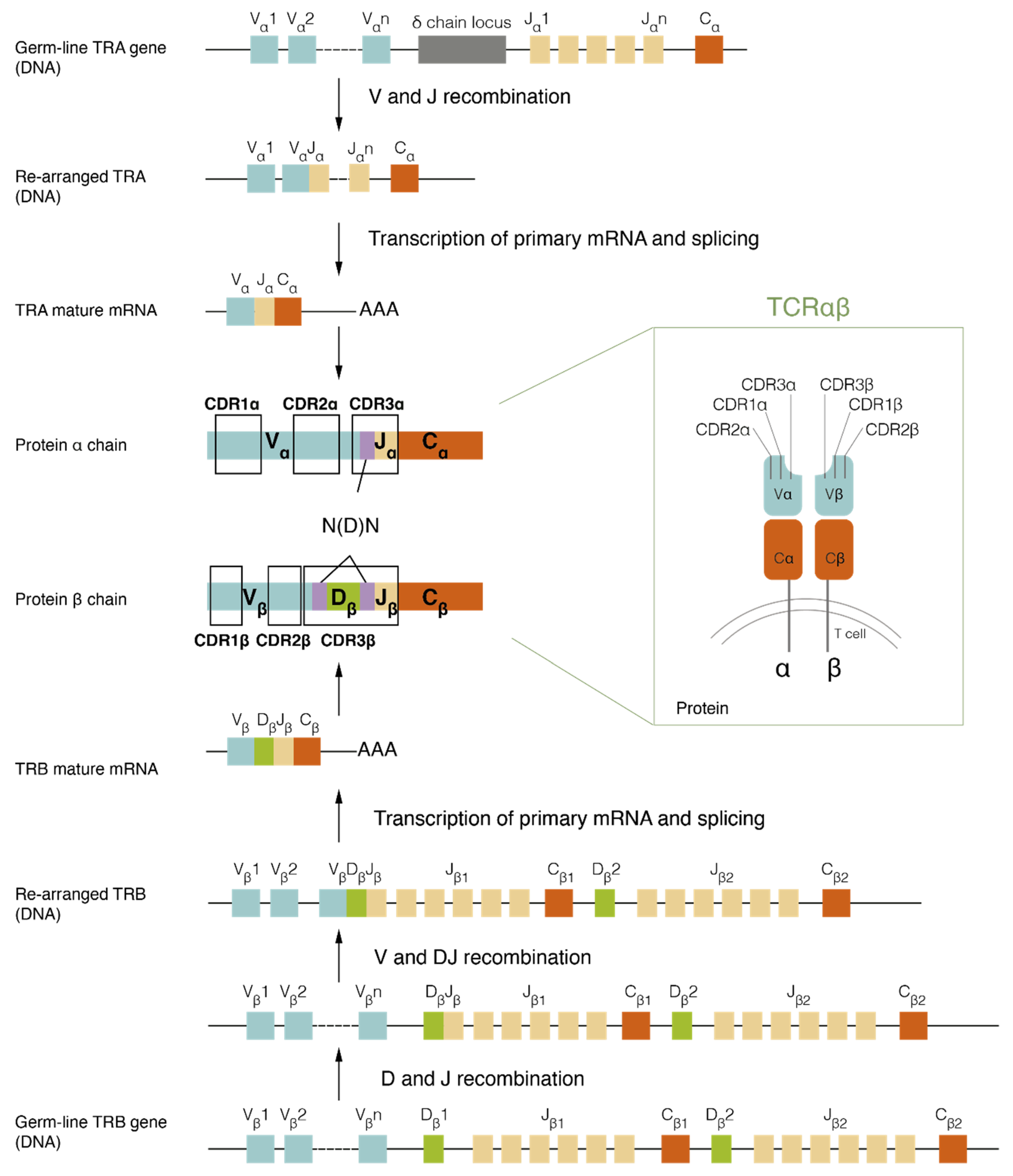
2.1. TCR Assembly and Structure
2.2. TCR Sequencing and Analysis
3. T Cell Response to Different Type of Tumoural Antigens
3.1. Tumour-Associated Antigens Recognition
3.2. Tumour-Specific Antigens Recognition
4. The TCR Repertoire as a Prognostic Biomarker in Cancer
4.1. TCR Repertoire in Patients with Cancer

{kind=link}
{kind=link}
| Disease | Compartment | TCR Repertoire Results | Prognosis Association | References |
|---|---|---|---|---|
| Melanoma | PBMC | No differences between age or clinical stage and diversity. | High diversity associated with longer PFS. | Charles et al. [67] |
| Metastatic LN | nr | High diversity of metastatic LN/PBMC ratio associated with better prognosis. | ||
| Breast Cancer | PBMC | Inverse correlation between TCR diversity and age. | Low diversity combined with lymphopenia in patients with elevated risk of early death | Manuel et al. [68] |
| Ovarian Carcinoma | PBMC | PBMC showing TCR repertoires quite distinct from the tumour tissue. | nr | Emerson et al. [72] |
| Tumoural tissue | ||||
| Cervical Cancer | PBMC | No differences between age and TCR diversity. | Diversity in PBMC decreasing as the carcinogenesis progressed. Lower diversity in the PBMC of CC, followed by CIN and healthy donors. | Cui et al. [69] |
| Sentinel LN | nr | Lower number of clones in the sentinel LN indicating a worse prognosis. | ||
| Breast Cancer | Tumoural and healthy tissue | Higher T cell infiltrates and TCR diversity in tumour than in healthy tissue. | nr | Wang et al. [75] |
| LN | Higher diversity in the LN than in tumours or healthy tissue. | |||
| NPC | PBMC | Higher diversity in NPC patients than healthy individuals. | Higher diversity in PBMC related with worse prognosis. | Jin et al. [70] |
| Tumoural and healthy tissue | No differences between healthy and tumoural tissue. | Lower tumour/healthy diversity ratio associated with worse prognosis. | ||
| HBV-associated HCC | Tumoural and healthy tissue | Higher diversity in tumoural tissue than in healthy tissue. | nr | Chen et al. [76] |
| Breast Cancer | Tumoural tissue | Lower diversity in tumoural tissue than in normal tissue. | nr | Beausang et al. [80] |
| Gastric Cancer | Tumoural tissue | Higher diversity in the adjacent mucosa than in tumoural tissue. | Diversity in the tumour not having an impact on the survival rate. | Jia et al. [81] |
| Adjacent mucosa | Low diversity in the adjacent mucosa related with a poor clinical prognosis. | |||
| PBMC | Higher diversity in PBMC than in tumoural tissue. | Diversity in PBMC not having an impact in the survival rate. | ||
| Diffuse Large B-Cell Lymphoma | DLBCL nodes and non-diseased nodes | Lower diversity in DLBCL nodes than in non-diseased nodes. | Lower diversity is associated with adverse outcomes. | Keane et al. [83] |
| Colorectal Cancer | Tumoural tissue and adjacent mucosa | Higher diversity in the adjacent mucosa than in tumoural tissue. | nr | Sherwood et al. [82] |
| HBV-associated HCC | Tumoural and adjacent tissue | No differences between tumoural and adjacent tissue. | Diversity not correlated with the progression of the disease. | Lin et al. [77] |
| Lower overlap between healthy and tumoural tissue observed in patients with shorter PFS. | ||||
| Gastric Cancer | Tumoural tissue and adjacent mucosa | No differences between tumoural and adjacent tissue. | Tumoural and adjacent mucosa overlap gradually decreasing during gastric carcinogenesis. | Kuang et al. [78] |
| OSCC | Tumoural and adjacent tissue | No differences between tumoural and adjacent tissue. | nr | Chen et al. [79] |
4.2. Determinant Factors of the TCR Repertoire
5. The TCR Repertoire as a Predictive Biomarker of Immune Checkpoint Inhibitor Treatments
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Mariuzza, R.A.; Agnihotri, P.; Orban, J. The Structural Basis of T-Cell Receptor (TCR) Activation: An Enduring Enigma. J. Biol. Chem. 2020, 295, 914–925. [Google Scholar] [CrossRef]
- Schatz, D.G.; Ji, Y. Recombination Centres and the Orchestration of V(D)J Recombination. Nat. Rev. Immunol. 2011, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Blom, B.; Verschuren, M.C.M.; Heemskerk, M.H.M.; Bakker, A.Q.; Gastel-Mol, E.J.V.; Wolvers-Tettero, I.L.M.; Dongen, J.J.M.V.; Spits, H. TCR Gene Rearrangements and Expression of the Pre-T Cell Receptor Complex during Human T-Cell Differentiation. Blood 1999, 93, 3033–3043. [Google Scholar] [CrossRef]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Kedzierska, K.; Koutsakos, M. The ABC of Major Histocompatibility Complexes and T Cell Receptors in Health and Disease. Viral Immunol. 2020, 33, 160–178. [Google Scholar] [CrossRef] [Green Version]
- Merkenschlager, M.; Graf, D.; Lovatt, M.; Bommhardt, U.; Zamoyska, R.; Fisher, A.G. How Many Thymocytes Audition for Selection? J. Exp. Med. 1997, 186, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Arstila, T.P.; Casrouge, A.; Baron, V.; Even, J.; Kanellopoulos, J.; Kourilsky, P. A Direct Estimate of the Human Aβ T Cell Receptor Diversity. Science 1999, 286, 958. [Google Scholar] [CrossRef]
- Attaf, M.; Huseby, E.; Sewell, A.K. Aβ T Cell Receptors as Predictors of Health and Disease. Cell Mol. Immunol. 2015, 12, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Buhler, S.; Bettens, F.; Dantin, C.; Ferrari-Lacraz, S.; Ansari, M.; Mamez, A.C.; Masouridi-Levrat, S.; Chalandon, Y.; Villard, J. Genetic T-Cell Receptor Diversity at 1 Year Following Allogeneic Hematopoietic Stem Cell Transplantation. Leukemia 2020, 34, 1422–1432. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L. Evaluation of TCR Repertoire Diversity in Patients after Hematopoietic Stem Cell Transplantation. Stem Cell Investig. 2015, 2, 17. [Google Scholar] [CrossRef]
- Robins, H.S.; Srivastava, S.K.; Campregher, P.V.; Turtle, C.J.; Andriesen, J.; Riddell, S.R.; Carlson, C.S.; Warren, E.H. Overlap and Effective Size of the Human CD8+ T Cell Receptor Repertoire. Sci. Trans. Med. 2010, 2, 47ra64. [Google Scholar] [CrossRef] [Green Version]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of Methodologies for T-Cell Receptor Repertoire Analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Pai, J.A.; Satpathy, A.T. High-Throughput and Single-Cell T Cell Receptor Sequencing Technologies. Nat. Methods 2021, 18, 881–892. [Google Scholar] [CrossRef]
- Chiffelle, J.; Genolet, R.; Perez, M.A.; Coukos, G.; Zoete, V.; Harari, A. T-Cell Repertoire Analysis and Metrics of Diversity and Clonality. Curr. Opin. Biotechnol. 2020, 65, 284–295. [Google Scholar] [CrossRef]
- Six, A.; Mariotti-Ferrandiz, E.; Chaara, W.; Magadan, S.; Pham, H.-P.; Lefranc, M.-P.; Mora, T.; Thomas-Vaslin, V.; Walczak, A.; Boudinot, P. The Past, Present, and Future of Immune Repertoire Biology—The Rise of Next-Generation Repertoire Analysis. Front. Immunol. 2013, 4, 413. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, W.; Zeng, X.; Zhang, R.; Du, Y.; Hong, X.; Cao, H.; Su, Z.; Wang, C.; Wu, J.; et al. Systematic Comparative Evaluation of Methods for Investigating the TCRβ Repertoire. PLoS ONE 2016, 11, e0152464. [Google Scholar] [CrossRef]
- Arunkumar, M.; Zielinski, C.E. T-Cell Receptor Repertoire Analysis with Computational Tools-An Immunologist’s Perspective. Cells 2021, 10, 3582. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, X.; Zhang, Y.; Zhang, Y.; Wang, M.; Ou, J.X.; Zhu, Y.; Zeng, H.; Wu, J.; Lan, C.; et al. Tools for Fundamental Analysis Functions of TCR Repertoires: A Systematic Comparison. Brief Bioinform. 2020, 21, 1706–1716. [Google Scholar] [CrossRef] [Green Version]
- Dupic, T.; Marcou, Q.; Walczak, A.M.; Mora, T. Genesis of the Aβ T-Cell Receptor. PLoS Comput. Biol. 2019, 15, e1006874. [Google Scholar] [CrossRef] [Green Version]
- Nature Publishing Group. Rapid Amplification of 5′ Complementary DNA Ends (5′ RACE). Nat. Methods 2005, 2, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Okino, S.T.; Kong, M.; Sarras, H.; Wang, Y. Evaluation of Bias Associated with High-Multiplex, Target-Specific Pre-Amplification. Biomol. Detect. Quantif. 2015, 6, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shugay, M.; Britanova, O.V.; Merzlyak, E.M.; Turchaninova, M.A.; Mamedov, I.Z.; Tuganbaev, T.R.; Bolotin, D.A.; Staroverov, D.B.; Putintseva, E.V.; Plevova, K.; et al. Towards Error-Free Profiling of Immune Repertoires. Nat. Methods 2014, 11, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiong, D.; Wang, X.; Liu, H.; Wang, T. Mapping the Functional Landscape of T Cell Receptor Repertoires by Single-T Cell Transcriptomics. Nat. Methods 2021, 18, 92–99. [Google Scholar] [CrossRef]
- Rath, J.A.; Bajwa, G.; Carreres, B.; Hoyer, E.; Gruber, I.; Martínez-Paniagua, M.A.; Yu, Y.-R.; Nouraee, N.; Sadeghi, F.; Wu, M.; et al. Single-Cell Transcriptomics Identifies Multiple Pathways Underlying Antitumor Function of TCR- and CD8αβ-Engineered Human CD4+ T Cells. Sci. Adv. 2020, 6, eaaz7809. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, A.; Lu, Y.-C. Single-Cell TCR and Transcriptome Analysis: An Indispensable Tool for Studying T-Cell Biology and Cancer Immunotherapy. Front. Immunol. 2021, 12, 689091. [Google Scholar] [CrossRef]
- Valkiers, S.; de Vrij, N.; Gielis, S.; Verbandt, S.; Ogunjimi, B.; Laukens, K.; Meysman, P. Recent Advances in T-Cell Receptor Repertoire Analysis: Bridging the Gap with Multimodal Single-Cell RNA Sequencing. ImmunoInformatics 2022, 5, 100009. [Google Scholar] [CrossRef]
- Lowery, F.J.; Krishna, S.; Yossef, R.; Parikh, N.B.; Chatani, P.D.; Zacharakis, N.; Parkhurst, M.R.; Levin, N.; Sindiri, S.; Sachs, A.; et al. Molecular Signatures of Antitumor Neoantigen-Reactive T Cells from Metastatic Human Cancers. Science 2022, 375, 877–884. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Liu, S.; Iorgulescu, B.; Li, S.; Morriss, J.; Borji, M.; Murray, E.; Braun, D.; Livak, K.; Wu, C.; Chen, F. 76 Spatial Mapping of T Cell Receptors and Transcriptomes in Renal Cell Carcinoma Following Immune Checkpoint Inhibitor Therapy. J. Immunother. Cancer 2021, 9, A1–A1054. [Google Scholar] [CrossRef]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—New Insights into Old Paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Ilyas, S.; Yang, J.C. Landscape of Tumor Antigens in T Cell Immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final Common Pathway’ of Human Cancer Immunotherapy: Targeting Random Somatic Mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Aung, P.P.; Liu, Y.-C.; Ballester, L.Y.; Robbins, P.F.; Rosenberg, S.A.; Lee, C.-C.R. Expression of New York Esophageal Squamous Cell Carcinoma-1 in Primary and Metastatic Melanoma. Hum. Pathol. 2014, 45, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, J.-P.; Robbins, P.F.; Raffeld, M.; Aung, P.P.; Tsokos, M.; Rosenberg, S.A.; Miettinen, M.M.; Lee, C.-C.R. NY-ESO-1 Expression in Synovial Sarcoma and Other Mesenchymal Tumors: Significance for NY-ESO-1-Based Targeted Therapy and Differential Diagnosis. Mod. Pathol. 2012, 25, 854–858. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Wang, Z.-F.; Lasota, J.; Park, T.; Patel, K.; Groh, E.; Rosenberg, S.A.; Miettinen, M.M. MAGE-A Is More Highly Expressed Than NY-ESO-1 in a Systematic Immunohistochemical Analysis of 3668 Cases. J. Immunother. 2016, 39, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1-Reactive T-Cell Receptor: Long-Term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A Gene Encoding an Antigen Recognized by Cytolytic T Lymphocytes on a Human Melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Kim, H.-R.; Oh, S.; Ko, J.Y.; Kim, Y.; Kang, K.; Yang, Y.; Kim, J.; Park, J.H.; Roe, J.-S.; et al. Epigenetic Upregulation of MAGE-A Isoforms Promotes Breast Cancer Cell Aggressiveness. Cancers 2021, 13, 3176. [Google Scholar] [CrossRef]
- Sharma, P.; Gnjatic, S.; Jungbluth, A.A.; Williamson, B.; Herr, H.; Stockert, E.; Dalbagni, G.; Donat, S.M.; Reuter, V.E.; Santiago, D.; et al. Frequency of NY-ESO-1 and LAGE-1 Expression in Bladder Cancer and Evidence of a New NY-ESO-1 T-Cell Epitope in a Patient with Bladder Cancer. Cancer Immun. 2003, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, T.; Noguchi, Y.; Saika, T.; Ono, T.; Nagata, Y.; Jungbluth, A.; Ritter, G.; Chen, Y.-T.; Stockert, E.; Tsushima, T.; et al. NY-ESO-1 Expression and Immunogenicity Associated with Transitional Cell Carcinoma: Correlation with Tumor Grade1. Cancer Res. 2001, 61, 4671–4674. [Google Scholar] [PubMed]
- Bolli, M.; Schultz-Thater, E.; Zajac, P.; Guller, U.; Feder, C.; Sanguedolce, F.; Carafa, V.; Terracciano, L.; Hudolin, T.; Spagnoli, G.C.; et al. NY-ESO-1/LAGE-1 Coexpression with MAGE-A Cancer/Testis Antigens: A Tissue Microarray Study. Int. J. Cancer 2005, 115, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Akcakanat, A.; Kanda, T.; Koyama, Y.; Watanabe, M.; Kimura, E.; Yoshida, Y.; Komukai, S.; Nakagawa, S.; Odani, S.; Fujii, H.; et al. NY-ESO-1 Expression and Its Serum Immunoreactivity in Esophageal Cancer. Cancer Chemother. Pharmacol. 2004, 54, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.L.; Jungbluth, A.A.; Patel, S.G.; Iversen, K.; Hoshaw-Woodard, S.; Busam, K.J. Expression and Significance of Cancer Testis Antigens in Primary Mucosal Melanoma of the Head and Neck. Head Neck 2004, 26, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Jungbluth, A.A.; Chen, Y.T.; Stockert, E.; Busam, K.J.; Kolb, D.; Iversen, K.; Coplan, K.; Williamson, B.; Altorki, N.; Old, L.J. Immunohistochemical Analysis of NY-ESO-1 Antigen Expression in Normal and Malignant Human Tissues. Int. J. Cancer 2001, 92, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Jungbluth, A.A.; Stockert, E.; Qian, F.; Gnjatic, S.; Tammela, J.; Intengan, M.; Beck, A.; Keitz, B.; Santiago, D.; et al. NY-ESO-1 and LAGE-1 Cancer-Testis Antigens Are Potential Targets for Immunotherapy in Epithelial Ovarian Cancer. Cancer Res. 2003, 63, 6076–6083. [Google Scholar]
- Fosså, A.; Berner, A.; Fosså, S.D.; Hernes, E.; Gaudernack, G.; Smeland, E.B. NY-ESO-1 Protein Expression and Humoral Immune Responses in Prostate Cancer. Prostate 2004, 59, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, A.; Picard, V.; LaRue, H.; Harel, F.; Hovington, H.; Lacombe, L.; Fradet, Y. High Frequency of MAGE-A4 and MAGE-A9 Expression in High-Risk Bladder Cancer. Int. J. Cancer 2009, 125, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Sang, M.; Lian, Y.; Zhou, X.; Shan, B. MAGE-A Family: Attractive Targets for Cancer Immunotherapy. Vaccine 2011, 29, 8496–8500. [Google Scholar] [CrossRef] [PubMed]
- Weon, J.L.; Potts, P.R. The MAGE Protein Family and Cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-Targeted Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, A.B.; Schreurs, M.W.; de Boer, A.J.; Kawakami, Y.; Rosenberg, S.A.; Adema, G.J.; Figdor, C.G. Melanocyte Lineage-Specific Antigen Gp100 Is Recognized by Melanoma-Derived Tumor-Infiltrating Lymphocytes. J. Exp. Med. 1994, 179, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Eliyahu, S.; Sakaguchi, K.; Robbins, P.F.; Rivoltini, L.; Yannelli, J.R.; Appella, E.; Rosenberg, S.A. Identification of the Immunodominant Peptides of the MART-1 Human Melanoma Antigen Recognized by the Majority of HLA-A2-Restricted Tumor Infiltrating Lymphocytes. J. Exp. Med. 1994, 180, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disis, M.L.; Wallace, D.R.; Gooley, T.A.; Dang, Y.; Slota, M.; Lu, H.; Coveler, A.L.; Childs, J.S.; Higgins, D.M.; Fintak, P.A.; et al. Concurrent Trastuzumab and HER2/Neu-Specific Vaccination in Patients With Metastatic Breast Cancer. J. Clin. Oncol. 2009, 27, 4685–4692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disis, M.L.; Knutson, K.L.; Schiffman, K.; Rinn, K.; McNeel, D.G. Pre-Existent Immunity to the HER-2/Neu Oncogenic Protein in Patients with HER-2/Neu Overexpressing Breast and Ovarian Cancer. Breast Cancer Res. Treat. 2000, 62, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.E.; Crawford, J.C.; Thomas, P.G. Hitting the Target: How T Cells Detect and Eliminate Tumors. J. Immunol. 2018, 200, 392–399. [Google Scholar] [CrossRef]
- Hoffmann, M.M.; Slansky, J.E. T-Cell Receptor Affinity in the Age of Cancer Immunotherapy. Mol. Carcinog. 2020, 59, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Bareke, H.; Juanes-Velasco, P.; Landeira-Viñuela, A.; Hernandez, A.-P.; Cruz, J.J.; Bellido, L.; Fonseca, E.; Niebla-Cárdenas, A.; Montalvillo, E.; Góngora, R.; et al. Autoimmune Responses in Oncology: Causes and Significance. Int. J. Mol. Sci. 2021, 22, 8030. [Google Scholar] [CrossRef] [PubMed]
- Prehn, R.T.; Main, J.M. Immunity to Methylcholanthrene-Induced Sarcomas. J. Natl. Cancer Inst. 1957, 18, 769–778. [Google Scholar] [PubMed]
- Schrama, D.; Ritter, C.; Becker, J.C. T Cell Receptor Repertoire Usage in Cancer as a Surrogate Marker for Immune Responses. Semin. Immunopathol. 2017, 39, 255–268. [Google Scholar] [CrossRef]
- Unanue, E.R.; Turk, V.; Neefjes, J. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu. Rev. Immunol. 2016, 34, 265–297. [Google Scholar] [CrossRef]
- Sette, A.; Sidney, J. Nine Major HLA Class I Supertypes Account for the Vast Preponderance of HLA-A and -B Polymorphism. Immunogenetics 1999, 50, 201–212. [Google Scholar] [CrossRef]
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddell, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive Assessment of T-Cell Receptor Beta-Chain Diversity in Alphabeta T Cells. Blood 2009, 114, 4099–4107. [Google Scholar] [CrossRef]
- Charles, J.; Mouret, S.; Challende, I.; Leccia, M.-T.; De Fraipont, F.; Perez, S.; Plantier, N.; Plumas, J.; Manuel, M.; Chaperot, L.; et al. T-Cell Receptor Diversity as a Prognostic Biomarker in Melanoma Patients. Pigment Cell Melanoma Res. 2020, 33, 612–624. [Google Scholar] [CrossRef]
- Manuel, M.; Tredan, O.; Bachelot, T.; Clapisson, G.; Courtier, A.; Parmentier, G.; Rabeony, T.; Grives, A.; Perez, S.; Mouret, J.-F.; et al. Lymphopenia Combined with Low TCR Diversity (Divpenia) Predicts Poor Overall Survival in Metastatic Breast Cancer Patients. Oncoimmunology 2012, 1, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.-H.; Lin, K.-R.; Yuan, S.-H.; Jin, Y.-B.; Chen, X.-P.; Su, X.-K.; Jiang, J.; Pan, Y.-M.; Mao, S.-L.; Mao, X.-F.; et al. TCR Repertoire as a Novel Indicator for Immune Monitoring and Prognosis Assessment of Patients With Cervical Cancer. Front. Immunol. 2018, 9, 2729. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.-B.; Luo, W.; Zhang, G.-Y.; Lin, K.-R.; Cui, J.-H.; Chen, X.-P.; Pan, Y.-M.; Mao, X.-F.; Tang, J.; Wang, Y.-J. TCR Repertoire Profiling of Tumors, Adjacent Normal Tissues, and Peripheral Blood Predicts Survival in Nasopharyngeal Carcinoma. Cancer Immunol. Immunother. 2018, 67, 1719–1730. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, Q.; Wu, S.; Zhang, X.; Wang, M.; He, F.; Wei, T.; Yang, J.; Lou, Y.; Cai, Z.; et al. Characteristics of Tumor Infiltrating Lymphocyte and Circulating Lymphocyte Repertoires in Pancreatic Cancer by the Sequencing of T Cell Receptors. Sci. Rep. 2015, 5, 13664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, R.O.; Sherwood, A.M.; Rieder, M.J.; Guenthoer, J.; Williamson, D.W.; Carlson, C.S.; Drescher, C.W.; Tewari, M.; Bielas, J.H.; Robins, H.S. High-Throughput Sequencing of T Cell Receptors Reveals a Homogeneous Repertoire of Tumor-Infiltrating Lymphocytes in Ovarian Cancer. J. Pathol. 2013, 231, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, C.; Tian, X.; Wu, J.; Zhang, C.; Tan, Q.; Guan, X.; Dong, B.; Zhao, M.; Lu, Z.; Hao, C. T Cell Receptor β-Chain Repertoire Analysis of Tumor-Infiltrating Lymphocytes in Pancreatic Cancer. Cancer Sci. 2019, 110, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Baars, A.; Gomez, R.; Weder, P.; Smits, M.; de Gruijl, T.D.; von Blomberg, B.M.E.; Bloemena, E.; Scheper, R.J.; van Ham, S.M.; et al. Melanoma-Specific Tumor-Infiltrating Lymphocytes but Not Circulating Melanoma-Specific T Cells May Predict Survival in Resected Advanced-Stage Melanoma Patients. Cancer Immunol. Immunother. 2006, 55, 451–458. [Google Scholar] [CrossRef]
- Wang, T.; Wang, C.; Wu, J.; He, C.; Zhang, W.; Liu, J.; Zhang, R.; Lv, Y.; Li, Y.; Zeng, X.; et al. The Different T-Cell Receptor Repertoires in Breast Cancer Tumors, Draining Lymph Nodes, and Adjacent Tissues. Cancer Immunol. Res. 2017, 5, 148–156. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Y.; Zhao, M.; Liu, Y.; Gong, M.; Xie, C.; Wu, H.; Wang, Z. High-Throughput T Cell Receptor Sequencing Reveals Distinct Repertoires between Tumor and Adjacent Non-Tumor Tissues in HBV-Associated HCC. Oncoimmunology 2016, 5, e1219010. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.-R.; Deng, F.-W.; Jin, Y.-B.; Chen, X.-P.; Pan, Y.-M.; Cui, J.-H.; You, Z.-X.; Chen, H.-W.; Luo, W. T Cell Receptor Repertoire Profiling Predicts the Prognosis of HBV-Associated Hepatocellular Carcinoma. Cancer Med. 2018, 7, 3755–3762. [Google Scholar] [CrossRef]
- Kuang, M.; Cheng, J.; Zhang, C.; Feng, L.; Xu, X.; Zhang, Y.; Zu, M.; Cui, J.; Yu, H.; Zhang, K.; et al. A Novel Signature for Stratifying the Molecular Heterogeneity of the Tissue-Infiltrating T-Cell Receptor Repertoire Reflects Gastric Cancer Prognosis. Sci. Rep. 2017, 7, 7762. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, C.; Pan, Y.; Xu, R.; Xu, C.; Chen, Z.; Lu, Z.; Ke, Y. T Cell Receptor β-Chain Repertoire Analysis Reveals Intratumour Heterogeneity of Tumour-Infiltrating Lymphocytes in Oesophageal Squamous Cell Carcinoma. J. Pathol. 2016, 239, 450–458. [Google Scholar] [CrossRef]
- Beausang, J.F.; Wheeler, A.J.; Chan, N.H.; Hanft, V.R.; Dirbas, F.M.; Jeffrey, S.S.; Quake, S.R. T Cell Receptor Sequencing of Early-Stage Breast Cancer Tumors Identifies Altered Clonal Structure of the T Cell Repertoire. Proc. Natl. Acad. Sci. USA 2017, 114, E10409–E10417. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Zhou, J.; Chen, G.; Shi, Y.; Yu, H.; Guan, P.; Lin, R.; Jiang, N.; Yu, P.; Li, Q.-J.; et al. Diversity Index of Mucosal Resident T Lymphocyte Repertoire Predicts Clinical Prognosis in Gastric Cancer. Oncoimmunology 2015, 4, e1001230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood, A.M.; Emerson, R.O.; Scherer, D.; Habermann, N.; Buck, K.; Staffa, J.; Desmarais, C.; Halama, N.; Jaeger, D.; Schirmacher, P.; et al. Tumor-Infiltrating Lymphocytes in Colorectal Tumors Display a Diversity of T Cell Receptor Sequences That Differ from the T Cells in Adjacent Mucosal Tissue. Cancer Immunol. Immunother. 2013, 62, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Keane, C.; Gould, C.; Jones, K.; Hamm, D.; Talaulikar, D.; Ellis, J.; Vari, F.; Birch, S.; Han, E.; Wood, P.; et al. The T-Cell Receptor Repertoire Influences the Tumor Microenvironment and Is Associated with Survival in Aggressive B-Cell Lymphoma. Clin. Cancer Res. 2017, 23, 1820–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-Cell Profiling of Breast Cancer T Cells Reveals a Tissue-Resident Memory Subset Associated with Improved Prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Phasouk, K.; Bossard, E.; Klock, A.; Jin, L.; Laing, K.J.; Johnston, C.; Williams, N.A.; Czartoski, J.L.; Varon, D.; et al. Distinct Populations of Antigen-Specific Tissue-Resident CD8+ T Cells in Human Cervix Mucosa. JCI Insight 2021, 6, 149950. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and Low Mutational Burden Tumors versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Chen, Y.; Yang, Q. Significance of Tumor Mutation Burden Combined with Immune Infiltrates in the Progression and Prognosis of Ovarian Cancer. Cancer Cell Int. 2020, 20, 373. [Google Scholar] [CrossRef]
- Jiang, F.; Wu, C.; Wang, M.; Wei, K.; Zhou, G.; Wang, J. Multi-Omics Analysis of Tumor Mutation Burden Combined with Immune Infiltrates in Melanoma. Clin. Chim. Acta 2020, 511, 306–318. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, J.; Dong, Z.; Hou, L.; Zhao, C.; Li, X.; Mao, B.; Zhu, W.; Guo, X.; Zhang, H.; et al. Genomic Landscape and Its Correlations with Tumor Mutational Burden, PD-L1 Expression, and Immune Cells Infiltration in Chinese Lung Squamous Cell Carcinoma. J. Hematol. Oncol. 2019, 12, 75. [Google Scholar] [CrossRef] [Green Version]
- Karn, T.; Denkert, C.; Weber, K.E.; Holtrich, U.; Hanusch, C.; Sinn, B.V.; Higgs, B.W.; Jank, P.; Sinn, H.P.; Huober, J.; et al. Tumor Mutational Burden and Immune Infiltration as Independent Predictors of Response to Neoadjuvant Immune Checkpoint Inhibition in Early TNBC in GeparNuevo. Ann. Oncol. 2020, 31, 1216–1222. [Google Scholar] [CrossRef]
- Marderstein, A.R.; Uppal, M.; Verma, A.; Bhinder, B.; Tayyebi, Z.; Mezey, J.; Clark, A.G.; Elemento, O. Demographic and Genetic Factors Influence the Abundance of Infiltrating Immune Cells in Human Tissues. Nat. Commun. 2020, 11, 2213. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.W.; Lin, Y.-C.; Yao, P.-L.; Yuan, A.; Chen, H.-Y.; Shun, C.-T.; Tsai, M.-F.; Chen, C.-H.; Yang, P.-C. Tumor-Associated Macrophages: The Double-Edged Sword in Cancer Progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasetto, A.; Gros, A.; Robbins, P.F.; Deniger, D.C.; Prickett, T.D.; Matus-Nicodemos, R.; Douek, D.C.; Howie, B.; Robins, H.; Parkhurst, M.R.; et al. Tumor- and Neoantigen-Reactive T-Cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol. Res. 2016, 4, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.-L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated Molecular Analysis of Tumor Biopsies on Sequential CTLA-4 and PD-1 Blockade Reveals Markers of Response and Resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Jen, J.; Vogelstein, B.; Hamilton, S.R. Clinical and Pathological Characteristics of Sporadic Colorectal Carcinomas with DNA Replication Errors in Microsatellite Sequences. Am. J. Pathol. 1994, 145, 148–156. [Google Scholar]
- Vilar, E.; Gruber, S.B. Microsatellite Instability in Colorectal Cancer-the Stable Evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune Checkpoint Inhibitors for the Treatment of MSI-H/MMR-D Colorectal Cancer and a Perspective on Resistance Mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macrì, E.; Fornasarig, M.; Boiocchi, M. High Prevalence of Activated Intraepithelial Cytotoxic T Lymphocytes and Increased Neoplastic Cell Apoptosis in Colorectal Carcinomas with Microsatellite Instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-Infiltrating Lymphocytes Are a Marker for Microsatellite Instability in Colorectal Carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosho, K.; Baba, Y.; Tanaka, N.; Shima, K.; Hayashi, M.; Meyerhardt, J.A.; Giovannucci, E.; Dranoff, G.; Fuchs, C.S.; Ogino, S. Tumour-Infiltrating T-Cell Subsets, Molecular Changes in Colorectal Cancer, and Prognosis: Cohort Study and Literature Review. J. Pathol. 2010, 222, 350–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Pamplona, R.; Melas, M.; Maoz, A.; Schmit, S.L.; Rennert, H.; Lejbkowicz, F.; Greenson, J.K.; Sanjuan, X.; Lopez-Zambrano, M.; Alonso, M.H.; et al. Lymphocytic Infiltration in Stage II Microsatellite Stable Colorectal Tumors: A Retrospective Prognosis Biomarker Analysis. PLoS Med. 2020, 17, e1003292. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Huang, F.; Goncalves, C.; Del Rincón, S.V.; Miller, W.H. Translation of Cancer Immunotherapy from the Bench to the Bedside. Adv. Cancer Res. 2019, 143, 1–62. [Google Scholar] [CrossRef]
- Van Nieuwenhuijze, A.; Liston, A. Chapter Four—The Molecular Control of Regulatory T Cell Induction. In Progress in Molecular Biology and Translational Science; Liston, A., Ed.; Regulatory T Cells in Health and Disease; Academic Press: Cambridge, MA, USA, 2015; Volume 136, pp. 69–97. [Google Scholar]
- Chambers, C.A.; Sullivan, T.J.; Allison, J.P. Lymphoproliferation in CTLA-4-Deficient Mice Is Mediated by Costimulation-Dependent Activation of CD4+ T Cells. Immunity 1997, 7, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Wang, G.; Wang, Y.; Riese, M.J.; You, M. Uncoupling Therapeutic Efficacy from Immune-Related Adverse Events in Immune Checkpoint Blockade. iScience 2020, 23, 101580. [Google Scholar] [CrossRef] [PubMed]
- Buder-Bakhaya, K.; Hassel, J.C. Biomarkers for Clinical Benefit of Immune Checkpoint Inhibitor Treatment—A Review From the Melanoma Perspective and Beyond. Front. Immunol. 2018, 9, 1474. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-Related Adverse Events of Checkpoint Inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yao, Z.; Yang, H.; Liang, N.; Zhang, X.; Zhang, F. Are Immune-Related Adverse Events Associated with the Efficacy of Immune Checkpoint Inhibitors in Patients with Cancer? A Systematic Review and Meta-Analysis. BMC Med. 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-H.; Zang, X.-Y.; Wang, J.-C.; Huang, S.-S.; Xu, J.; Zhang, P. Diagnosis and Management of Immune Related Adverse Events (IrAEs) in Cancer Immunotherapy. Biomed. Pharmacother. 2019, 120, 109437. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Vollmer, S.; Tietze, J.; Galinski, A.; Heppt, M.V.; Bürdek, M.; Berking, C.; Prinz, J.C. Clonality of CD4+ Blood T Cells Predicts Longer Survival With CTLA4 or PD-1 Checkpoint Inhibition in Advanced Melanoma. Front. Immunol. 2019, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Klinger, M.; Hou, Y.; Cummings, C.; Ribas, A.; Faham, M.; Fong, L. Improved Survival with T Cell Clonotype Stability after Anti-CTLA-4 Treatment in Cancer Patients. Sci. Transl. Med. 2014, 6, 238ra70. [Google Scholar] [CrossRef] [Green Version]
- Hogan, S.A.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.-F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T Cell Receptor Repertoire Features Associated with Survival in Immunotherapy-Treated Pancreatic Ductal Adenocarcinoma. JCI Insight 2018, 3, 122092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postow, M.A.; Manuel, M.; Wong, P.; Yuan, J.; Dong, Z.; Liu, C.; Perez, S.; Tanneau, I.; Noel, M.; Courtier, A.; et al. Peripheral T Cell Receptor Diversity Is Associated with Clinical Outcomes Following Ipilimumab Treatment in Metastatic Melanoma. J. Immunother. Cancer 2015, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 Blockade Broadens the Peripheral T Cell Receptor Repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, L.; Harview, C.; Emerson, R.; Wang, X.; Mok, S.; Homet, B.; Comin-Anduix, B.; Koya, R.C.; Robins, H.; Tumeh, P.C.; et al. Distinct Immunological Mechanisms of CTLA-4 and PD-1 Blockade Revealed by Analyzing TCR Usage in Blood Lymphocytes. Oncoimmunology 2014, 3, e29244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subudhi, S.K.; Aparicio, A.; Gao, J.; Zurita, A.J.; Araujo, J.C.; Logothetis, C.J.; Tahir, S.A.; Korivi, B.R.; Slack, R.S.; Vence, L.; et al. Clonal Expansion of CD8 T Cells in the Systemic Circulation Precedes Development of Ipilimumab-Induced Toxicities. Proc. Natl. Acad. Sci. USA 2016, 113, 11919–11924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khunger, A.; Rytlewski, J.A.; Fields, P.; Yusko, E.C.; Tarhini, A.A. The Impact of CTLA-4 Blockade and Interferon-α on Clonality of T-Cell Repertoire in the Tumor Microenvironment and Peripheral Blood of Metastatic Melanoma Patients. Oncoimmunology 2019, 8, e1652538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looney, T.J.; Topacio-Hall, D.; Lowman, G.; Conroy, J.; Morrison, C.; Oh, D.; Fong, L.; Zhang, L. TCR Convergence in Individuals Treated With Immune Checkpoint Inhibition for Cancer. Front. Immunol. 2019, 10, 2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, D.Y.; Cham, J.; Zhang, L.; Fong, G.; Kwek, S.S.; Klinger, M.; Faham, M.; Fong, L. Immune Toxicities Elicted by CTLA-4 Blockade in Cancer Patients Are Associated with Early Diversification of the T-Cell Repertoire. Cancer Res. 2017, 77, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Duan, J.; Bai, H.; Wang, Y.; Wan, R.; Wang, X.; Chen, S.; Tian, Y.; Wang, D.; Fei, K.; et al. TCR Repertoire Diversity of Peripheral PD-1+CD8+ T Cells Predicts Clinical Outcomes after Immunotherapy in Patients with Non-Small Cell Lung Cancer. Cancer Immunol Res. 2020, 8, 146–154. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Naidus, E.; Bouquet, J.; Oh, D.Y.; Looney, T.J.; Yang, H.; Fong, L.; Standifer, N.E.; Zhang, L. Early Changes in the Circulating T Cells Are Associated with Clinical Outcomes after PD-L1 Blockade by Durvalumab in Advanced NSCLC Patients. Cancer Immunol. Immunother. 2021, 70, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Nathanson, T.; Funt, S.A.; Ahuja, A.; Buros Novik, J.; Hellmann, M.D.; Chang, E.; Aksoy, B.A.; Al-Ahmadie, H.; Yusko, E.; et al. Contribution of Systemic and Somatic Factors to Clinical Response and Resistance to PD-L1 Blockade in Urothelial Cancer: An Exploratory Multi-Omic Analysis. PLoS Med. 2017, 14, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Page, D.B.; Yuan, J.; Redmond, D.; Wen, Y.H.; Durack, J.C.; Emerson, R.; Solomon, S.; Dong, Z.; Wong, P.; Comstock, C.; et al. Deep Sequencing of T-Cell Receptor DNA as a Biomarker of Clonally Expanded TILs in Breast Cancer after Immunotherapy. Cancer Immunol Res. 2016, 4, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Park, J.-H.; Kiyotani, K.; Zewde, M.; Miyashita, A.; Jinnin, M.; Kiniwa, Y.; Okuyama, R.; Tanaka, R.; Fujisawa, Y.; et al. Intratumoral Expression Levels of PD-L1, GZMA, and HLA-A along with Oligoclonal T Cell Expansion Associate with Response to Nivolumab in Metastatic Melanoma. Oncoimmunology 2016, 5, e1204507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Spassova, I.; Ugurel, S.; Terheyden, P.; Sucker, A.; Hassel, J.C.; Ritter, C.; Kubat, L.; Habermann, D.; Farahpour, F.; Saeedghalati, M.; et al. Predominance of Central Memory T Cells with High T-Cell Receptor Repertoire Diversity Is Associated with Response to PD-1/PD-L1 Inhibition in Merkel Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2257–2267. [Google Scholar] [CrossRef]
- Yusko, E.; Vignali, M.; Wilson, R.K.; Mardis, E.R.; Hodi, F.S.; Horak, C.; Chang, H.; Woods, D.M.; Robins, H.; Weber, J. Association of Tumor Microenvironment T-Cell Repertoire and Mutational Load with Clinical Outcome after Sequential Checkpoint Blockade in Melanoma. Cancer Immunol. Res. 2019, 7, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; De Luca, C.D.; Locatelli, F.; Velardi, E. Thymic Function and T-Cell Receptor Repertoire Diversity: Implications for Patient Response to Checkpoint Blockade Immunotherapy. Front. Immunol. 2021, 12, 752042. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Semeraro, M.D.; Herrmann, M.; Absenger, G.; Gerger, A.; Renner, W. Immune Aging and Immunotherapy in Cancer. Int. J. Mol. Sci. 2021, 22, 7016. [Google Scholar] [CrossRef]
- Pawelec, G. Does Patient Age Influence Anti-Cancer Immunity? Semin. Immunopathol. 2019, 41, 125–131. [Google Scholar] [CrossRef]
- Improved Survival with Ipilimumab in Patients with Metastatic Melanoma|NEJM. Available online: https://www.nejm.org/doi/full/10.1056/NEJMoa1003466 (accessed on 21 March 2022).
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 Blockade Enhances Polyfunctional NY-ESO-1 Specific T Cell Responses in Metastatic Melanoma Patients with Clinical Benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the Anti-PD-1 and Anti-PD-L1 Immune Checkpoint Antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef]
- Wang, J.; Yang, T.; Xu, J. Therapeutic Development of Immune Checkpoint Inhibitors. In Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy; Xu, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2020; pp. 619–649. ISBN 9789811532665. [Google Scholar]
- Luoma, A.M.; Suo, S.; Williams, H.L.; Sharova, T.; Sullivan, K.; Manos, M.; Bowling, P.; Hodi, F.S.; Rahma, O.; Sullivan, R.J.; et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020, 182, 655–671.e22. [Google Scholar] [CrossRef]
- Jing, Y.; Liu, J.; Ye, Y.; Pan, L.; Deng, H.; Wang, Y.; Yang, Y.; Diao, L.; Lin, S.H.; Mills, G.B.; et al. Multi-Omics Prediction of Immune-Related Adverse Events during Checkpoint Immunotherapy. Nat. Commun. 2020, 11, 4946. [Google Scholar] [CrossRef]
| Association with ICI Response | |||||||
|---|---|---|---|---|---|---|---|
| Disease | Compartment | ICI | Effect of ICI | TCR Repertoire at Baseline | TCR Repertoire after Treatment | Development of irAEs | References |
| Melanoma | PBMC | a-CTLA-4 | nr | High diversity at baseline in LTS. | nr | Highly restricted TCR repertoire in patients developing irAEs. | Arakawa et al. [117] |
| a-PD-1 | nr | High diversity at baseline in LTS. | nr | nr | |||
| Melanoma | PBMC | a-CTLA-4 | Increase in diversity. | High diversity at baseline associated with improved survival. | Reduced clonotype loss associated with improved clinical outcome. | nr | Cha et al. [118] |
| Prostate Cancer | PBMC | a-CTLA-4 | Increase in diversity. | High diversity at baseline associated with improved survival. | Reduced clonotype loss associated with improved clinical outcome. | nr | |
| Melanoma | PBMC | a-CTLA-4 | nr | High diversity at baseline associated with improved survival. | nr | nr | Hogan et al. [119] |
| a-PD-1 | nr | Higher clonality at baseline associated with major pathological response. | nr | nr | |||
| Pancreatic Ductal Adenocarcinoma | PBMC | a-CTLA-4 | No significant changes. | High diversity at baseline associated with improved survival. | LTS showing a higher number of expanded clones after treatment. | nr | Hopkins et al. [120] |
| a-PD-1 | No significant changes. | nr | LTS showing higher clonality after treatment. | nr | |||
| Melanoma | PBMC | a-CTLA-4 | nr | High diversity at baseline associated with improved survival. | nr | nr | Postow et al. [121] |
| Melanoma | PBMC | a-CTLA-4 | Increase in diversity. | nr | Responders exhibiting an increase in TCR richness. | Higher diversity associated with increased toxicities. | Robert et al. [122] |
| Melanoma | PBMC | a-PD-1 | No significant changes. | nr | Responders exhibiting both increase and decrease in richness indifferently. | nr | Robert et al. [123] |
| Prostate Cancer | PBMC | a-CTLA-4 | nr | nr | nr | High CD8+ clonality related with irAEs. | Subudhi et al. [124] |
| Melanoma | PBMC | a-CTLA-4 | nr | No association between pre-treatment and response. | Patients with higher diversity having longer PFS and OS. | nr | Khunger et al. [125] |
| Tumoural tissue | a-CTLA-4 | nr | Higher clonality at baseline associated with longer PFS and OS. | nr | nr | ||
| Clear Cell Adenocarcinoma, Melanoma and Prostate Cancer | PBMC | a-CTLA-4 | nr | nr | A trend toward higher clonality in responders. | nr | Looney et al. [126] |
| Prostate Cancer | PBMC | a-CTLA-4 | Increase in diversity. | nr | nr | Higher diversity post-ICI/baseline ratio associated with irAEs. | Oh et al. [127] |
| NSCLC | PBMC | a-PD-1 | nr | High diversity of PD-1+CD8+ at baseline associated with improved survival. | High clonality after treatment associated with increased PFS. | nr | Han et al. [128] |
| NSCLC | PBMC | a-PD-1 | nr | nr | Responders exhibiting a higher expansion of peripheral clones previously found in the tumour. | nr | Forde et al. [129] |
| Tumoural tissue | a-PD-1 | nr | Higher clonality at baseline associated with major pathological response. | nr | nr | ||
| NSCLC | PBMC | a-PD-L1 | nr | nr | High diversity after treatment associated with longer OS. | nr | Naidus et al. [130] |
| Urothelial Carcinoma | PBMC | a-PD-L1 | nr | High diversity at baseline associated with improved survival. | High clonality after treatment associated with increased PFS. | nr | Snyder et al. [131] |
| Breast Cancer | Tumoural tissue | a-CTLA-4 | a-CTLA-4 alone expands intra-tumoural lymphocytes, increasing clonality. Cryoablation inducing polyclonality, independently from a-CTLA-4. | nr | nr | nr | Page et al. [132] |
| Melanoma | Tumoural tissue | a-CTLA-4 | nr | Not significant results. | nr | nr | Roh et al. [96] |
| a-PD-1 + a-CTLA-4 | nr | Responders exhibiting a higher clonality at pre-a-CTLA-4 treatment. | nr | nr | |||
| Melanoma | Tumoural tissue | a-PD-1 | nr | nr | Responders exhibiting more oligoclonal expansions. | nr | Inoue et al. [133] |
| Glioblastoma | Tumoural tissue | a-PD-1 | Increase in diversity. | nr | nr | nr | Schalper et al. [134]. |
| Melanoma | Tumoural tissue | a-PD-1 | nr | High clonality at baseline associated with improved survival. | Responders exhibiting more oligoclonal expansions. | Tumeh et al. [95] | |
| Merkel Cell Carcinoma | Tumoural tissue | a-PD1 or | nr | nr | Responders exhibiting higher diversity. | nr | Spassova et al. [135] |
| a-PD-L1 | |||||||
| Melanoma | Tumoural tissue | a-PD-1+ | nr | Responders exhibiting a higher clonality at baseline. | Higher clonality correlating with clinical benefit. | nr | Yusko et al. [136] |
| a-CTLA-4 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aran, A.; Garrigós, L.; Curigliano, G.; Cortés, J.; Martí, M. Evaluation of the TCR Repertoire as a Predictive and Prognostic Biomarker in Cancer: Diversity or Clonality? Cancers 2022, 14, 1771. https://doi.org/10.3390/cancers14071771
Aran A, Garrigós L, Curigliano G, Cortés J, Martí M. Evaluation of the TCR Repertoire as a Predictive and Prognostic Biomarker in Cancer: Diversity or Clonality? Cancers. 2022; 14(7):1771. https://doi.org/10.3390/cancers14071771
Chicago/Turabian StyleAran, Andrea, Laia Garrigós, Giuseppe Curigliano, Javier Cortés, and Mercè Martí. 2022. "Evaluation of the TCR Repertoire as a Predictive and Prognostic Biomarker in Cancer: Diversity or Clonality?" Cancers 14, no. 7: 1771. https://doi.org/10.3390/cancers14071771
APA StyleAran, A., Garrigós, L., Curigliano, G., Cortés, J., & Martí, M. (2022). Evaluation of the TCR Repertoire as a Predictive and Prognostic Biomarker in Cancer: Diversity or Clonality? Cancers, 14(7), 1771. https://doi.org/10.3390/cancers14071771