Simple Summary
A substantial disparity in breast cancer incidence and mortality exists between African American (AA) and European American (EA) women. However, the basis for these disparities is poorly understood. In this article, we describe that gene–environment interactions mediated through epigenetic modifications may play a significant role in racial disparities in BC incidence and outcomes. Our in silico analyses and an in-depth literature survey suggest that there exists a significant difference in epigenetic patterns between AA and EA women with breast cancer. Herein, we describe the environmental factors that contribute to these epigenetic changes, which may underlie the disparate racial burden in patients with breast cancer. We suggest that AA women with higher basal epigenetic changes, may have higher pre-disposition to cancer onset, and an aggressive disease course. Pre-existing racial differences in epigenetic profiles of breast tissues raises the possibility of examining these profiles for early diagnosis.
Abstract
Breast cancer (BC) is the most commonly diagnosed cancer in women. Despite advancements in BC screening, prevention, and treatment, BC incidence and mortality remain high among African American (AA) women. Compared with European American (EA) women, AA women tend to be diagnosed with more advanced and aggressive tumors and exhibit worse survival outcomes. Most studies investigating the determinants of racial disparities in BC have focused on genetic factors associated with African ancestry. However, various environmental and social stressors over an individual’s life course can also shape racial stratification in BC. These social and environmental exposures result in long-term changes in gene expression mediated by epigenetic mechanisms. Epigenetics is often portrayed as an intersection of socially patterned stress and genetic expression. The enduring nature of epigenetic changes makes them suitable for studying the effects of different environmental exposures over an individual’s life course on gene expression. The role of differential social and environmental exposures in racial disparities in BC suggests varied epigenetic profiles or signatures associated with specific BC subtypes in AA and EA women. These epigenetic profiles in EA and AA women could be used as biomarkers for early BC diagnosis and disease prognosis and may prove valuable for the development of targeted therapies for BC. This review article discusses the current state of knowledge regarding epigenetic differences between AA and EA women with BC. We also discuss the role of socio-environmental factors, including psychosocial stress, environmental toxicants, and dietary factors, in delineating the different epigenetic profiles in AA and EA patients with BC.
1. Introduction
Breast cancer (BC) is the second most common cause of cancer-related deaths among women in the United States [1,2,3,4]. Although a significant reduction in BC mortality has been achieved by increasing awareness and advancing BC diagnosis and treatment, a vast gap in BC mortality exists between African American (AA) and European American (EA) women. Studies have consistently shown higher BC mortality rates in AA women than in EA women. Although the overall BC incidence rate is slightly higher in EA women than in AA women, the BC incidence rate in women under the age of 45 is higher in AA women [5,6]. Furthermore, AA patients with BC exhibit more aggressive tumor phenotypes and worse overall survival than EA patients [1,2]. Racial and ethnic disparities in BC incidence and survival are significant public health concerns; however, the basis for these disparities is poorly understood.
The reasons behind the racial disparities in BC incidence and outcomes are complex and multifactorial, involving both non-biological and biological factors. Gene–environment interactions play a substantial role in the development and progression of BC. Specifically, environmental factors, including pollutants, social stressors, and cultural and behavioral factors (e.g., dietary lifestyle and physical activity), play a pivotal role in the etiology of BC through modulation of gene expression. These factors also contribute to racial disparities in BC [7] (Figure 1).
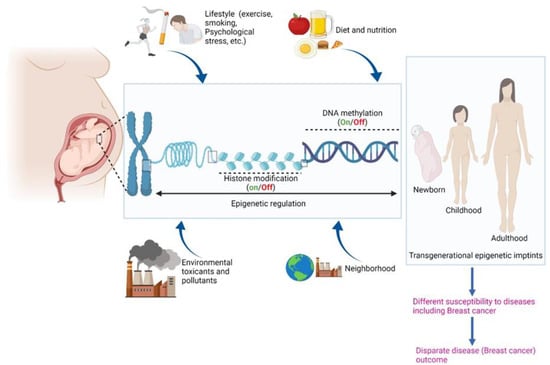
Figure 1.
Schematic depicting role of epigenetics and various biological and non-biological factors in etiology of BC. Created with BioRender.com (accessed on 22 January 2022).
Due to systemic inequalities, including structural racism, individuals in the US are exposed to different physical and social milieus through their life course according to their race or socioeconomic status [8]. These social determinants (i.e., structural inequities) play a significant role in disparities in health outcomes. A vast proportion of AAs is poorer than EAs, resulting in limited access to health care due to a lack of insurance or sufficient health coverage. Furthermore, low socioeconomic status is associated with behavioral risk factors, such as smoking, drinking, and physical inactivity, contributing to increased mortality [9]. AAs also face tremendous stress due to racial discrimination, which determines their access to housing and employment [10]. As a result of residential segregation along racial lines, AAs and minority communities tend to inhabit disadvantaged neighborhoods with high poverty, air pollution, and limited essential resources [11]. Factors associated with race and low socioeconomic status, including racial discrimination [12], neighborhood disadvantage, health behaviors, and exposure to pollutants [13], are associated with an increased risk of BC. Therefore, racial differences in social and environmental exposures over an individual’s lifetime can contribute to racial disparities in BC by influencing gene expression patterns. Structurally rooted biopsychosocial processes that the social patterning of stressors may activate in a historically unequal society could also influence cellular physiology and gene expression patterns in BC. In other words, gene–environment interactions mediated through epigenetic modifications may play a significant role in racial disparities in BC incidence and outcomes.
Epigenetic modifications (e.g., DNA methylation, histone modifications, and non-coding RNAs) can modulate gene expression without altering the DNA sequence (i.e., the inherited genome) [14]. Even though epigenetic modifications induced by environmental changes can be reversed, histone modifications and DNA methylation tend to be durable or transgenerational. As such, DNA methylation and histone modifications can serve as reliable molecular biomarkers to evaluate the cumulative effects of exposure to various physical and social environments over an individual’s life course. Lifetime exposure to specific environments associated with socioeconomic position and race may differentially imprint the epigenome, producing exposure-specific epigenetic signatures [15]. Consistent with this, specific DNA methylation patterns have been associated with low socioeconomic status, neighborhood disadvantage, and exposure to stressors [16]. In this review, we discuss the current state of knowledge about the differences in epigenetic patterns between AA and EA patients with BC. We also describe the environmental factors that contribute to these epigenetic changes.
2. Epigenome: A Tutor of the Genome
If we consider genes as the body’s hardware, then the epigenome is the software that instructs them how to work. Essentially, the epigenome is the instruction guide on top of the genome, and it serves as a dimmer switch that promotes a gene to turn on slightly, turn on fully, or turn off completely [17] (Figure 2). BC is a collusion of genetic and epigenetic alterations. Genetic and epigenetic changes intertwine and take advantage of each other during the BC initiation and progression, ultimately leading to aberrant gene expression. Changes in epigenetic regulators acquire genetic mutations, and genetic mutations in epigenetic regulators lead to an altered epigenome. It is believed that genetic changes acquired during BC progression are somewhat straightforward, but epigenetic pathway is much more intricate, which involves genome-wide loss of DNA methylation (hypomethylation), frequent hypermethylation in CpG islands of the promoter, changes in nucleosome occupancy, and modification profiles. Recent whole exome sequencing of various cancers including BC revealed that mutations in genes that regulate the epigenome are remarkably widespread [18]. Given that the epigenome works at the apex of the pyramid of gene control mechanisms, this suggests that the mutations in genes regulating the epigenome probably have impacts on multiple pathways of BC. Thus, in-depth research into how epigenetic and genetic modifiers communicate with each other to alter the expression is warranted.
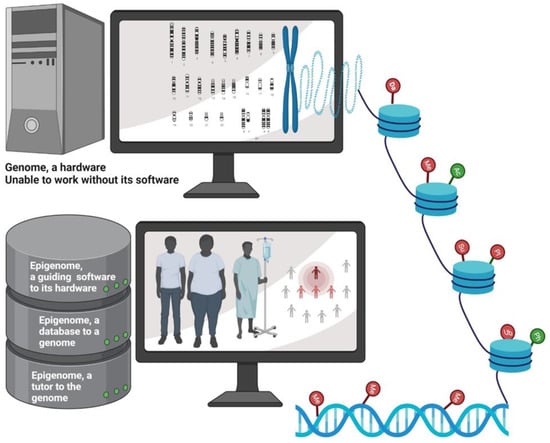
Figure 2.
Schematic depicting epigenetics as a software which guides the genome. Epigenetic regulation determines the phenotype of the person. Epigenetic imprints are transgenerational and impacts a group of population. Da: deamination; Me: methylation; Ub: ubiquitination; Su: Sumoylation; PI: protein isomerization; Ph: phosphorylation; Ac: acetylation. Symbols represented in green indicate activation of target gene due to epigenetic modification, while those represented in red indicate repression of target gene upon epigenetic modification. Created with BioRender.com (accessed on 22 January 2022).
Given the notion that genes are governed by biological memories of experiences acquired earlier in life or inherited by recent ancestors, studying epigenetic modifications serves as a potent alternative to the simple model of genetic determinism. Although genetic divergence between different ethnic groups could result in health disparities, evidence supports that BC inequalities are strongly influenced by various environmental stressors rather than being driven by genetic factors alone [19].
Studies have shown that social models of racial group identity might predict an individuals’ health status rather than genetic ancestry. Thus, comparing the experiences (e.g., social, physical, and nutritional experiences) of the present generation with those of their ancestors to identify epigenetic imprints may provide further insight into racial disparities in BC. Thus, a deep understanding of how genetic factors and the socially and economically structured environments that we inhabit interact to influence patterns in racially diverse BC patients is an unmet need.
3. Differences in DNA Methylation Patterns May Underpin Racial Disparity in Breast Cancer
Specific epigenetic changes involving DNA methylation and histone modifications have been associated with BC development and progression [20]. Various findings suggest that different epigenetic signatures may exist in AA and EA patients with BC. The presence of such epigenetic signatures is supported by recent studies demonstrating different methylation patterns in BC tissues from AA and EA patients [21]. Identifying epigenetic signatures and the corresponding environmental factors associated with these epigenetic patterns in AA and EA patients with BC will improve our understanding of BC etiology. Additionally, epigenetic patterns at specific sites may serve as diagnostic or prognostic biomarkers in AA and EA patients and guide the development of personalized therapies for different BC subtypes.
Due to its potentially enduring effects, DNA methylation is one of the most commonly studied epigenetic modifications in the context of cancer disparities [21]. In humans, DNA methylation typically occurs at cytosine nucleotides that are immediately followed by a guanine nucleotide (i.e., CpG dinucleotides). CpG dinucleotides are frequently found in the form of CpG repeats in the 5′ regulatory region of genes. These CpG repeats are known as CpG islands, and their methylation can modulate gene transcription. Although DNA methylation can either induce or suppress their expression, hypermethylation is often associated with gene silencing. Similarly, hypomethylation is associated with increased gene expression [22]. Aberrant methylation of CpG islands is a frequent and early event in BC [22]. Additionally, the methylation status of CpG islands is emerging as a diagnostic biomarker for BC [22].
Repression of tumor suppressor gene (TSG) expression has been observed in various cancers and is associated with poor prognosis [23]. Silencing of TSGs in cancer is often caused by hypermethylation of CpG islands in their promoter regions. Interestingly, differences in DNA methylation profiles of various TSGs have been observed between AA and EA patients with BC (Table 1) [21]. Mehrotra et al. investigated the methylation status of five TSGs (SCGB3A1, TWIST1, CCND2, RARB, and RASSF1) in AA and EA patients with BC [21]. SCGB3A, TWIST1, CCND2, and RASSF1 were hypermethylated in AA but not in EA patients with BC. These racial differences in methylation patterns were more pronounced in patients under the age of 50 with ER−/PR− tumors than in younger patients with ER+/PR+ tumors [21]. TSG hypermethylation in AAs with ER−/PR− BC may contribute to the aggressive tumor phenotypes observed in AA women. Wang et al. showed that the TSG CDH13 (encoding cadherin 13) was differentially methylated between AA and EA women with ER- BC [24]. Nevertheless, CDH13 was not differentially methylated between AA and EA patients with ER+ BC [24].

Table 1.
Hypermethylated TSGs in racially diverse AA and EA patients with BC (reported data).
Other cancer-related genes are also differentially methylated between AAs and EAs with BC. Conway et al. compared samples from AA and EA patients with BC and identified differential methylation patterns in several cancer-related genes, including DSC2, KCNK4, GSTM1, AXL, DNAJC15, SNORD115, TUSC3, and TES [27]. TES was hypermethylated in AA patients regardless of tumor type, whereas TUSC3 was hypermethylated only in AA patients with hormone receptor-negative BC [27]. These findings indicate that the differential methylation of TSGs and other cancer-related genes between AA and EA patients with BC is strongly associated with BC subtype, receptor status, and patient’s age. Genes involved in transcriptional regulation, metabolism, and signal transduction have also been found to be differentially methylated between BC patients and healthy individuals (Table 2 and Table 3) [28]. Whether these genes also display racial discrepancies in their methylation status merits further investigation.
In a genome-wide DNA methylation profiling study, Ambrosone et al. identified 157 CpG sites that were differentially methylated in normal breast, ER− BC, and ER+ BC tissues from EA and AA women [29]. Almost twice as many CpG sites were differentially methylated (EA vs. AA) in ER- tumors, indicating that different etiological pathways may be involved in the development of ER− BC in AA and EA women. Racial differences in gene methylation patterns have also been observed in breast tissues from healthy women. In breast tissues from healthy AA and EA women, Song et al. identified 485 differentially methylated genes, including AHRR, OPCML, PACS2, HIPK2, and TNK2 [30].
Table 2.
Hypermethylated TSGs in patients with BC (reported data, race wise difference in methylation pattern not studied).
Table 2.
Hypermethylated TSGs in patients with BC (reported data, race wise difference in methylation pattern not studied).
| Tumor Suppressor Genes | Regular Function | Effect after Hypermethylation | Comments | References |
|---|---|---|---|---|
| BRCA1 |
|
|
| [31] |
| GSTP1 |
|
|
| [32] |
| NKX-2 NKX-5 |
|
|
| [33] |
| GARL2 |
|
|
| [33] |
| CDKN2A |
|
|
| [34] |
| RIL |
|
|
| [35] |
| PTEN |
|
|
| [36] |
| ARH1 |
|
|
| [37] |
| 14-3-3 sigma |
|
|
| [37] |
| RIZ1 |
|
|
| [37] |
| DAPK1 |
|
|
| [38] |
These differences in epigenetic patterns may predispose AA and EA women to specific BC subtypes or affect disease outcomes. As such, DNA methylation patterns may serve as diagnostic markers or help guide personalized treatment of different BC subtypes in individuals with a particular ancestry.
Table 3.
Hypermethylated genes (other than TSGs) in patients with BC (reported data, race wise difference in methylation pattern not studied).
Table 3.
Hypermethylated genes (other than TSGs) in patients with BC (reported data, race wise difference in methylation pattern not studied).
| Genes | Regular Function | Effect after Promoter Hypermethylation | Comments | References |
|---|---|---|---|---|
| CXCL12 |
|
|
| [39] |
| CXCR4 |
|
|
| [40] |
| TOX |
|
|
| [41] |
| SPOCK2 |
|
|
| [33] |
| DPY5 |
|
|
| [33] |
| PITX2 |
|
|
| [42] |
| TP53 |
|
|
| [43] |
| MDM2 |
|
|
| [28] |
4. Differentially Methylated Genes in AA and EA Breast Cancer Patients: Unpublished Data from In Silico Analysis
We performed an in silico analysis of data obtained from 738 BC patients (AAs, n = 160; EAs, n = 578) to identify differences in methylation patterns between AA and EA BC patients using the UALCAN portal [44]. We determined the methylation status of genes regulating various functions in BC based on the β-value ranging from 0 (unmethylated) to 1 (fully methylated). Different β-value cutoffs have been considered to indicate hypermethylation (β-value: 0.5–0.7) or hypomethylation (β-value: 0.25–0.3). Out of ~150 genes, 50 genes were found to be significantly (p ≤ 0.05) differentially methylated between AA and EA BC patients; 36 genes were hypermethylated, and 14 were hypomethylated (Figure 3a,b, Table 4 and Table 5). Notably, hypermethylation of most TSGs (including MIR663A, ZNF208, and NKAPL) was more profound in AAs than in EAs. Furthermore, the TSG S100A2 was hypomethylated in EA patients with BC. TSG hypermethylation in AA patients may contribute to aggressive tumor phenotypes and poor disease outcomes.
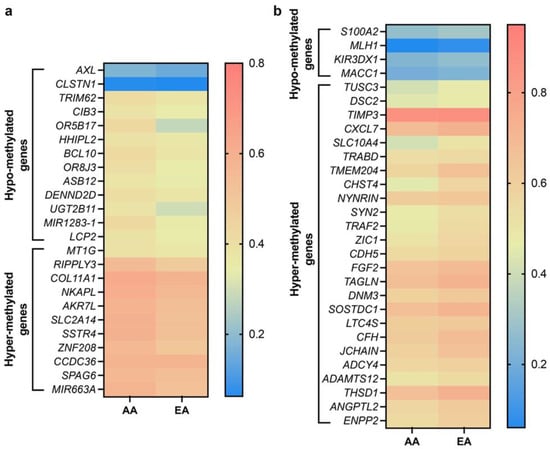
Figure 3.
Heatmaps showing differential methylation pattern of various genes in AA and EA BC patients. (a) Heatmap showing significantly hypo- and hyper-methylated genes higher in AA over EA BC patients. (b) Heatmap showing significantly hypo- and hyper-methylated genes higher in EA over AA BC patients. AA, n = 160, EA, n = 578, p ≤ 0.05. Data were analyzed using graph pad prism 8 software.
Table 4.
Hypermethylated genes in AA and EA patients with BC (in silico analysis, unpublished data).
Table 5.
Hypomethylated genes in AA and EA patients with BC (in silico analysis, unpublished data).
However, some TSGs were hypermethylated in EAs or hypomethylated in AAs. The methylation levels of the TSGs TIMP3 and TUSC3 were higher in EA than in AA patients, whereas DENDD2D and TRIM62 were hypomethylated in AA patients with BC. The methylation levels of genes involved in tumorigenesis, metastasis, invasion, angiogenesis, and cell proliferation (e.g., ENPP2, ANGPTL2, THSD1, LTC4S, DNM3, TAGLN, FGF2, CDH5, ZIC1, TANK/TRAF2, CXCL7) were higher in EA patients with BC than in their AA counterparts. These differences in methylation patterns may contribute to the more aggressive disease course observed in AA BC patients than EA women. Although these differential methylation patterns may contribute to racial disparities in BC outcomes, further in vitro validation of these data is necessary.
5. Plausible Role of Histone Modifications in Breast Cancer Racial Disparity
Gene silencing by DNA hypermethylation is closely related to chromatin modifications involving acetylation, methylation, phosphorylation, sumoylation, deamination, proline isomerization, and ubiquitination. These chromatin modifications play a substantial role in the organization of DNA in nucleosomes [72]. Alterations in chromatin remodeling factors and histone-modifying enzymes are frequently observed in BC [73]. Histone modifications, along with DNA methylation, modulate the transcriptional state of many genes regulating cancer progression [73]. In general, histone acetylation, methylation, phosphorylation, and ubiquitination are associated with gene activation. In contrast, histone methylation, ubiquitination, sumoylation, deamination, and proline isomerization tend to repress gene expression. However, the role of these modifications in repressing or activating gene expression is context-dependent [74].
Although distinct histone modification patterns have been identified in BC samples and BC subtypes, these patterns have not been studied in racially diverse patients. Alteration in the methylation levels of histone 3, lysine 27 (H3K27me), a modification associated with gene repression, is a hallmark of transformation in BC [75]. In vitro and in vivo data suggest that H3K27me may be a suitable target for anti-neoplastic therapy [75]. Differential expression of H3K27me3 (trimethylation of H3K27) has also been reported among the different BC subtypes [76]. Holm et al. reported higher H3K27me3 levels in luminal A, HER2-enriched, and normal-like tumors than in basal-like, triple-negative, and luminal B subtypes. Furthermore, higher H3K27Me3 levels have been associated with better BC outcomes [76]. Genome-wide sequencing studies have shown that H3K27me deregulation can result from mutations in the H3K27 methyltransferase complex consisting of PRC2, EZH2, and accessory proteins [77]. These mutations alter H3K27me function and are linked to poor clinical outcomes in cancer patients; however, their role in BC racial disparities merits further investigation.
Changes in the global methylation status of histone H3 lysine 4 (H3K4) [78] and enzymes regulating these modifications [79] are also associated with tumorigenesis. The presence of the H3K4me3 mark in the promoter of the ERBB2 (i.e., HER2) gene is associated with HER2-overexpressing breast carcinomas [80]. HER2 overexpression is associated with an increased risk of all-cause mortality and BC-specific mortality in EA women with low Native American (NA) ancestry [81]. Messier et al. reported a global increase in H3K4 methylation (H3K4me) and acetylation (H3K4ac) in BC cell lines relative to normal breast epithelial cells. An increase in H3K4ac marks was associated with early stage cancer progression. On the other hand, H3K4me was predominant in the metastatic cell line MDA-MB-231, indicating its potential role in epithelial-mesenchymal transition in late-stage cancer [82]. Considering the importance of the H3K4me and H3K4ac mark in BC progression, its potential role in BC disparities between AAs and EAs should be investigated.
Another chromatin-modifying enzyme involved in BC progression is lysine-specific demethylase 1(KDM1A). KDM1A catalyzes the removal of methylated groups from H3K4 and H3K9 [83]. Altered KDM1A expression is associated with tumorigenesis and is upregulated in various cancers [84]. KDM1A is involved in BC cell proliferation [85], invasion, and metastasis [86], and that KDM1A overexpression is associated with aggressive and poorly differentiated BC [83]. KDM1A also plays a critical role in BC chemoresistance by maintaining a pool of cancer stem cells [83]. Lim et al. identified KDM1A expression level as a predictive biomarker for aggressive tumor phenotypes biology in patients with BC, with ER- tumors exhibiting higher KDM1A expression levels [87]. Our survival analysis from UALCAN (AA, n = 46; EA, n = 182) confirmed the prognostic value of KDM1A expression level (Figure 4). For this analysis, samples were categorized into two groups: (1) High expression (with TPM values above upper quartile) and (2) Low/Medium expression (with TPM values below upper quartile).The Kaplan–Meier survival plot was generated for every gene in each TCGA cancer type, using “survival” package [88] and “survminer” package [89]. Our analysis revealed that high expression of KDM1A is associated with lower survival rates, particularly in AA than EA BC patients (Figure 4a–d). Thus, downstream genes epigenetically regulated by KDM1A may contribute to the aggressive BC phenotypes observed in AA patients. Variation in global levels of histone marks, such as lysine acetylation (H3K9ac, H3K18ac, H4K12ac, and H4K16ac), lysine methylation (H3K4me2 and H4K20me3), and arginine methylation (H4R3me2), are also linked with different BC subtypes and outcomes [20], but their role in racially diverse BC population is largely unexplored. Delineating these histone modifications and their effect on gene expression could help us understand the biological mechanisms underpinning BC racial disparities.
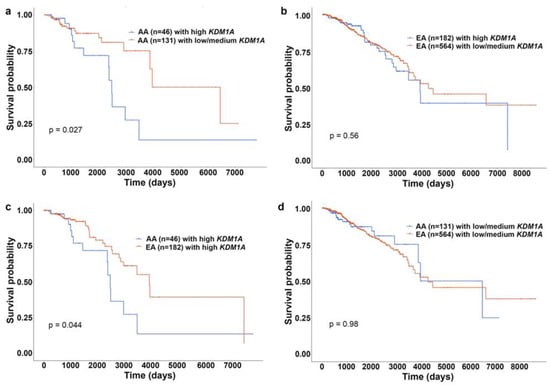
Figure 4.
KM plots showing effect of KDM1A expression on survival of AA and EA patients with BC. (a) KM plot showing survival rates of AA patients with BC with high (blue) and low/medium (red) expression of KDM1A. (b) KM plot showing survival rates of EA patients with BC with high (blue) and low/medium (red) expression of KDM1A. (c) KM plot showing survival rates of AA (blue) EA (red) patients with BC with high expression of KDM1A. (d) KM plot showing survival rates of AA (blue) EA (red) patients with BC with low/medium expression of KDM1A.
6. RNA Modifications—Another Layer of Epigenetic Regulation
The discovery of the first RNA modification enzyme, such as fat mass and obesity associated protein (FTO), which promotes oxidative demethylation of abundant N6-methyladenosine (m6A) residues in RNA, underscores the idea that alteration in RNA may serve as an epigenetic marker [90]. Gene regulation by miRNAs is another critical epigenetic event, and its role in cancer health disparities is evolving [91]. ncRNAs, a cluster of RNAs that do not encode a functional protein, have recently garnered attention of scientists for their role in regulation of gene expression through epigenetic network [92]. The hypermethylation of CpG islands near the miRNA genes causes transcription repression by epigenetic silencing. A study by Nassar FJ [93] performed a comparative miRNA microarray profile analysis of Lebanese and matched American BC patients. Their study demonstrated 21 exclusively dysregulated miRNA (miR-31, 362-3p, and 663) and 4 miRNA (miR-1288-star and 324-3p) with different expression pattern in Lebanese than American patients with BC. A study by Lewis K et al.,2018 [94] suggested that a combinatorial treatment with suberoylanilide (HDAC inhibitor) and epigallocatechin gallate (EGCG) (DNMT inhibitor) down-regulated the expression of oncogenic miRNA-221/222 in AA TNBC cell lines, such as MDA-MB-157 and HCC 1806. However, this study failed to incorporate EA TNBC cell lines and other BC specific AA and EA cell lines. Thus, a comprehensive miRNA profiling analysis is warranted in AA and EA patients with BC to understand the race signatures that are responsible for differential disease course and therapeutic outcomes.
Furthermore, an aberrant miRNA activity is also responsible for ectopic expression of human telomerase reverse transcriptase (hTERT) that regulates the proliferative span of cells. Various studies have reported that hTERT reactivation by aberrant miRNA expression is an early molecular event during BC neoplastic transformation and enhances genomic instability [95,96,97]. The miRNAs control hTERT expression by either directly acting on 3′-UTR/ORF of hTERT mRNA or indirectly intervening with other genes that control hTERT expression. Wu et al. have demonstrated that miRNA-4458 suppressed expression of hTERT in selected BC cell lines [98]. Hypermethylation of the CpG sites within the hTERT promoter regulatory region causes transcriptional activation, and hypomethylation enables transcription repression [99]. Apart from CpG methylation, hTERT expression is also regulated by histone modification. Various studies have reported a decrease in repressive H3K9me3 and H3K27me3 mark while an increase in activator marks, such as ac-H3, H3K9ac, and ac-H4 acetylation on the hTERT promoter in various cancer cell lines [100,101]. A thorough analysis of differential miRNA signatures and their hTERT regulation could help delineate the disparate racial burden in BC.
7. Role of Diet and Nutrition in Breast Cancer Racial Disparities through Epigenetic Modifications
Diet plays a significant role in BC development and progression [102]. For example, a Mediterranean diet rich in fiber and consisting of vegetables, fruits, fish, and soy can significantly reduce BC risk. The epidemiological link between BC and dietary factors is also supported by studies describing the impact of diet on methylation patterns of genes involved in cancer-related pathways [103].
DNA methyltransferases use methyl donor groups, including S-adenosyl methionine (SAM), to catalyze DNA methylation [104]. The production of SAM is dependent on dietary sources of methyl groups, such as foods containing methionine and folate (vitamin B9). The amino acid methionine is abundant in fish, meat, and dairy products, whereas folate is present in green leafy vegetables and fruits. Mikol et al. reported that folate and methionine deficiencies significantly altered DNA methylation patterns and induced liver cancer in rats in the absence of a carcinogen [105]. Although the evidence linking folate deficiency with an increased BC risk has been mixed [106], the data are more conclusive for BC subtypes. Recent population-based studies suggest that higher folate intake may lower the risk of ER−/PR− [107] and ER− BC [107]. Similarly, impaired folate absorption and metabolism due to alcohol consumption are associated with an increased risk of BC [108]. Although the evidence for an association between folate intake and overall BC risk remains inconclusive, folate intake may be especially critical in reducing the risk of BC in women who consume moderate-to-high alcohol levels [107]. Sustained deficiency of methyl-group-containing foods [109] or intake of high levels of methyl antagonists, such as alcohol [110], can result in aberrant methylation patterns. Christensen et al. reported changes in methylation patterns in breast tumor tissues, with increased folate intake associated with lower global hypomethylation and higher alcohol consumption linked with increased global hypomethylation [110]. They also found an association between folate intake and IL17RB, a protein implicated in BC development and progression [111].
Obesity is also associated with an increased risk of BC incidence and mortality [112]. Munsell et al. reported a positive association between obesity and risk of ER+/PR+ BC but not ER−/PR− BC [95]. Consistent with the relationship between obesity and BC, several studies have also revealed a positive association between dietary fat intake and BC risk [113]. Some of the effects of obesity and dietary behaviors on BC risk may be mediated by changes in DNA methylation patterns. Delgado-Cruzata et al. found that weight-loss interventions in overweight BC survivors could increase global methylation levels in long interspersed element-1 (LINE-1) [96]. However, the study did not investigate whether these interventions led to a reduction in BC recurrence. Several population-based studies in BC patients have also revealed a positive association between obesity and hypermethylation of tumor suppressor genes, such as RASSF1 [97], BRCA1 [97], RARB [114], ECAD [114], and CDKN2A [114].
Differences in dietary patterns and the consequent epigenetic changes may, in part, explain the racial disparities in BC occurrence and outcomes. Poor and minority communities do not have access to a healthy, fiber-rich diet due to the lack of access to supermarkets that sell affordable healthy and fresh food [115]. Instead, these neighborhoods tend to have access to cheaper high-fat food and alcohol [116]. Such areas, referred to as food deserts, are associated with higher rates of tobacco use, diabetes, and high BC mortality rates [117]. Compared with EAs, AAs are more likely to have a diet high in cholesterol and low in dietary fiber and folate [118]. Furthermore, AA women are less likely to engage in physical activity or meet the suggested physical activity guidelines [119]. The lack of access to healthy food and physical inactivity may contribute to the high risk of BC among AA women [119,120].
9. Potential Role of Environmental Toxicants in Racial Disparities in Breast Cancer through Epigenetic Changes
Emerging evidence suggests that air pollutants can also increase the risk of BC [149]. Studies show that individuals residing in the proximity of chemical facilities and high-traffic urban zones are more susceptible to BC [150]. Traffic- and industry-related air pollutants, including nitrogen dioxide, sulfur dioxide, polycyclic aromatic hydrocarbons (PAHs), particulate matter, and carbon monoxide, are associated with an increased risk of BC [151]. PAHs [152] and metallic pollutants [153] exert their carcinogenic effects by mimicking or antagonizing the actions of estrogen and, thus, acting as endocrine disruptors. Emerging evidence also suggests that air pollutants may enhance the risk of BC through epigenetic mechanisms [149,154].
Individuals belonging to minority communities or having low socioeconomic status are at a disproportionately high risk of exposure to air pollutants. For example, AAs are more likely to inhabit localities in the vicinity of polluting industrial facilities [155]. These inequalities may be attributed to residential segregation along racial lines, with disenfranchised communities often having limited power to influence decisions where the industries are located [156]. Moreover, discriminatory zoning policies or those designed in the past with such intent often allow polluting industries to enter such neighborhoods. Furthermore, non-white, and low-income children are more likely to reside in high-traffic areas [157]. A recent prospective study showed that childhood residence in the proximity of a road with a median or other form of barrier suggestive of high vehicular traffic was associated with an increased risk of BC [158]. Thus, AAs are more likely to be exposed to air pollutants, and aberrant epigenetic alterations due to air pollutants may contribute to racial disparities in BC.
9.1. Metallic Pollutants
Although there are inconsistencies in the literature regarding the specific metallic pollutants associated with an elevated risk of BC, accumulating evidence suggests that long-term exposure to at least some of these pollutants can promote BC development. Recent studies showed that metallic air pollutants, including arsenic, lead, mercury, antimony, and cadmium, can increase the Torisk of BC [159,160]. The California Teachers Study revealed an association between cadmium and arsenic exposure and hormone receptor-negative BC among non-smokers [161]. Metallic air pollutants accumulating in the adipose tissue of the breast can have endocrine-disrupting effects [162]. Furthermore, the concentrations of cadmium, mercury, and lead in the breast tissue are higher in women with BC than in healthy women [162]. The study found that active concentrations of these metals are comparable to biomarker concentrations in AA women. In a methylomic analysis, Hanna et al. showed that lead and mercury exposure were associated with hypomethylation of the COL1A2 promoter and hypermethylation of the GSTM1 promoter, respectively [163]. Importantly, changes in COL1A2 and GSTM1 [164] expression have been associated with BC development and progression. These studies should further be extrapolated to racially diverse BC population to see if any difference in methylation patterns exist due to the higher concentration of metallic pollutants in AA than EA women.
9.2. Polycyclic Aromatic Hydrocarbons
PAHs are present in fossil fuel emissions, tobacco smoke, and smoked meats [165], and exposure to PAHs is linked to increased BC risk [166]. Furthermore, exposure to PAHs is associated with aberrant methylation patterns in promoters of cancer-related genes in BC cell lines and tissues. Exposure of the BC cell lines to the PAH benzo [a]pyrene (BaP) was associated with aberrant methylation of specific genes, but global methylation patterns remained unaltered [165]. Specifically, the TSG TSC2 was significantly hypomethylated in MCF-7 cells exposed to BaP. Changes in TSC2 expression have been implicated in BC metastasis [167], suggesting that exposure to PAHs may affect BC outcomes due to aberrant DNA methylation. A population-based study demonstrated that polycyclic hydrocarbon exposure was associated with changes in the methylation status of various BC-related genes in peripheral blood cells. For example, synthetic log burning-related exposure to PAH was associated with hypermethylation of the tumor suppressor genes HIN1 and CDH1 and hypomethylation of the oncogene CCND2. In contrast, smoking and vehicular traffic exposure were associated with hypermethylation of the metastasis-related gene TWIST1 and hypomethylation of the TSG DAPK1 (encoding death-associated protein kinase 1) [166]. Callahan et al. investigated the association between lifetime PAH exposure and epigenetic changes in BC by estimating the exposure to particulate matter and traffic emissions (as a proxy for PAH) at the subject’s address at birth, menarche, and the birth of the first child [168]. They found a positive association between the methylation of BC-related genes, depending upon the source of PAHs and age at exposure. For example, exposure to traffic-related PAHs at birth was associated with increased methylation of the TSG SYK in breast tumor tissues [168]. To date there are no studies showing differential PAH exposure in AA and EA women with BC. However, such studies are warranted in future to understand disparate BC burden among AA and EA.
9.3. Nitrogen Dioxide
Nitrogen dioxide (NO2) is another air pollutant released by vehicles and industries as a by-product of fuel combustion. Exposure to NO2 in ambient air is associated with an increased risk of BC [169,170] and can alter methylation patterns in BC-related genes. Plusquin et al. investigated the association between lifelong exposure to air pollutants and methylation patterns in the blood samples of two cohorts from the European Prospective Investigation into Cancer and Nutrition (EPIC) study [171]. NO2 exposure was associated with global hypomethylation, especially at CpG island shelves and shores. The TSG EPHB2 encoding ephrin type-B receptor 2 was among the significantly hypomethylated genes in response to NO2 exposure. Lower expression of EPHB2, (member of the Eph tyrosine kinase receptor family), is associated with tumor invasiveness, metastasis, and poor survival outcomes in BC [172,173]. Although these studies do not establish direct causation between various air pollutants and BC, they suggest that air pollutants may contribute to BC development and progression by inducing epigenetic changes.
10. Potential Role of Smoking in Racial Disparities in BC through Epigenetic Changes
Cigarettes contain a wide array of carcinogenic compounds and endocrine disruptors that can cause cancer [174]. Although the role of smoking in BC is inconclusive, recent studies revealed a modest association between chronic smoking and BC development [175,176]. Moreover, evidence exists that suggests that smoking may elevate the risk of ER+ BC but not of triple-negative BC [177,178]. In addition, smoking at the time of BC diagnosis has been associated with poor survival outcomes [179]. A large population-based study showed that smoking at the time of BC diagnosis was associated with higher BC mortality in the long term (13 years) but not in the short term (5 years) [180]. The study also showed that the negative impact of smoking on long-term survival was more profound in AA women than in EA women. Furthermore, smoking during BC diagnosis was associated with poor survival in women with ER- tumors but not in those with ER+ BC [180]. Although the prevalence of smoking is slightly higher in EA women than in AA women in the general population [181], a similar smoking rate has been reported at the time of BC diagnosis [180]. Therefore, racial differences in BC-associated mortality may be partly attributed to preexisting genetic variation and differences in ER status between AAs and EAs. However, it is also plausible that smoking could potentiate the effects of these genetic differences by modulating gene-environment interactions.
Epigenome-wide association studies showed that nearly 1000 CpG sites were differentially methylated in peripheral blood cells of active smokers and non-smokers [182]. Hypomethylation of CpG sites at the gene AHRR (encoding aryl hydrocarbon receptor repressor) and the intergenic locus 2q37.1 have been consistently observed in these studies [182]. In an epigenome-wide association study, Shenker et al. found that hypomethylation of 2q37.1 locus but not AHRR was associated with increased BC risk [183]. Furthermore, specific methylation patterns have been observed in breast tumors from smokers and non-smokers with different BC subtypes. Conway et al. reported that in hormone receptor-negative (HR−) tumors, hypomethylation of CpG sites in cancer-related genes was more profound in smokers than in non-smokers [184]. In contrast, smoking was associated with hypermethylation of CpG sites in BC-related genes in HR+ tumors [184]. Additionally, HR+ tumors from AA smokers showed higher hypermethylation levels compared with those from EA smokers. In HR- tumors, no differences in methylation levels were observed between AA and EA smokers.
Furthermore, the Carolina Breast Cancer study revealed significant racial differences in the methylation patterns of CpG sites in the promoter regions of several cancer-related genes in breast tumors [27]. Differentially methylated genes between AA and non-AA patients included the gene AXL, which was hypermethylated in tumor tissues from AAs. AXL encodes a receptor tyrosine kinase implicated in BC initiation and progression [185]. Notably, AXL hypermethylation has been associated with prenatal smoke exposure [186]. Chronic smoking has also been associated with lower oral [187] and serum folate levels [188]. In turn, folate deficiency can increase the risk of BC by impairing DNA methylation [189]. These smoking-associated differences in epigenetic patterns between AAs and EAs may contribute to racial disparities in BC incidence and clinical outcomes.
11. Conclusions
In this review article, we have outlined differences in epigenetic patterns between AA and EA patients with BC that may contribute to racial disparities in BC incidence and clinical outcomes. Despite evidence supporting the presence of racial differences in DNA methylation patterns in BC, such differences in histone modification patterns are underexplored. Some of the differentially methylated genes between AA and EA patients with BC have been identified as biomarkers of cancer development and progression. These genes could be used as diagnostic markers and to guide the development of personalized treatments. Differences in methylation patterns have also been observed between healthy AA and EA women. Although epigenetic regulation of genes can increase the risk of cancer in individuals of any race/ethnicity, AA women with higher basal epigenetic changes, might be disadvantaged and are, therefore, much more pre-disposed to cancer onset, and an aggressive disease course. The presence of preexisting racial differences in epigenetic profiles of breast tissues indicates that these profiles could be used for early diagnosis. Classification of the different BC subtypes in AA and EA BC patients with the help of detailed epigenetic profiles may allow for early diagnosis, prediction of prognosis, and development of personalized treatments. Characterizing the epigenetic patterns associated with BC in AA and EA patients may also improve our better understanding of the complex tumor biology of BC. Most current epigenetic profiling approaches involve the analysis of bulk tumor cells consisting of a diverse cell population. Thus, the use of epigenetic profiling as a tool for understanding tumor biology would require more fine-tuned approaches to capture the heterogeneity of cells populating tumors and the tumor microenvironment. Epigenetic changes due to social and environmental factors may also mediate racial disparities in BC outcomes. Differences in dietary habits, exposure to environmental pollutants, and psychosocial stress associated with elevated BC risk are, to a large extent, a consequence of racial and economic inequalities. Although it may be tempting to suggest mitigating measures on the community or local level to reduce BC disparities, the structural inequalities at the core of these disparities necessitate urgent broader systemic changes that complement local action.
Author Contributions
Conceptualization, S.J., C.G. and R.A.; methodology, S.J. and C.G.; software, S.J. and C.G.; validation, S.J., C.G. and R.A.; formal analysis, S.J., C.G.; investigation, S.J., C.G. and R.A.; resources, R.A.; data curation, S.J. and C.G.; writing—original draft preparation, S.J. and C.G.; writing—review and editing, S.J., C.G. and R.A.; visualization, S.J. and C.G..; supervision, R.A.; project administration, R.A.; funding acquisition, R.A. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a grant from the National Cancer Institutes of Health (R01CA239120) to RA.
Data Availability Statement
The in-silico results of BC AA and EA samples shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga (accessed on 22 January 2022). The data underlying this article will be shared on a reasonable request to the corresponding author (R.A.).
Acknowledgments
Schematics were created with BioRender.com (accessed on 22 January 2022). Editorial support for this manuscript was provided by Deep Shukla.
Conflicts of Interest
The authors declare no conflict of interest.
References
- DeSantis, C.E.; Fedewa, S.A.; Sauer, A.G.; Kramer, J.L.; Smith, R.A.; Jemal, A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J. Clin. 2016, 66, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlapati, C.; Joshi, S.; Sahoo, B.; Kapoor, S.; Aneja, R. The persisting puzzle of racial disparity in triple negative breast cancer: Looking through a new lens. Front. Biosci. 2019, 11, 75–88. [Google Scholar]
- Saini, G.; Joshi, S.; Garlapati, C.; Li, H.; Kong, J.; Krishnamurthy, J.; Reid, M.D.; Aneja, R. Polyploid giant cancer cell charac-terization: New frontiers in predicting response to chemotherapy in breast cancer. Semin. Cancer Biol. 2021. [Google Scholar]
- Ghimire, H.; Garlapati, C.; Janssen, E.A.M.; Krishnamurti, U.; Qin, G.; Aneja, R.; Perera, A.G.U. Protein Conformational Changes in Breast Cancer Sera Using Infrared Spectroscopic Analysis. Cancers 2020, 12, 1708. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.K.; Agurs-Collins, T.; Browne, D.; Lubet, R.; Johnson, K.A. Health disparities in breast cancer: Biology meets socio-economic status. Breast Cancer Res. Treat. 2010, 121, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.; Garlapati, C.; Li, X.; Turaga, R.C.; Oprea-Ilies, G.; Wright, N.; Bhattarai, S.; Mittal, K.; Wetherilt, C.S.; Krishnamurti, U.; et al. Multi-institutional study of nuclear KIFC1 as a biomarker of poor prognosis in African American women with triple-negative breast cancer. Sci. Rep. 2017, 7, 42289. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, M.C.; Grayson, J.; Lie, A.; Yu, C.H.; Shiao, S.-Y.P.K. Gene-environment interactions and predictors of breast cancer in family-based multi-ethnic groups. Oncotarget 2018, 9, 29019–29035. [Google Scholar] [CrossRef] [Green Version]
- Bailey, Z.D.; Krieger, N.; Agénor, M.; Graves, J.; Linos, N.; Bassett, M.T. Structural racism and health inequities in the USA: Evidence and interventions. Lancet 2017, 389, 1453–1463. [Google Scholar] [CrossRef]
- Nandi, A.; Glymour, M.M.; Subramanian, S.V. Association Among Socioeconomic Status, Health Behaviors, and All-Cause Mortality in the United States. Epidemiology 2014, 25, 170–177. [Google Scholar] [CrossRef]
- Williams, D.R.; Mohammed, S.A.; Shields, A.E. Understanding and effectively addressing breast cancer in African American women: Unpacking the social context. Cancer 2016, 122, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.R.; Collins, C. Racial residential segregation: A fundamental cause of racial disparities in health. Public Health Rep. 2001, 116, 404–416. [Google Scholar] [CrossRef]
- Taylor, T.R.; Williams, C.D.; Makambi, K.H.; Mouton, C.; Harrell, J.P.; Cozier, Y.; Palmer, J.R.; Rosenberg, L.; Adams-Campbell, L.L. Racial Discrimination and Breast Cancer Incidence in US Black Women: The Black Women’s Health Study. Am. J. Epidemiol. 2007, 166, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Labrèche, F.; Weichenthal, S.; Lavigne, E.; Valois, M.-F.; Hatzopoulou, M.; Van Ryswyk, K.; Shekarrizfard, M.; Villeneuve, P.; Crouse, D.; et al. The association between the incidence of postmenopausal breast cancer and concentrations at street-level of nitrogen dioxide and ultrafine particles. Environ. Res. 2017, 158, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, H.; Samadani, A.A. DNA Methylation and Cancer Development: Molecular Mechanism. Cell Biochem. Biophys. 2013, 67, 501–513. [Google Scholar] [CrossRef]
- Olden, K.; Olden, H.A.; Lin, Y.-S. The Role of the Epigenome in Translating Neighborhood Disadvantage into Health Dis-parities. Curr. Environ. Health Rep. 2015, 2, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Lam, L.L.; Emberly, E.; Fraser, H.B.; Neumann, S.M.; Chen, E.; Miller, G.E.; Kobor, M.S. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA 2012, 109 (Suppl. 2), 17253–17260. [Google Scholar] [CrossRef] [Green Version]
- Bonetta, L. Epigenomics: The New Tool in Studying Complex Diseases. Nat. Educ. 2008, 1, 178. [Google Scholar]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Thayer, Z.M.; Kuzawa, C.W. Biological memories of past environments: Epigenetic pathways to health disparities. Epigenetics 2011, 6, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Elsheikh, S.E.; Green, A.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, J.; Ganpat, M.M.; Kanaan, Y.; Fackler, M.J.; McVeigh, M.; Lahti-Domenici, J.; Polyak, K.; Argani, P.; Naab, T.; Garrett, E.; et al. Estrogen Receptor/Progesterone Receptor-Negative Breast Cancers of Young African-American Women Have a Higher Frequency of Methylation of Multiple Genes than Those of Caucasian Women. Clin. Cancer Res. 2004, 10, 2052–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yan, L.; Davidson, N.E. DNA methylation in breast cancer. Endocr. Relat. Cancer 2001, 8, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.-K.; Yu, J. Promoter hypermethylation of tumour suppressor genes as potential biomarkers in colorectal cancer. Int. J. Mol. Sci. 2015, 16, 2472–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Dorsey, T.H.; Terunuma, A.; Kittles, R.A.; Ambs, S.; Kwabi-Addo, B. Relationship between Tumor DNA Methylation Status and Patient Characteristics in African-American and European-American Women with Breast Cancer. PLoS ONE 2012, 7, e37928. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, J.; Vali, M.; McVeigh, M.; Kominsky, S.L.; Fackler, M.J.; Lahti-Domenici, J.; Polyak, K.; Sacchi, N.; Garrett-Mayer, E.; Argani, P.; et al. Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin. Cancer Res. 2004, 10, 3104–3109. [Google Scholar] [CrossRef] [Green Version]
- Klopocki, E.; Kristiansen, G.; Wild, P.J.; Klaman, I.; Castaños-Vélez, E.; Singer, G.; Stöhr, R.; Simon, R.; Sauter, G.; Leibiger, H.; et al. Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int. J. Oncol. 2004, 25, 641–649. [Google Scholar] [CrossRef]
- Conway, K.; Edmiston, S.N.; Tse, C.-K.; Bryant, C.; Kuan, P.F.; Hair, B.Y.; Parrish, E.A.; May, R.; Swift-Scanlan, T. Racial Var-iation in Breast Tumor Promoter Methylation in the Carolina Breast Cancer Study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Tanas, A.S.; Sigin, V.O.; Kalinkin, A.I.; Litviakov, N.V.; Slonimskaya, E.M.; Ibragimova, M.K.; Ignatova, E.O.; Simonova, O.A.; Kuznetsova, E.B.; Kekeeva, T.V.; et al. Genome-wide methylotyping resolves breast cancer epigenetic heterogeneity and suggests novel therapeutic perspectives. Epigenomics 2019, 11, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Ambrosone, C.B.; Young, A.C.; Sucheston, L.E.; Wang, D.; Yan, L.; Liu, S.; Tang, L.; Hu, Q.; Freudenheim, J.L.; Shields, P.G.; et al. Genome-wide methylation patterns provide insight into differences in breast tumor biology between American women of African and European ancestry. Oncotarget 2013, 5, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Song, M.-A.; Brasky, T.M.; Marian, C.; Weng, D.Y.; Taslim, C.; Dumitrescu, R.G.; A Llanos, A.; Freudenheim, J.L.; Shields, P.G. Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics 2015, 10, 1177–1187. [Google Scholar] [CrossRef]
- Zhang, L.; Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 2015, 5, 17869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Wei, X.-M.; Zeng, X.-T.; Wang, F.-B.; Weng, H.; Long, X. Aberrant GSTP1 promoter methylation is associated with increased risk and advanced stage of breast cancer: A meta-analysis of 19 case-control studies. BMC Cancer 2015, 15, 920. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Kwabi-Addo, B.; Ittmann, M.; Jelinek, J.; Shen, L.; Yu, Y.; Issa, J.P.J. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS ONE 2008, 3, e2079. [Google Scholar] [CrossRef]
- Bae, Y.K.; Brown, A.; Garrett, E.; Bornman, D.; Fackler, M.J.; Sukumar, S.; Herman, J.G.; Gabrielson, E. Hypermethylation in Histologically Distinct Classes of Breast Cancer. Clin. Cancer Res. 2004, 10, 5998–6005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Shetty, P.B.; Feng, W.; Chenault, C.; Bast, R.C.; Issa, J.-P.J.; Hilsenbeck, S.G.; Yu, Y. Methylation of HIN-1, RASSF1A, RIL and CDH13 in breast cancer is associated with clinical characteristics, but only RASSF1A methylation is associated with outcome. BMC Cancer 2012, 12, 243. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Chen, J.; Mo, X. The association of PTEN hypermethylation and breast cancer: A meta-analysis. OncoTargets Ther. 2016, 9, 5643–5650. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef] [Green Version]
- Yadav, P.; Masroor, M.; Nandi, K.; Kaza, R.C.M.; Jain, S.K.; Khuarana, N.; Saxena, A. Promoter Methylation of BRCA1, DAPK1 and RASSF1A is Associated with Increased Mortality among Indian Women with Breast Cancer. Asian Pac. J. Cancer Prev. 2018, 19, 443–448. [Google Scholar]
- Wendt, M.K.; Cooper, A.N.; Dwinell, M.B. Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene 2008, 27, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Ramos, E.A.S.; Grochoski, M.; Prado, K.B.; Seniski, G.G.; Cavalli, I.J.; Ribeiro, E.M.S.F.; Camargo, A.A.; Costa, F.F.; Klassen, G. Epigenetic Changes of CXCR4 and Its Ligand CXCL12 as Prognostic Factors for Sporadic Breast Cancer. PLoS ONE 2011, 6, e29461. [Google Scholar] [CrossRef] [Green Version]
- Tessema, M.; Yingling, C.M.; Grimes, M.J.; Thomas, C.L.; Liu, Y.; Leng, S.; Joste, N.; Belinsky, S.A. Differential Epigenetic Regulation of TOX Subfamily High Mobility Group Box Genes in Lung and Breast Cancers. PLoS ONE 2012, 7, e34850. [Google Scholar] [CrossRef] [PubMed]
- Absmaier, M.; Napieralski, R.; Schuster, T.; Aubele, M.; Walch, A.; Magdolen, V.; Dorn, J.; Gross, E.; Harbeck, N.; Noske, A.; et al. PITX2 DNA-methylation predicts response to anthracycline-based adjuvant chemotherapy in triple-negative breast cancer patients. Int. J. Oncol. 2018, 52, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.H.; Kim, S.J.; Noh, D.-Y.; Park, I.A.; Choe, K.J.; Yoo, O.J.; Kang, H.-S. Methylation in the p53 Promoter Is a Supple-mentary Route to Breast Carcinogenesis: Correlation between CpG Methylation in the p53 Promoter and the Mutation of the p53 Gene in the Progression from Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Lab. Investig. 2001, 81, 573–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashetl, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Va-rambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Azare, J.; Doane, A.; Leslie, K.; Chang, Q.; Berishaj, M.; Nnoli, J.; Mark, K.; Al-Ahmadie, H.; Gerald, W.; Hassimi, M.; et al. Stat3 Mediates Expression of Autotaxin in Breast Cancer. PLoS ONE 2011, 6, e27851. [Google Scholar] [CrossRef] [Green Version]
- Stracke, L.M.; Clair, T.; Liotta, L.A. Autotaxin, tumor motility-stimulating exophosphodiesterase. Adv. Enzym. Regul. 1997, 37, 135–144. [Google Scholar] [CrossRef]
- Fan, Y.; Mu, J.; Huang, M.; Imani, S.; Wang, Y.; Lin, S.; Fan, J.; Wen, Q. Epigenetic identification of ADCY4 as a biomarker for breast cancer: An integrated analysis of adenylate cyclases. Epigenomics 2019, 11, 1561–1579. [Google Scholar] [CrossRef]
- Wang, H.; Yu, J.; Guo, Y.; Zhang, Z.; Liu, G.; Li, J.; Zhang, X.; Jin, T.; Wang, Z. Genetic variants in the ZNF208 gene are associated with esophageal cancer in a Chinese Han population. Oncotarget 2016, 7, 86829–86835. [Google Scholar] [CrossRef] [Green Version]
- Kumar, U.; Grigorakis, S.I.; Watt, H.L.; Sasi, R.; Snell, L.; Watson, P.; Chaudhari, S. Somatostatin receptors in primary human breast cancer: Quantitative analysis of mRNA for subtypes 1--5 and correlation with receptor protein expression and tumor pathology. Breast Cancer Res. Treat. 2005, 92, 175–186. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Helland, Å.; Fleischer, T.; Haug, K.M.; Alnaes, G.I.G.; Nebdal, D.; Syljuåsen, R.G.; Touleimat, N.; Busato, F.; Tost, J.; et al. Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int. J. Cancer 2014, 135, 2085–2095. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Gong, H.; Wang, T.; Wang, J.; Han, Z.; Bai, G.; Han, S.; Yang, X.; Zhou, W.; Liu, T.; et al. SOSTDC1 inhibits bone metastasis in non-small cell lung cancer and may serve as a clinical therapeutic target. Int. J. Mol. Med. 2018, 42, 3424–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, K.A.; Blish, K.R.; Birse, C.E.; Triplette, M.A.; Kute, T.E.; Russell, G.B.; D’Agostino, R.B.; Miller, L.D.; Torti, F.M.; Torti, S.V. SOSTDC1 differentially modulates Smad and beta-catenin activation and is down-regulated in breast cancer. Breast Cancer Res. Treat. 2011, 129, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, D.; Kimler, B.F.; Nothnick, W.B.; Davis, M.K.; Fan, F.; Tawfik, O. Transgelin: A potentially useful diagnostic marker differentially expressed in triple-negative and non-triple-negative breast cancers. Hum. Pathol. 2015, 46, 876–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Cao, F.; Gao, X.-J.; Wang, H.-B.; Chen, F.; Cai, S.-J.; Zhang, C.; Hu, Y.-W.; Ma, J.; Gu, X.; et al. ZIC1 acts a tumor suppressor in breast cancer by targeting survivin. Int. J. Oncol. 2018, 53, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Peramuhendige, P.; Marino, S.; Bishop, R.T.; De Ridder, D.; Khogeer, A.; Baldini, I.; Capulli, M.; Rucci, N.; Idris, A.I. TRAF2 in osteotropic breast cancer cells enhances skeletal tumour growth and promotes osteolysis. Sci. Rep. 2018, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Jin, F. Screening of differentially methylated genes in breast cancer and risk model construction based on TCGA database. Oncol. Lett. 2018, 16, 6407–6416. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ren, Y.; Qian, C.; Liu, J.; Li, G.; Li, Z. Over-expression of CDX2 alleviates breast cancer by up-regulating microRNA let-7b and inhibiting COL11A1 expression. Cancer Cell Int. 2020, 20, 13. [Google Scholar] [CrossRef]
- Maneerat, Y.; Prasongsukarn, K.; Benjathummarak, S.; Dechkhajorn, W. PPBP and DEFA1/DEFA3 genes in hyperlipidaemia as feasible synergistic inflammatory biomarkers for coronary heart disease. Lipids Health Dis. 2017, 16, 80. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-W.; Lin, C.-W.; Yang, W.-E.; Yang, S.-F. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919864247. [Google Scholar] [CrossRef] [Green Version]
- Kolegraff, K.; Nava, P.; Helms, M.N.; Parkos, C.A.; Nusrat, A. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol. Biol. Cell 2011, 22, 1121–1134. [Google Scholar] [CrossRef]
- Kratochvílová, K.; Horak, P.; Ešner, M.; Souček, K.; Pils, D.; Anees, M.; Tomasich, E.; Dráfi, F.; Jurtíková, V.; Hampl, A.; et al. Tumor suppressor candidate 3 (TUSC3) prevents the epithelial-to-mesenchymal transition and inhibits tumor growth by modulating the endoplasmic reticulum stress response in ovarian cancer cells. Int. J. Cancer 2015, 137, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.-F.; Fan, Y.-C.; Gao, S.; Yang, Y.; Zhang, J.-J.; Wang, K. MT1M and MT1G promoter methylation as biomarkers for hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 4723–4729. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Xie, X.; Li, L.; Tang, H.; Ye, X.; Chen, L.; Tang, W.; Gao, J.; Pan, L.; Zhang, X.; et al. Diagnostic and prognostic value of serum MACC1 in breast cancer patients. Oncotarget 2016, 7, 84408–84415. [Google Scholar] [CrossRef] [PubMed]
- Miaskowski, C.; Dodd, M.; Paul, S.M.; West, C.; Hamolsky, D.; Abrams, G.; Cooper, B.A.; Elboim, C.; Neuhaus, J.; Schmidt, B.L.; et al. Lymphatic and Angiogenic Candidate Genes Predict the Development of Secondary Lymphedema following Breast Cancer Surgery. PLoS ONE 2013, 8, e60164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Shidfar, A.; Ivancic, D.; Ranjan, M.; Liu, L.; Choi, M.-R.; Parimi, V.; Gursel, D.B.; Sullivan, M.E.; Najor, M.S.; et al. Overexpression of lipid metabolism genes and PBX1 in the contralateral breasts of women with estrogen receptor-negative breast cancer. Int. J. Cancer 2017, 140, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Ju, J.; Hou, L.; Li, Z.; Xiao, D.; Li, Y.; Yao, J.; Wang, C.; Zhang, Y.; et al. Downregulation of miR-522 suppresses proliferation and metastasis of non-small cell lung cancer cells by directly targeting DENN/MADD domain containing 2D. Sci. Rep. 2016, 6, 19346. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Dronyk, A.; Hu, X.; Hendzel, M.; Shaw, A.R. BCL10 is recruited to sites of DNA damage to facilitate DNA dou-ble-strand break repair. Cell Cycle 2016, 15, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Post, S.M.; Solis, L.M.; Xiong, S.; Yang, P.; Chen, N.; Wistuba, I.I.; Killary, A.M.; Lozano, G. Loss of the novel tumour suppressor and polarity gene Trim62 (Dear1) synergizes with oncogenic Ras in invasive lung cancer. J. Pathol. 2014, 234, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Harkness, E.F.; Barrow, E.; Newton, K.; Green, K.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: A cohort study. J. Med. Genet. 2015, 52, 553–556. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.-P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Wicki, R.; Franz, C.; Scholl, F.A.; Heizmann, C.W.; Schäfer, B.W. Repression of the candidate tumor suppressor gene S100A2 in breast cancer is mediated by site-specific hypermethylation. Cell Calcium 1997, 22, 243–254. [Google Scholar] [CrossRef]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrescu, R.G.; Verma, M. Cancer Epigenetics: Methods and Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2021; Volume 863. [Google Scholar]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichol, J.N.; Dupéré-Richer, D.; Ezponda, T.; Licht, J.D.; Miller, W.H., Jr. H3K27 methylation: A focal point of epigenetic deregulation in cancer. Adv. Cancer Res. 2016, 131, 59–95. [Google Scholar] [PubMed] [Green Version]
- Holm, K.; Grabau, D.; Lövgren, K.; Aradottir, S.; Gruvberger-Saal, S.; Howlin, J.; Saal, L.H.; Ethier, S.P.; Bendahl, P.-O.; Stål, O.; et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol. Oncol. 2012, 6, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezponda, T.; Licht, J.D. Molecular Pathways: Deregulation of Histone H3 Lysine 27 Methylation in Cancer—Different Paths, Same Destination. Clin. Cancer Res. 2014, 20, 5001–5008. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Lee, H.; Yoon, J.-G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 Is an H3K4 Demethylase Involved in Transcriptional Repression and Breast Cancer Cell Proliferation. Mol. Cell 2007, 25, 801–812. [Google Scholar] [CrossRef]
- Mungamuri, S.K.; Murk, W.; Grumolato, L.; Bernstein, E.; Aaronson, S.A. Chromatin modifications sequentially enhance ErbB2 expression in ErbB2-positive breast cancers. Cell Rep. 2013, 5, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; John, E.M.; Stern, M.C.; Herrick, J.; Lundgreen, A.; Giuliano, A.R.; Hines, L.; Baumgartner, K.B.; Torres-Mejia, G.; Wolff, R.K. Associations with growth factor genes (FGF1, FGF2, PDGFB, FGFR2, NRG2, EGF, ERBB2) with breast cancer risk and survival: The Breast Cancer Health Disparities Study. Breast Cancer Res. Treat. 2013, 140, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Messier, T.L.; Gordon, J.A.R.; Boyd, J.R.; Tye, C.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/ΚDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, A.; Minucci, S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 2017, 9, 1123–1142. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Dell’Aversana, C.; De Marchi, T.; Rotili, D.; Liu, N.Q.; Novakovic, B.; Boccella, S.; Di Maro, S.; Cosconati, S.; Baldi, A.; et al. Inhibition of histone demethylases LSD1 and UTX regulates ERα signaling in breast cancer. Cancers 2019, 11, 2027. [Google Scholar] [CrossRef] [Green Version]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef]
- Therneau, M.T.; Grambsch, P.M.; Fleming, T. A Package for Survival Analysis in S; Mayo Foundation: Rochester, MN, USA, 1994. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves Using ’ggplot2’; R Package Version 0.3; R Core Team: Vienna, Austria, 2017. [Google Scholar]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Ahmad, A.; Azim, S.; Zubair, H.; Khan, M.A.; Singh, S.; Carter, J.E.; Rocconi, R.P.; Singh, A.P. Epigenetic basis of cancer health disparities: Looking beyond genetic differences. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 16–28. [Google Scholar] [CrossRef]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Nassar, F.; Talhouk, R.; Zgheib, N.K.; Tfayli, A.; El Sabban, M.; El Saghir, N.S.; Boulos, F.; Jabbour, M.N.; Chalala, C.; Boustany, R.-M.; et al. microRNA Expression in Ethnic Specific Early Stage Breast Cancer: An Integration and Comparative Analysis. Sci. Rep. 2017, 7, 16829. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.K.; Jordan, H.R.; Tollefsbol, T.O. Effects of SAHA and EGCG on Growth Potentiation of Triple-Negative Breast Cancer Cells. Cancers 2018, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munsell, M.F.; Sprague, B.L.; Berry, D.A.; Chisholm, G.; Trentham-Dietz, A. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol. Rev. Rev. 2014, 36, 114–136. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Cruzata, L.; Zhang, W.; McDonald, J.A.; Tsai, W.Y.; Valdovinos, C.; Falci, L.; Wang, Q.; Crew, K.D.; Santella, R.M.; Hershman, D.L.; et al. Dietary modifications, weight loss, and changes in metabolic markers affect global DNA methylation in Hispanic, African American, and Afro-Caribbean breast cancer survivors. J. Nutr. 2015, 145, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naushad, S.M.; Hussain, T.; Al-Attas, O.S.; Prayaga, A.; Digumarti, R.R.; Gottumukkala, S.R.; Kutala, V.K. Molecular insights into the association of obesity with breast cancer risk: Relevance to xenobiotic metabolism and CpG island methylation of tumor suppressor genes. Mol. Cell. Biochem. 2014, 392, 273–280. [Google Scholar] [CrossRef]
- Wu, J.; Miao, J.; Ding, Y.; Zhang, Y.; Huang, X.; Zhou, X.; Tang, R. MiR-4458 inhibits breast cancer cell growth, migration, and invasiveness by targeting CPSF4. Biochem. Cell Biol. 2019, 97, 722–730. [Google Scholar] [CrossRef]
- Zinn, R.L.; Pruitt, K.; Eguchi, S.; Baylin, S.B.; Herman, J.G. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007, 67, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.S.; Kala, R.; Tollefsbol, T.O. Mechanisms for the Inhibition of Colon Cancer Cells by Sulforaphane through Epigenetic Modulation of MicroRNA-21 and Human Telomerase Reverse Transcriptase (hTERT) Down-regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Liu, Y.B.; Zhang, Y.; Shaw, J.; Valeriote, F.A.; Gautam, S.C. Inhibition of hTERT in pancreatic cancer cells by pristimerin involves suppression of epigenetic regulators of gene transcription. Oncol. Rep. 2017, 37, 1914–1920. [Google Scholar] [CrossRef]
- Barlési, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non–small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef]
- Sapienza, C.; Issa, J.-P. Diet, nutrition, and cancer epigenetics. Annu. Rev. Nutr. 2016, 36, 665–681. [Google Scholar] [CrossRef]
- Kim, Y. Folate and carcinogenesis: Evidence, mechanisms, and implications. J. Nutr. Biochem. 1999, 10, 66–88. [Google Scholar] [CrossRef]
- Mikol, Y.B.; Hoover, K.L.; Creasia, D.; Poirier, L.A. Hepatocarcinogenesis in rats fed methyl-deficient, amino acid-defined diets. Carcinogenesis 1983, 4, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.S.; Giovannucci, E.; Wolk, A. Folate and Risk of Breast Cancer: A Meta-analysis. JNCI J. Natl. Cancer Inst. 2007, 99, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Wang, K.; Ye, F.; Lei, L.; Zhou, Y.; Chen, J.; Zhao, G.; Chang, H. Folate intake and the risk of breast cancer: An up-to-date meta-analysis of prospective studies. Eur. J. Clin. Nutr. 2019, 73, 1657–1660. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Hodgson, S.; Omar, R.Z.; Jensen, T.K.; Thompson, S.G.; Boobis, A.R.; Davies, D.S.; Elliott, P. Meta-analysis of studies of alcohol and breast cancer with consideration of the methodological issues. Cancer Causes Control 2006, 17, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Davidson, N.E.; Hunter, S.; Yang, X.; Payne-Wilks, K.; Roland, C.L.; Phillips, D.; Bentley, C.; Dai, M.; Williams, S.M. Methyl-group dietary intake and risk of breast cancer among African-American women: A case–control study by methylation status of the estrogen receptor alpha genes. Cancer Causes Control 2003, 14, 827–836. [Google Scholar] [CrossRef]
- Christensen, B.C.; Kelsey, K.T.; Zheng, S.; Houseman, E.A.; Marsit, C.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.; Karagas, M.R.; Kushi, L.; et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010, 6, e1001043. [Google Scholar] [CrossRef] [Green Version]
- Alinejad, V.; Dolati, S.; Motallebnezhad, M.; Yousefi, M. The role of IL17B-IL17RB signaling pathway in breast cancer. Biomed. Pharmacother. 2017, 88, 795–803. [Google Scholar] [CrossRef]
- Niraula, S.; Ocana, A.; Ennis, M.; Goodwin, P.J. Body size and breast cancer prognosis in relation to hormone receptor and menopausal status: A meta-analysis. Breast Cancer Res. Treat. 2012, 134, 769–781. [Google Scholar] [CrossRef]
- Cao, Y.; Hou, L.; Wang, W. Dietary total fat and fatty acids intake, serum fatty acids and risk of breast cancer: A meta-analysis of prospective cohort studies. Int. J. Cancer 2016, 138, 1894–1904. [Google Scholar] [CrossRef]
- Tao, M.-H.; Marian, C.; Nie, J.; Ambrosone, C.; Krishnan, S.S.; Edge, S.B.; Trevisan, M.; Shields, P.G.; Freudenheim, J.L. Body mass and DNA promoter methylation in breast tumors in the Western New York Exposures and Breast Cancer Study1–3. Am. J. Clin. Nutr. 2011, 94, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, I.N.; Story, M.T.; Nelson, M.C. Neighborhood environments: Disparities in access to healthy foods in the U.S. Am. J. Prev. Med. 2009, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Zenk, S.N.; Schulz, A.J.; Israel, B.A.; James, S.A.; Bao, S.; Wilson, M.L. Fruit and vegetable access differs by community racial composition and socioeconomic position in Detroit, Michigan. Ethn. Dis. 2006, 16, 275–280. [Google Scholar] [PubMed]
- Fong, A.J.; Lafaro, K.; Ituarte, P.H.G.; Fong, Y. Association of Living in Urban Food Deserts with Mortality from Breast and Colorectal Cancer. Ann. Surg. Oncol. 2021, 28, 1311–1319. [Google Scholar] [CrossRef]
- Forshee, A.R.; Storey, M.L.; Ritenbaugh, C. Breast cancer risk and lifestyle differences among premenopausal and post-menopausal African-American women and white women. Cancer 2003, 97 (Suppl. 1), 280–288. [Google Scholar] [CrossRef]
- Yedjou, C.G.; Sims, J.N.; Miele, L.; Noubissi, F.; Lowe, L.; Fonseca, D.D.; Alo, R.A.; Payton, M.; Tchounwou, P.B. Health and Racial Disparity in Breast Cancer. Adv. Exp. Med. Biol. 2019, 1152, 31. [Google Scholar]
- Stolley, M.R.; Sharp, L.; Wells, A.M.; Simon, N.; Schiffer, L. Health Behaviors and Breast Cancer: Experiences of Urban African American Women. Health Educ. Behav. 2006, 33, 604–624. [Google Scholar] [CrossRef]
- Michael, Y.L.; Carlson, N.E.; Chlebowski, R.T.; Aickin, M.; Weihs, K.L.; Ockene, J.K.; Bowen, D.J.; Ritenbaugh, C. Influence of stressors on breast cancer incidence in the Women’s Health Initiative. Health Psychol. Off. J. Div. Health Psychol. Am. Psychol. Assoc. 2009, 28, 137–146. [Google Scholar]
- Lin, Y.; Wang, C.; Zhong, Y.; Huang, X.; Peng, L.; Shan, G.; Wang, K.; Sun, Q. Striking life events associated with primary breast cancer susceptibility in women: A meta-analysis study. J. Exp. Clin. Cancer Res. 2013, 32, 53. [Google Scholar] [CrossRef] [Green Version]
- Chiriac, V.-F.; Baban, A.; Dumitrascu, D.L. Psychological stress and breast cancer incidence: A systematic review. Clujul Med. 2018, 91, 18–26. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S. Stress, Adaptation, and Disease: Allostasis and Allostatic Load. Ann. N Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.M.; Butler, L.K. Endocrinology of Stress. Int. J. Comp. Psychol. 2007, 20, 89–95. [Google Scholar]
- Akinyemiju, T.; Wilson, L.E.; Deveaux, A.; Aslibekyan, S.; Cushman, M.; Gilchrist, S.; Safford, M.; Judd, S.; Howard, V. Asso-ciation of Allostatic Load with All-Cause and Cancer Mortality by Race and Body Mass Index in the REGARDS Cohort. Cancers 2020, 12, 1695. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Doorenbos, A.Z.; Li, H.; Jang, M.K.; Park, C.G.; Bronas, U.G. Allostatic Load in Cancer: A Systematic Review and Mini Meta-Analysis. Biol. Res. Nurs. 2020, 23, 1099800420969898. [Google Scholar] [CrossRef]
- Sephton, S.E.; Sapolsky, R.M.; Kraemer, H.C.; Spiegel, D. Diurnal Cortisol Rhythm as a Predictor of Breast Cancer Survival. JNCI J. Natl. Cancer Inst. 2000, 92, 994–1000. [Google Scholar] [CrossRef]
- Dai, S.; Mo, Y.; Wang, Y.; Xiang, B.; Liao, Q.; Zhou, M.; Li, X.; Li, Y.; Xiong, W.; Li, G.; et al. Chronic Stress Promotes Cancer Development. Front. Oncol. 2020, 10, 10. [Google Scholar] [CrossRef]
- Xing, C.Y.; Doose, M.; Qin, B.; Lin, Y.; Plascak, J.J.; Omene, C.; He, C.; Demissie, K.; Hong, C.-C.; Bandera, E.V.; et al. Predi-agnostic Allostatic Load as a Predictor of Poorly Differentiated and Larger Sized Breast Cancers among Black Women in the Women’s Circle of Health Follow-Up Study. Cancer Epidemiol. Prev. Biomark. 2020, 29, 216–224. [Google Scholar] [CrossRef]
- Sternthal, J.M.; Slopen, N.; Williams, D.R. Racial Disparities in Health. Du Bois Rev. Soc. Sci. Res. Race 2011, 8, 95–113. [Google Scholar] [CrossRef]
- Tomfohr, M.L.; Pung, M.A.; Dimsdale, J.E. Mediators of the relationship between race and allostatic load in African and White Americans. Health Psychol. 2016, 35, 322–332. [Google Scholar] [CrossRef]
- Geronimus, A.T.; Hicken, M.; Keene, D.; Bound, J. “Weathering” and age patterns of allostatic load scores among blacks and whites in the United States. Am. J. Public Health 2006, 96, 826–833. [Google Scholar] [CrossRef]
- Kroenke, C.H.; Kubzansky, L.D.; Schernhammer, E.S.; Holmes, M.D.; Kawachi, I. Social networks, social support, and survival after breast cancer diagnosis. J. Clin. Oncol. 2006, 24, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.; Boyd, P.T.; Blacklow, R.S.; Jackson, J.S.; Greenberg, R.S.; Austin, D.F.; Chen, V.W.; Edwards, B.K. The relationship between social ties and survival among black and white breast cancer patients. National Cancer Institute Black/White Cancer Survival Study Group. Cancer Epidemiol. Prev. Biomark. 1994, 3, 253–259. [Google Scholar]
- Weinstock, M.; Matlina, E.; Maor, G.I.; Rosen, H.; McEwen, B.S. Prenatal stress selectively alters the reactivity of the hypo-thalamic-pituitary adrenal system in the female rat. Brain Res. 1992, 595, 195–200. [Google Scholar] [CrossRef]
- Louvart, H.; Maccari, S.; Darnaudery, M. Prenatal stress affects behavioral reactivity to an intense stress in adult female rats. Brain Res. 2005, 1031, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.G.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- Shields, A.E.; Wise, L.A.; Ruiz-Narvaez, E.A.; Seddighzadeh, B.; Byun, H.; Cozier, Y.C.; Rosenberg, L.; Palmer, J.R.; Baccarelli, A.A. Childhood abuse, promoter methylation of leukocyte NR3C1 and the potential modifying effect of emotional support. Epigenomics 2016, 8, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesset, A.K.; Perri, A.M.; Mueller, C.R. Frequent promoter hypermethylation and expression reduction of the glucocorticoid receptor gene in breast tumors. Epigenetics 2014, 9, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Melas, P.A.; Wei, Y.; Wong, C.C.Y.; Sjöholm, L.K.; Åberg, E.; Mill, J.; Schalling, M.; Forsell, Y.; Lavebratt, C. Genetic and ep-igenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int. J. Neuropsychopharmacol. 2013, 16, 1513–1528. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef] [Green Version]
- Needham, B.L.; Smith, J.A.; Zhao, W.; Wang, X.; Mukherjee, B.; Kardia, S.L.R.; Shively, C.A.; Seeman, T.E.; Liu, Y.; Roux, A.V.D. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: The multiethnic study of atherosclerosis. Epigenetics 2015, 10, 958–969. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Zhao, W.; Wang, X.; Ratliff, S.M.; Mukherjee, B.; Kardia, S.L.R.; Liu, Y.; Roux, A.V.D.; Needham, B.L. Neighborhood characteristics influence DNA methylation of genes involved in stress response and inflammation: The Multi-Ethnic Study of Atherosclerosis. Epigenetics 2017, 12, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lou, Z.; Wang, L. The role of FKBP5 in cancer aetiology and chemoresistance. Br. J. Cancer 2011, 104, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwynne, W.D.; Hallett, R.M.; Girgis-Gabardo, A.; Bojovic, B.; Dvorkin-Gheva, A.; Aarts, C.; Dias, K.; Bane, A.; Hassell, J.A. Serotonergic system antagonists target breast tumor initiating cells and synergize with chemotherapy to shrink human breast tumor xenografts. Oncotarget 2017, 8, 32101–32116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hix, L.M.; Shi, Y.H.; Brutkiewicz, R.R.; Stein, P.L.; Wang, C.-R.; Zhang, M. CD1d-Expressing Breast Cancer Cells Modulate NKT Cell-Mediated Antitumor Immunity in a Murine Model of Breast Cancer Metastasis. PLoS ONE 2011, 6, e20702. [Google Scholar] [CrossRef] [PubMed]
- Kuehnemuth, B.; Piseddu, I.; Wiedemann, G.; Lauseker, M.; Kuhn, C.; Hofmann, S.; Schmoeckel, E.; Endres, S.; Mayr, D.; Jeschke, U.; et al. CCL1 is a major regulatory T cell attracting factor in human breast cancer. BMC Cancer 2018, 18, 1278. [Google Scholar] [CrossRef]
- Sahay, D.; Terry, M.B.; Miller, R. Is breast cancer a result of epigenetic responses to traffic-related air pollution? A review of the latest evidence. Epigenomics 2019, 11, 701–714. [Google Scholar] [CrossRef]
- Lewis-Michl, E.L.; Melius, J.M.; Kallenbach, L.R.; Ju, C.L.; Talbot, T.O.; Orr, M.F. Breast cancer risk and residence near industry or traffic in Nassau and Suffolk Counties, Long Island, New York. Arch. Environ. Health Int. J. 1996, 51, 255–265. [Google Scholar] [CrossRef]
- Wei, Y.; Davis, J.; Bina, W.F. Ambient air pollution is associated with the increased incidence of breast cancer in US. Int. J. Environ. Health Res. 2012, 22, 12–21. [Google Scholar] [CrossRef]
- Arcaro, K.F. Antiestrogenicity of environmental polycyclic aromatic hydrocarbons in human breast cancer cells. Toxicology 1999, 133, 115–127. [Google Scholar] [CrossRef]
- Choe, S.-Y.; Kim, S.-J.; Kim, H.-G.; Lee, J.H.; Choi, Y.; Lee, H.; Kim, Y. Evaluation of estrogenicity of major heavy metals. Sci. Total Environ. 2003, 312, 15–21. [Google Scholar] [CrossRef]
- Natarajan, R.; Aljaber, D.; Au, D.; Thai, C.; Sanchez, A.; Nunez, A.; Resto, C.; Chavez, T.; Jankowska, M.M.; Benmarhnia, T.; et al. Environmental exposures during puberty: Window of breast cancer risk and epigenetic damage. Int. J. Environ. Res. Public Health 2020, 17, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohai, P.; Lantz, P.M.; Morenoff, J.; House, J.S.; Mero, R.P. Racial and socioeconomic disparities in residential proximity to polluting industrial facilities: Evidence from the Americans’ Changing Lives Study. Am. J. Public Health 2009, 99 (Suppl. 3), S649–S656. [Google Scholar] [CrossRef] [PubMed]
- Woo, B.; Kravitz-Wirtz, N.; Sass, V.; Crowder, K.; Teixeira, S.; Takeuchi, D.T. Residential Segregation and Racial/Ethnic Dis-parities in Ambient Air Pollution. Race Soc. Probl. 2019, 11, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; Zandbergen, P.A. Children at risk: Measuring racial/ethnic disparities in potential exposure to air pollution at school and home. J. Epidemiol. Community Health 2007, 61, 1074–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmuel, S.; White, A.J.; Sandler, D.P. Residential Exposure to Vehicular Traffic-Related Air Pollution During Childhood and Breast Cancer Risk. Environ. Res. 2017, 159, 257–263. [Google Scholar] [CrossRef]
- White, A.J.; O’Brien, K.M.; Niehoff, N.M.; Carroll, R.; Sandler, D.P. Metallic Air Pollutants and Breast Cancer Risk in a Na-tionwide Cohort Study. Epidemiology 2019, 30, 20–28. [Google Scholar] [CrossRef]
- Kresovich, J.K.; Erdal, S.; Chen, H.Y.; Gann, P.H.; Argos, M.; Rauscher, G.H. Metallic air pollutants and breast cancer heter-ogeneity. Environ. Res. 2019, 177, 108639. [Google Scholar] [CrossRef]
- Liu, R.; Nelson, D.; Hurley, S.; Hertz, A.; Reynolds, P. Residential Exposure to Estrogen Disrupting Hazardous Air Pollutants and Breast Cancer Risk: The California Teachers Study. Epidemiology 2015, 26, 365–373. [Google Scholar] [CrossRef]
- Ionescu, J.G.; Novotny, J.; Stejskal, V.; Lätsch, A.; Blaurock-Busch, E.; Eisenmann-Klein, M. Increased levels of transition metals in breast cancer tissue. Neuro Endocrinol. Lett. 2006, 27 (Suppl. 1), 36–39. [Google Scholar]
- Hanna, C.W.; Bloom, M.S.; Robinson, W.; Kim, D.; Parsons, P.; Saal, F.S.V.; Taylor, J.A.; Steuerwald, A.J.; Fujimoto, V.Y. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol A, in women undergoing ovarian stimulation for IVF. Hum. Reprod. 2012, 27, 1401–1410. [Google Scholar] [CrossRef]
- Yu, K.-D.; Fan, L.; Di, G.-H.; Yuan, W.-T.; Zheng, Y.; Huang, W.; Chen, A.-X.; Yang, C.; Wu, J.; Shen, Z.-Z.; et al. Genetic variants in GSTM3 gene within GSTM4-GSTM2-GSTM1-GSTM5-GSTM3 cluster influence breast cancer susceptibility depending on GSTM1. Breast Cancer Res. Treat. 2010, 121, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Rodenhiser, D.I. Benzopyrene exposure disrupts DNA methylation and growth dynamics in breast cancer cells. Toxicol. Appl. Pharmacol. 2006, 216, 458–468. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Chen, J.; Teitelbaum, S.L.; McCullough, L.E.; Xu, X.; Cho, Y.H.; Conway, K.; Beyea, J.; Stellman, S.D.; Steck, S.E.; et al. Sources of polycyclic aromatic hydrocarbons are associated with gene-specific promoter methylation in women with breast cancer. Environ. Res. 2016, 145, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, M.S.; Vazquez, A.; Kulkarni, D.A.; Kerrigan, J.E.; Atwal, G.; Metsugi, S.; Toppmeyer, D.L.; Levine, A.J.; Hirshfield, K.M. Polymorphic variants in TSC1 and TSC2 and their association with breast cancer phenotypes. Breast Cancer Res. Treat. 2011, 125, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, C.L.; Bonner, M.R.; Nie, J.; Han, D.; Wang, Y.; Tao, M.-H.; Shields, P.G.; Marian, C.; Eng, K.H.; Trevisan, M.; et al. Lifetime exposure to ambient air pollution and methylation of tumor suppressor genes in breast tumors. Environ. Res. 2018, 161, 418–424. [Google Scholar] [CrossRef]
- Keramatinia, A.; Hassanipour, S.; Nazarzadeh, M.; Wurtz, M.; Monfared, A.B.; Khayyamzadeh, M.; Bidel, Z.; Mhrvar, N.; Mosavi-Jarrahi, A. Correlation Between Nitrogen Dioxide as an Air Pollution Indicator and Breast Cancer: A Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2016, 17, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S.; Villeneuve, P.J.; Crouse, D.; To, T.; Weichenthal, S.A.; Wall, C.; Miller, A.B. Associations between incident breast cancer and ambient concentrations of nitrogen dioxide from a national land use regression model in the Canadian National Breast Screening Study. Environ. Int. 2019, 133, 105182. [Google Scholar] [CrossRef]
- Plusquin, M.; Guida, F.; Polidoro, S.; Vermeulen, R.; Raaschou-Nielsen, O.; Campanella, G.; Hoek, G.; Kyrtopoulos, S.; Georgiadis, P.; Naccarati, A.; et al. DNA methylation and exposure to ambient air pollution in two prospective cohorts. Environ. Int. 2017, 108, 127–136. [Google Scholar] [CrossRef]
- Chukkapalli, S.; Amessou, M.; Dilly, A.K.; Dekhil, H.; Zhao, J.; Liu, Q.; Bejna, A.; Thomas, R.D.; Bandyopadhyay, S.; Bismar, T.A.; et al. Role of the EphB2 receptor in autophagy, apoptosis and invasion in human breast cancer cells. Exp. Cell Res. 2014, 320, 233–246. [Google Scholar] [CrossRef]
- Husa, A.-M.; Magić, Ž.; Larsson, M.; Fornander, T.; Pérez-Tenorio, G. EPH/ephrin profile and EPHB2 expression predicts patient survival in breast cancer. Oncotarget 2016, 7, 21362–21380. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Tobacco smoke carcinogens and breast cancer. Environ. Mol. Mutagen. 2002, 39, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Band, P.R.; Le, N.D.; Fang, R.; Deschamps, M. Carcinogenic and endocrine disrupting effects of cigarette smoke and risk of breast cancer. Lancet 2002, 360, 1044–1049. [Google Scholar] [CrossRef]
- Macacu, A.; Autier, P.; Boniol, M.; Boyle, P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2015, 154, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabat, G.C.; Kim, M.; Phipps, A.I.; Li, C.I.; Messina, C.R.; Wactawski-Wende, J.; Kuller, L.; Simon, M.S.; Yasmeen, S.; Was-sertheil-Smoller, S.; et al. Smoking and alcohol consumption in relation to risk of triple-negative breast cancer in a cohort of postmenopausal women. Cancer Causes Control CCC 2011, 22, 775–783. [Google Scholar] [CrossRef]
- Kawai, M.; Malone, K.E.; Tang, M.T.C.; Li, C.I. Active smoking and risk of estrogen receptor positive and triple-negative breast cancer among women 20–44 years of age. Cancer 2014, 120, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, S.; Lemieux, J.; Moore, L.; Maunsell, E.; Brisson, J. Smoking at time of diagnosis and breast cancer-specific survival: New findings and systematic review with meta-analysis. Breast Cancer Res. BCR 2014, 16, R42. [Google Scholar] [CrossRef] [Green Version]
- Parada, H.; Sun, X.; Tse, C.-K.; Olshan, A.F.; Troester, M.A.; Conway, K. Active smoking and survival following breast cancer among African-American and non-African-American women in the Carolina Breast Cancer Study. Cancer Causes Control CCC 2017, 28, 929–938. [Google Scholar] [CrossRef]
- Jamal, A.; King, B.A.; Neff, L.J.; Whitmill, J.; Babb, S.D.; Graffunder, C.M. Current Cigarette Smoking Among Adults—United States, 2005–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: A systematic review of DNA methylation studies. Clin. Epigenetics 2015, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013, 22, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.; Edmiston, S.N.; Parrish, E.; Bryant, C.; Tse, C.-K.; Swift-Scanlan, T.; McCullough, L.E.; Kuan, P.F. Breast tumor DNA methylation patterns associated with smoking in the Carolina Breast Cancer Study. Breast Cancer Res. Treat. 2017, 163, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Őrfi, L.; Szabadkai, I.; Daub, H.; Kéri, G.; Ullrich, A. AXL Is a Potential Target for Therapeutic Intervention in Breast Cancer Progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, C.V.; Byun, H.-M.; Wenten, M.; Pan, F.; Yang, A.; Gilliland, F.D. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.E.; Crott, J.W.; Ghandour, H.; E Dallal, G.; Choi, S.-W.; Keyes, M.K.; Jang, H.; Liu, Z.; Nadeau, M.; Johnston, A.; et al. Chronic cigarette smoking is associated with diminished folate status, altered folate form distribution, and increased genetic damage in the buccal mucosa of healthy adults. Am. J. Clin. Nutr. 2006, 83, 835–841. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Linardakis, M.K.; Hatzis, C.M.; Malliaraki, N.; Saris, W.H.; Kafatos, A.G. Smoking status in relation to serum folate and dietary vitamin intake. Tob. Induc. Dis. 2008, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Shrubsole, M.J.; Jin, F.; Dai, Q.; O Shu, X.; Potter, J.; Hebert, J.R.; Gao, Y.T.; Zheng, W. Dietary Folate Intake and Breast Cancer Risk: Results from the Shanghai Breast Cancer Study. Cancer Res. 2001, 61, 7136–7141. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).