1. Introduction
Gene therapy for the treatment of malignancies remains an area of particular interest. Previous attempts to deliver gene-coding DNA using replication-deficient viral vectors showed only limited success in preclinical and clinical studies, and have also raised serious safety concerns [
1,
2]. In comparison, non-viral delivery systems tend to be simple to manufacture, show lower immunogenicity, and are associated with fewer regulatory issues when translated into clinical settings. However, the broad application of therapeutic DNA using non-viral delivery systems is hampered by non-specific toxic side-effects, poor pharmacokinetics, low DNA transfection efficiency, and in most cases only transient gene expression (for review, see [
3]).
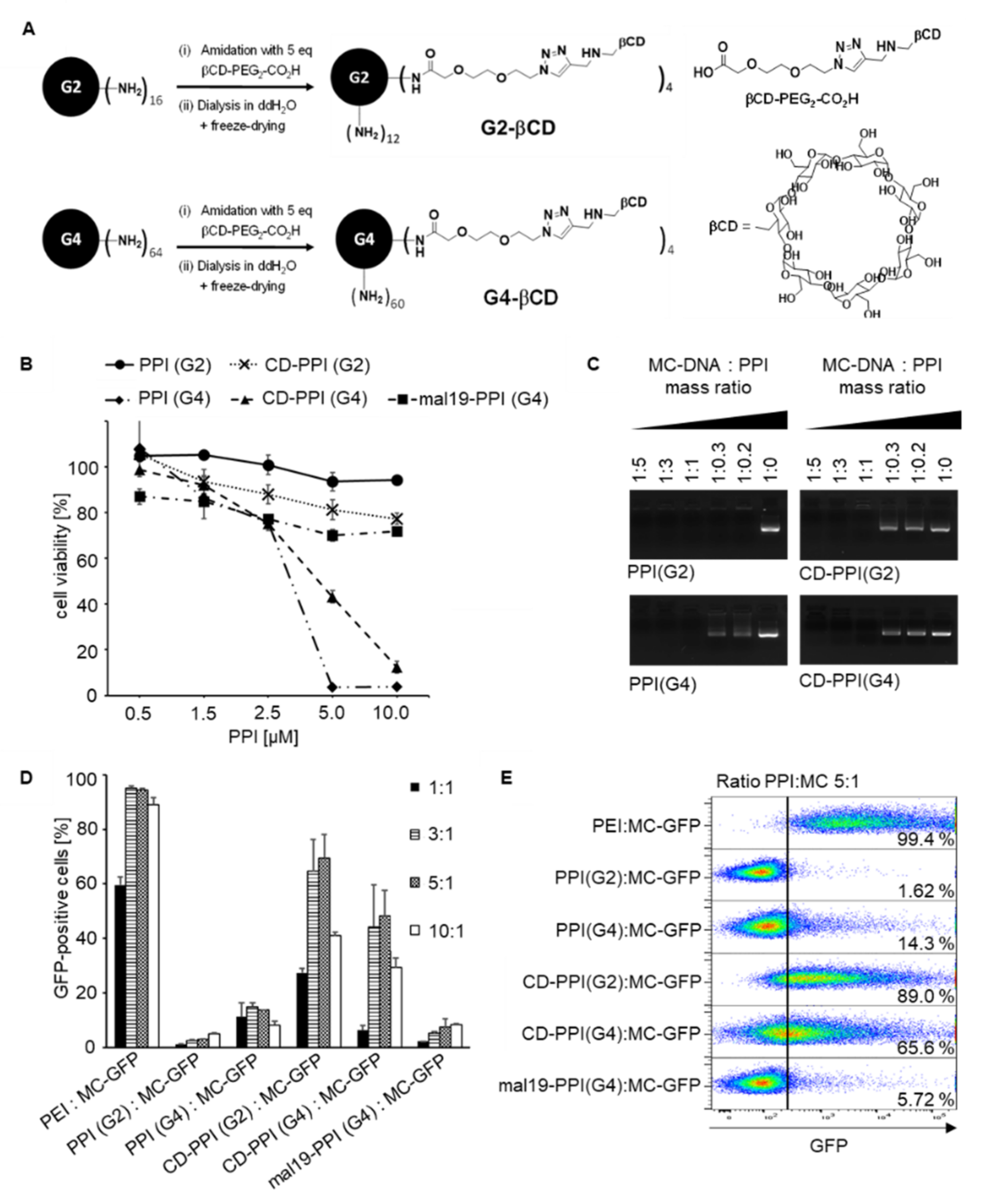
Among non-viral gene transfer systems, poly(propylene imine) (PPI) and its surface-modified derivatives have emerged as promising DNA carriers. The high rate of positively charged amino groups on the surface of the PPI dendrimers enables electrostatic interaction with the negatively charged DNA [
4] and results in the formation of compact nano-sized particles, designated “dendriplexes” [
5]. Tuning PPI by surface modifications with for instance poly(ethylene glycol) (PEG) reduces cytotoxicity and inhibits intermolecular aggregation. In addition, surface modifications provide a hydrophilic shell that avoids interaction with the reticuloendothelial system and prolongs circulation time in the bloodstream [
6]. Recently, we showed that surface charge shielding by maltose modification of peripheral amino groups greatly improves the biocompatibility of PPI glycodendrimers (mal19-PPI) while simultaneously reducing transfection efficiency. However, upon bioconjugation of mono-biotinylated single-chain antibodies (scFv-P-BAP) for targeting, PPI glycodendrimers, referred to as “polyplexes”, became competent for delivering siRNA to target cells in vitro and in vivo expressing the cognate antigen/receptor [
7,
8].
We sought to exploit this polyplex system for targeted delivery of gene-encoding therapeutic DNA. As a target on cancer cells, we chose the prostate stem cell antigen (PSCA), a glycophosphatidylinositol (GPI)-anchored tumor-associated antigen which is found in a variety of urogenital-related cancers [
9,
10,
11,
12,
13], and various other malignancies such as pancreatic adenocarcinoma [
14] and glioblastoma [
15]. Yet, preliminary studies using targeted polyplexes containing mal19-PPI revealed selective but only moderate transfection efficiency of DNA in target cells, which is likely related to the attenuated endosomal escape of the DNA payload. To improve DNA transfection efficiency of polyplexes, we developed a modified polyplex system, designated hybrid polyplex, combining transfection-incompetent antibody-conjugated mal19-PPIs with β-cyclodextrin-modified PPIs (CD-PPI). β-cyclodextrins are natural cyclic oligosaccharides with seven glucose units in their structure linked by α-(1,4) glucoside bonds [
16]. The donut-shaped β-cyclodextrins are characterized by a hydrophilic outer surface coated with hydroxyl groups and a hydrophobic inner cavity coated with ether groups of anomeric oxygen atoms [
17]. Noteworthily, β-cyclodextrins are approved by the FDA as excipients in pharmaceutical products [
18] that, in addition to their role as solubilizers, stabilizers of colloids, or modifiers for controlled release, can enhance the permeation of drugs through biological membranes (for review, see [
19]).
To achieve sustained transgene expression of the transgene, we flanked its expression cassette with Sleeping Beauty (SB) inverted terminal repeats (ITRs); these SB transposons are mobile genetic elements that efficiently integrate DNA into the genome. To enable transposition, we employed simultaneous delivery of DNA encoding a hyperactive form of SB transposase (SB100X). In targeted cells, the transposase recognizes the ITRs of the transposon and enables genomic integration via a cut-and-paste mechanism [
20,
21].
A major concern when considering the clinical application of gene-coding DNA delivery is the non-specific cytotoxic effects on cells and enhanced degradation of introduced foreign DNA caused by unmethylated CG dinucleotides located in the bacterial backbone of plasmid DNA [
22]. To address this safety concern and to optimize packaging of DNA encoding genes into polyplexes, we genetically engineered the content and size of plasmids. More specifically, the transposon as well as the expression cassettes for SB100X were flanked by attB and attP recognition sites for bacterial PhiC31 integrase to produce minicircle (MC) DNA devoid of bacterial backbones [
22].
To this end, PSCA-specific hybrid polyplexes efficiently delivered MC encoding a GFP transposon in PSCA-positive 293T cells, whereas control hybrid polyplexes conjugated to non-specific control scFv-P-BAPs showed low gene transfer efficiency. In an experimental gene therapy approach, delivery of MC encoding a TP53 transposon by PSCA-specific hybrid polyplexes into p53-deficient HCT116p53−/−/PSCA colon cancer cells and PTEN-deficient H4PSCA glioma cells led to decreased clonogenic survival. Remarkably, surviving glioma clones failed to proliferate further. Interestingly, a notable number of colon cancer clones that escaped transgenic p53 showed loss of transgene expression. Noteworthily, in such colon cancer cells, transgenic p53 expression could be induced by treatment with the DNA-damaging antibiotic zeocin. In summary, our results demonstrate the feasibility of combining tumor-targeting hybrid polyplexes and Sleeping Beauty gene transposition for gene therapy, which due to the modular design can be extended to other target genes and tumor entities.
2. Materials and Methods
2.1. Synthesis of PPIs
Synthesis of 4th generation maltose-modified PPI dendrimers (mal19-PPI (G4) 14,900 g/mol) and biotinylated mal19-PPIs (mal19-PPI-biotin (G4), 15,490 g/mol) were described previously [
7,
8]. For synthesis of β-cyclodextrin-modified PPIs, the complete reactions were carried out under an argon protection atmosphere. In a first reaction flask (heated, degassed, and filled with argon), 2nd and 4th generation PPI dendrimers were added and degassed for around 1 h under high vacuum, followed by the addition of 2 mL of degassed anhydrous DMSO and triethylamine (Et3N). In a second reaction flask (heated, degassed, and filled with argon) βCD-PEG-CO
2H and BOP were dissolved in 2 mL of degassed anhydrous DMSO. Subsequently, the resulting reaction mixture was stirred for 2 h. PPI dendrimer solution was slowly added to the activated βCD-PEG-CO
2H solution, stirred for 2–2.5 days at room temperature, and dialyzed for 2 days in 5 L water (membrane tube with MWCO 2000 g/mol) with exchanging aqueous solution. After the freeze-drying, a viscous liquid was obtained. Used quantities for CD-PPI (G2) and CD-PPI (G4) synthesis are presented in
Appendix A (
Table A1) and results of
1H NMR characterization are presented in
Appendix A as well (
Figure A1,
Figure A2,
Figure A3,
Figure A4 and
Figure A5), including the synthesis and NMR characterization of βCD-PEG-CO
2H.
2.2. Cell Lines
The hypodiploid colorectal carcinoma-derived HCT116 cells with homozygous knockout of p53, designated HCT116
p53−/− (kindly provided by B. Vogelstein, Johns Hopkins University, Baltimore, U.S.), the near triploid (+/−3n) glioma cell line H4, and the human embryonic kidney cell lines 293T, 293T
PSCA, and 293T
huBirA have been described previously [
7,
23,
24,
25]. HCT116
p53−/−/PSCA cells with ectopic expression of PSCA were generated by lentiviral transduction of HCT116
p53−/− cells using the lentiviral p6NST53-PSCA vector, described previously [
26], followed by geneticin (Invitrogen, Waltham, MA, USA) selection. Packaging of viral particles and transduction were performed using a three vector system described previously [
27]. HCT116
p53−/− and HCT116
p53−/−/PSCA cells were maintained in RPMI-1640 completed with 10%
v/v heat-inactivated FCS, 2 mM L-glutamine, 100 µg/mL streptomycin, 100 U/mL penicillin, and 10 mM HEPES (all from Life Technologies, Carlsbad, CA, USA). H4, H4
PSCA, 293T, and 293T
PSCA cell lines were cultured in DMEM completed with 4.5 g/L glucose, 10%
v/
v heat-inactivated FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10 mM HEPES (all from Life Technologies). 293T
huBirA cells were maintained in DMEM complete supplemented with 50 μM Biotin-C6 (Sigma-Aldrich, St. Louis, MO, USA) for scFv production. Cells were cultured at 37 °C with 5% CO
2 in a humidified incubator. All cell lines were authenticated (Multiplexion, Heidelberg, Germany).
2.3. Electrophoretic Mobility Gel Shift Assay
MC-DNA (1 µg) was incubated for 30 min at room temperature with increasing amounts of PPI or CD-PPI corresponding to mass ratios of 1:5 to 1:0.2. The dendriplexes were then separated by agarose gel electrophoresis [1% (w/v)] and analyzed under UV light (G:Box Chemi XX9).
2.4. Measurement of Cell Viability
2 × 104 293TPSCA cells were plated in 96-well plates and grown in 200 μL DMEM medium until 70% confluency, before adding different concentrations of PPI(G2), PPI(G4), mal19-PPI, CD-PPI (G2), or CD-PPI (G4). After 24 h, all wells of the assay were incubated with 20 μL AlamarBlue solution (Thermo Fisher Scientific, Waltham, MA, USA) for an additional 4 h. For normalization, untreated cells were included as the negative control. Cells lysed with 5% Triton X-100 (Sigma-Aldrich) served as the positive control. Finally, the fluorescence intensities of the reduced AlamarBlue in the wells were detected by a fluorescence imaging system (Synergy 2, BioTek, Winooski, VT, USA) and 560EX nm/590EM nm filter settings. The cytotoxicity of PPI glycodendrimers on cells was calculated in relation to untreated controls, which was set to 100% viability.
2.5. Plasmids for Minicircle Production and Sleeping Beauty Transposition
To generate a minicircle transposon encoding GFP, the synthetic SB transposase restriction sites IR/DR(L) and IR/DR(R) were ligated to the corresponding restriction sites SmaI—ClaI and StuI—EcoRV of the parental minicircle vector pMC.CMV-GFP (System Biosciences, Palo Alto, CA, USA).
A synthetic codon-optimized cDNA encoding the full 393 amino acids of p53 (Eurofins MWG Biotech, Ebersberg, Germany) fused to a T2A endoproteolytic cleavage site and a puromycin resistance gene was ligated to the corresponding restriction sites ClaI and HindIII of the parental minicircle vector pMC.CMV (System Biosciences), resulting in pMC-p53-puroR. In pMC-p53-puroR, the p53/puroR transgene was flanked by transposase restriction sites IR/DR(L) and IR/DR(R). pMC-puroR lacking the p53 coding sequence was used as mock control. The pCMV(CAT)T7-SB100 (Addgene, Watertown, MA, USA, plasmid # 34879) encoding hyperactive SB100X Sleeping Beauty transposase has been described previously [
28]. The SB100X gene sequence was amplified by PCR adding XbaI restriction sites and ligated to the XbaI restriction sites in the MCS of the parental minicircle vector pMC.CMV-MCS (System Biosciences). All vector inserts were confirmed by sequencing (Microsynth Seqlab, Göttingen, Germany).
2.6. Production of scFv-P-BAP
The DNA sequence and features of the single-chain antibody-derivative scFv(AM1)-P-BAP have been described previously [
8,
23]. The construct includes an N terminal Igκ leader sequence, a biotin acceptor peptide (P-BAP), and a C-terminal c-myc epitope and a 6x histidine (His)-tag. The biotinylated scFvs were expressed in transiently transfected 293T
huBirA producer cells and purified from the harvested cell culture supernatant by Ni2+-NTA affinity chromatography and Avidin-biotin affinity chromatography as described previously.
Briefly, the harvested medium was spun down and clarified supernatant was passed through a Ni2+-NTA spin column (Qiagen, Hilden, Germany) and subsequently washed with PBS containing 150 mM NaCl and 10 mM/20 mM imidazole. To elute bound scFv-P-BAPs the column was loaded with 500 μL 1x PBS containing 150 mM NaCl and 350 mM imidazole. Eluted scFv-P-BAPs were dialyzed in PBS twice for 2 h and for 12 h at 4 °C. Biotinylated scFv-P-BAPs were further purified using monomeric avidin affinity chromatography (Thermo Fisher Scientific, Waltham, MA, USA) according to the protocol of the manufacturer. Eluted scFv-P-BAPs were subsequently dialyzed again, as described previously. When needed, the recombinant scFv-P-BAPs were concentrated by employing Ultra-15 Amicon tubes (Merck Millipore, Burlington, VT, USA) and stored in aliquots at −20 °C.
2.7. Production of Minicircles
Minicircles were produced using the MC-Easy™ Minicircle DNA Production Kit (System Biosciences) according to the manufacturer’s protocol. Briefly, pMC-GFP, pMC-puroR, pMC-p53-puroR, and pMC-SB100 were grown in E. coli bacterial strain ZYCY10P3S2T harboring an arabinose-inducible system for simultaneous expression of PhiC31 integrase and Sce-I endonuclease. After incubation with induction medium, intramolecular (cis-) recombination generated MC from the parental plasmid mediated by PhiC31 integrase. The remaining parental plasmid-DNA backbone was degraded by Sce-I endonuclease. MC-GFP (3.7 kb), MC-puroR (2.6 kb), MC-p53-puroR (3.7 kb), and MC-SB100 (4.5 kb) were purified from the medium using Plasmid Plus Maxi Kit (Qiagen) according to the manufacturer’s protocol.
2.8. Assembly of Tumor-Specific Hybrid Polyplexes and Targeted Transfection of Cells
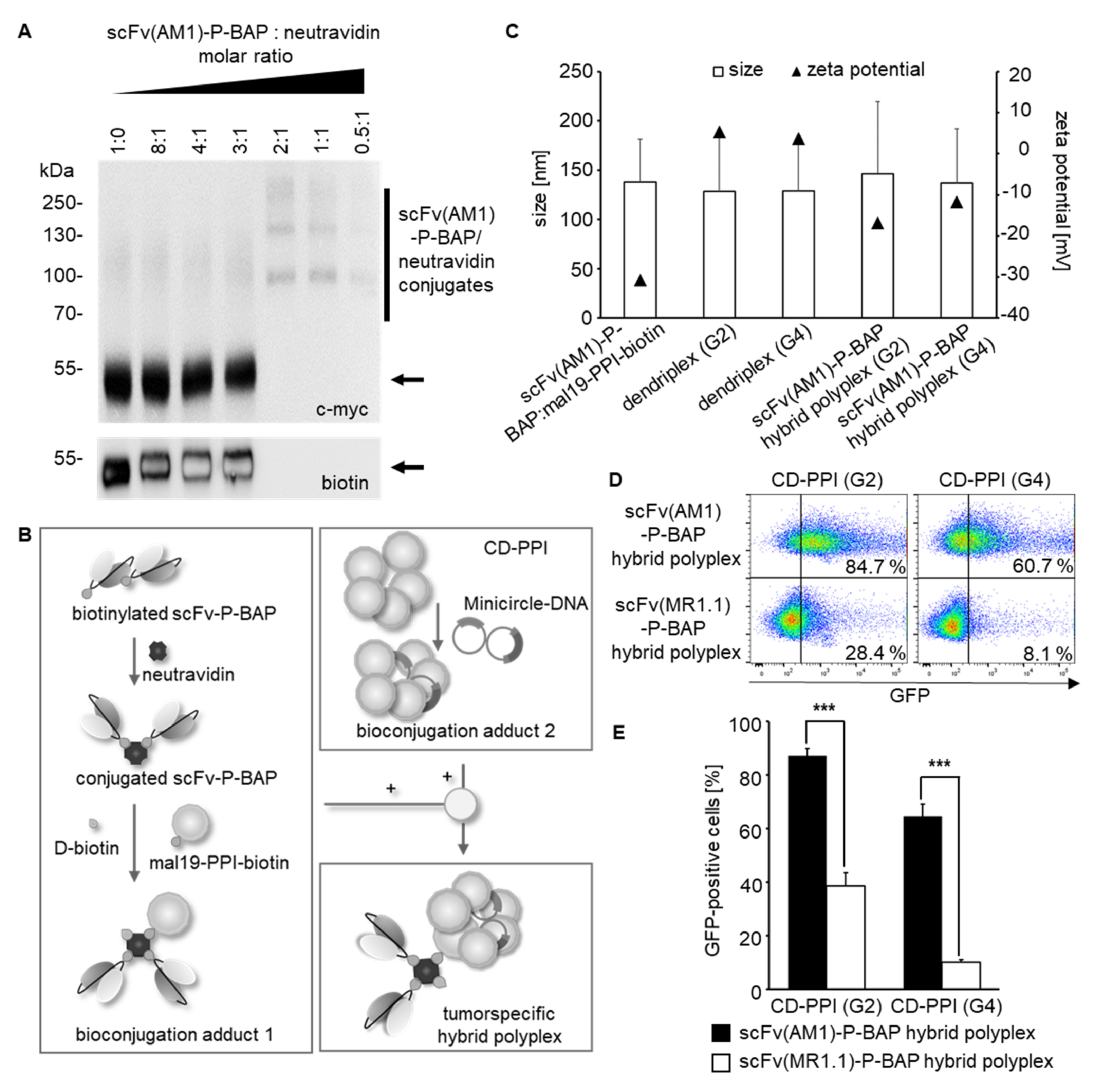
For the assembly of tumor-specific hybrid polyplexes, a two-step conjugation protocol was used. First, CD-PPIs were mixed with MC at a mass ratio of 5:1, resulting in bioconjugation adduct 1. In parallel, neutravidin (Thermo Fisher Scientific), scFv-P-BAP, and mal19-PPI-biotin were incubated for 30 min at room temperature at a molar ratio of 2:1:1, resulting in bioconjugation adduct 2. After saturation of the remaining free biotin binding sites of neutravidin with 0.3 mM D-biotin (Sigma-Aldrich), the intermediate conjugates were mixed for 30 min at room temperature and subsequently used for further studies. Hybrid polyplexes with 1 µg MC-DNA contained 100 pmol scFv-P-BAP, 50 pmol neutravidin, and 50 pmol mal19-PPI-biotin. Twenty-four hours before transfection, the indicated amounts of target cells were plated in a medium containing heat-inactivated FCS and treated with 6.5–7.5 × 103 hybrid polyplex nanoparticles/cell for 12 h before exchanging with a fresh medium.
2.9. Multiparameter Nanoparticle Tracking Analysis
Multiparameter nanoparticle tracking analysis was performed using the ZetaView® PMX120 (Particle Metrix, Inning am Ammersee, Germany) according to the manufacturer’s instructions to determine the particle quantities, zeta potential, and particle size of scFv(AM1)-P-BAP-hybrid polyplexes with MC. Hybrid polyplexes were prepared as described above. Data were analyzed using the manufacturer’s software (ZetaView 8.05.05).
2.10. Western Blot Analysis
The produced scFv-P-BAP and scFv-P-BAP:neutravidin conjugates were investigated using immunoblot analysis. Therefore, 1 μg of scFv-P-BAP was separated by SDS PAGE (12% polyacrylamide gel) under reducing conditions. Neutravidin conjugates including 1 μg of scFv-P-BAP were separated under non-reducing conditions. Subsequently, separated proteins were transferred by semi-dry Western Blot to a PVDF membrane (Whatman plc, Maidstone, UK). The PVDF membrane was blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) during a 1 h incubation followed by washing with TBS-T and TBS. Immune detection was performed using a primary monoclonal murine anti-c-myc antibody (1:5000, Invitrogen) and a secondary polyclonal rabbit anti-mouse IgG HRP conjugate (1:1000; Dako Agilent, Santa Clara, CA, USA). Biotinylated scFv-P-BAPs were detected by HRP-conjugated anti-biotin antibody (1:3000, Sigma-Aldrich). For immunoblot analysis of the conjugates, scFv(AM1)-P-BAP, and neutravidin were mixed in advanced at various molar ratios from 8:1 to 0.5:1 in 1 × PBS and incubated for 30 min at room temperature.
For analysis of targeted TP53 transposition, HCT116p53−/−/PSCA cell clones were pooled after transfection with scFv(AM1)-P-BAP-guided hybrid polyplexes, containing MC-SB100 and MC-p53-puroR or MC-puroR as a mock control, grown to 80% confluence in DMEM complete containing 2 µg/mL puromycin and incubated with 500 µg/mL zeocin (Invitrogen) for 4 h. Zeocin-treated and-non-treated cells were lysed in lysis buffer (10 mM Tris-HCl; pH 8.0; 140 mM NaCl; 1% Triton X-100). Equal amounts of total protein were subjected to SDS-PAGE under reducing conditions and blotted on a PVDF membrane using semi-dry Western Blotting. After blocking PVDF membrane with 5% BSA in TBS-T, p53, phospho-p53 (Ser 15) and p21waf/cip were detected with a polyclonal rabbit-anti-human p53 antibody (1:500; 7F5; Cell Signaling Technology, Danvers, MA, USA), a polyclonal rabbit-anti-human phospho-p53 antibody (1:500; ab1431; Abcam, Cambridge, UK) and a polyclonal rabbit-anti-human p21 antibody (1:500; 12D1; Cell Signaling Technology), followed by an HRP-conjugated anti-rabbit IgG secondary antibody (1:1000; Dako Agilent). To detect equal loading, PVDF membranes were stripped and subsequently stained with an anti-α tubulin antibody (1:5000; Sigma-Aldrich), followed by a secondary polyclonal rabbit anti-mouse IgG HRP conjugate (1:1000; Dako Agilent). Visual capturing of proteins was performed by a chemiluminescent method using Luminata Forte Western HRP substrate (Merck Millipore) and the G:Box Chemi XX9 (VWR, Radnor, PA, USA) gel doc documentation system. Analysis of immunoblots was performed using Fiji software (ImageJ 1.51 k, National Institute of Health, Bethesda, MD, USA).
2.11. Polymerase Chain Reaction
Analysis of transposon integration was performed by polymerase chain reaction (PCR) using PhusionTM High-Fidelity DNA Polymerase (Thermo Fisher Scientific). 1 × 105 293T cells polyethylenimine (PEI)-transfected with a total amount of 2 µg DNA containing MC-transposon-GFP alone or MC-transposon-GFP together with MC-SB at a molar ratio of 3:1, were harvested 28 days after transfection. Genomic DNA was prepared using the QIAamp DNA Mini Kit (Qiagen). Primers for the transposon (T) were MC-GFP-inside-For 5′-ccaacaagatgaagagcacc-3′ and MC-GFP-inside-Rev 5′-aagggacgtagcagaaggac-3′, for the minicircle backbone (MC) MC-GFP-outside-For 5′-gacggcgacaagcaaacatg-3′ and MC-GFP-outside-Rev 5′-tcgccttctatcgccttcttg-3′ and for the transposase SB100X (SB) MC-SB100X-For 5′-gtctggttcatccttgggag-3′ and MC-SB100X-Rev 5′-gggtcattgtcgtgttggaag-3′. The amplification protocol was: 98 °C denaturation 30 s, followed by 35 cycles at 98 °C denaturation 10 s, 59 °C annealing 30 s and 72 °C extension 90 s 5 µL PCR-product were separated by agarose gel electrophoresis [1% (w/v)] and analyzed under UV light (G:Box Chemi XX9, Syngene, Cambridge, UK).
2.12. Clonogenic Survival Assay
For analysis of direct effects after targeted p53 transposition, 2 × 104 HCT116p53−/−PSCA or H4PSCA cells plated in 6-well plates in DMEM complete were incubated with scFv(AM1)-P-BAP MC-SB100/MC-p53-puroR hybrid polyplexes. Cells treated with scFv(AM1)-P-BAP MC-SB100/MC-puroR hybrid polyplexes were included as mock control. Treated cells were continuously grown in a medium containing 2 µg/mL puromycin starting 24 h after transfection. Clonogenic survival was analyzed after 14 days. In order to investigate the long-term replicative potential of the treated cells, parallel experiments were performed and surviving HCT116p53−/−PSCA and H4PSCA cell clones were pooled. 1 × 103 cells thereof were plated on 10 cm dishes and analyzed after 14 days or grown until 80% confluency for Western blot analysis of transgenic p53 (see above). In all clonogenic survival experiments, cell clones were stained with crystal violet staining solution (Merck Millipore), photographed and counted using ImageJ software (National Institute of Health, USA).
2.13. Flow Cytometry
Binding of biotinylated scFv(AM1)-P-BAP to PSCA-positive target cells and isogenic parental cells was analyzed by flow cytometry. 2 × 105 cells were stained with 5 μg scFv-P-BAP for 1 h at 4 °C, followed by secondary anti-biotin-VioBlue antibody (Miltenyi Biotec, Cologne, Germany). Cells stained with the secondary antibody alone served as the control. To evaluate the targeted delivery of DNA, 2 × 105 293TPSCA cells were incubated with scFv-P-BAP hybrid polyplexes loaded with Cy3-labelled plasmid DNA (Mirus Bio, Madison, WI, USA, plasmid # MIRUMIR7904, 2.7 kb) for 4 h at 37 °C. Cy3-plasmid-loaded hybrid polyplexes conjugated with control scFv(MR1.1)-P-BAP were included as a negative control. Subsequently, the cells were washed with 0.1% Heparin/PBS (Sigma-Aldrich). To evaluate the transfection efficiency, 1 × 105 293TPSCA were incubated with PPI:MC-GFP dendriplexes containing 1 µg MC-DNA and PPI at different mass ratios for 48 h. At least 2 × 104 cells were measured by MACSQuant Analyser 10 flow cytometer (Miltenyi Biotec) and analyzed by FlowJo software version 10.1 (TreeStar, Ashland, OR, USA).
2.14. Confocal Laser Scanning Microscopy
For visualization of cellular DNA uptake, 6 × 105 293TPSCA cells grown on a coverslip were incubated with scFv(AM1)-P-BAP hybrid polyplexes loaded with Cy3-labelled plasmid DNA at 37 °C. After 24 h, cells were fixated with 4% paraformaldehyde in PBS (w/v). Subsequently, cell membranes and nuclei were stained with Alexa Fluor 647-conjugated wheat germ agglutinin (WGA, Life Technologies) and Hoechst 33,342 (Invitrogen). The early endosomes were stained with EEA1 (E-8) Alexa Fluor 647 (Santa Cruz Biotechnology, Dallas, TX, USA) according to the manufacturer’s protocols. The coverslips were placed upside down in a mounting medium (Vector Laboratories, Burlingame, CA, USA) on a microscope slide. Cells treated with Cy3-labelled hybrid polyplexes conjugated with non-binding control scFv(MR1.1)-P-BAP were included as negative control. Fluorescence microscopy images were captured by a confocal laser scanning microscope (Leica SP5, Leica Microsystems, Wetzlar, Germany) and analyzed by Fiji software (ImageJ 1.51k, National Institute of Health).
2.15. Statistical Analysis
All experiments were performed at least two times in at least triplicates. Differences between groups were examined for statistical significance using Student’s t-test. Values of p < 0.05 were considered statistically significant: * p < 0.05, ** p < 0.01, *** p < 0.001.
4. Discussion
Successful therapeutic gene delivery depends on the vector used for overcoming the main obstacles to DNA transfer: low uptake by the plasma membrane and insufficient release of DNA molecules into the cytoplasm. The vectors currently used can be roughly divided into viral vectors and non-viral vectors. Non-viral techniques for gene delivery are direct physical methods such as microinjection and particle bombardment, i.e., gene gun, electroporation, sonoporation, laser beam, and magnetofection, and the chemical-based approaches such as non-viral carrier systems (liposomes, lipoplexes, polymers, peptides, nanoparticles; for a review, see [
33]). Despite the great advantages of non-viral vectors—they are neither immunogenic nor carcinogenic and can provide large quantities of therapeutic DNA efficiently and cost-effectively—none of these vectors has yet proven more efficient than viral vectors for the transfer of therapeutic genetic material.
Recently, we established a novel tumor-specific siRNA delivery system, consisting of maltose-modified PPI/PPI-biotin, neutravidin, and a biotinylated scFv for targeted siRNA delivery to tumor cells [
7]. Yet, subsequent studies using polyplexes containing mal19-PPI revealed selective but only modest transfection efficiency of gene-coding DNA into corresponding target cells. In the present study, we therefore further modified this modular platform technology by combining transfection-incompetent antibody-conjugated mal19-PPIs with transfection-competent β-cyclodextrin-modified PPIs (CD-PPI) to reduce cytotoxicity of PPI while increasing transfection efficiency. It is well known that dendritic poly(amido amine) dendrimers with unshielded cationic surface groups exhibit generation-dependent high cytotoxicity in vitro. Moreover, the number of free amine groups is linearly related to their cytotoxicity [
34,
35]. Recently, we showed that partial surface modifications by direct maltose coupling to PPI-dendrimers are an effective way to optimize cytotoxic profiles [
7]. We obtained comparable results using partial surface modifications with β-cyclodextrin. Remarkably, in contrast to maltose-modified PPIs, β-cyclodextrin-modified PPIs showed high non-specific transfection efficiency of gene-coding DNA. This is consistent with previous reports describing the incorporation of β-cyclodextrin into polycationic dendrimer vectors for enhanced DNA transfer [
36]. Yet, the assembly of hybrid polyplexes consisting of β-cyclodextrin-modified PPI, maltose-modified PPI-biotin, neutravidin, and a mono-biotinylated scFv significantly reduced the non-specific transfection efficiency. Since a positive surface charge has been described as a prerequisite for efficient in vitro transfection [
37,
38], we hypothesize that the reduced zeta potential of the PSCA-specific hybrid polyplexes compared to dendriplexes containing only β-cyclodextrin-modified PPIs prevents non-specific in vitro transfection. Consequently, targeted gene delivery to PSCA-positive target cells with PSCA-specific hybrid polyplexes was mediated by the biotinylated scFv-P-BAP via receptor-mediated uptake. We chose PSCA as a target antigen on tumor cells, since it is found on a broad range of tumor entities [
13,
14,
39,
40]. Recent studies from our group have established PSCA as a targetable antigen, which after receptor-crosslinking by anti-PSCA immunoconjugates induces endocytosis [
23]. In this study, we identify a mixed clathrin- and caveolae-mediated uptake as the mechanism of PSCA-specific hybrid polyplex internalization. These results are similar to our recent report, which revealed a mixed clathrin- and caveolae-mediated uptake of nanoparticle-like immunoconjugates comprising mono-biotinylated anti-PSCA-scFv conjugated via neutravidin to mono-biotinylated Toll-like receptor 3 (TLR3) agonist [
23]. However, when using hybrid poylplexes, it cannot be completely ruled out that their β-cyclodextrin moieties, when brought in close proximity to the cell membrane, augment direct uptake of hybrid polyplexes.
In our concept of hybrid polyplexes for targeted gene therapy, we furthermore sought to avoid delivery of bacterial non-methylated CpG-motifs and to simultaneously implement a transposable element for stable expression of the delivered transgene. This combination should prevent unwanted activation of Toll-like receptor 9 or other pattern recognition receptors in treated cells, eventually leading to inflammation and activation of defense mechanisms, which ultimately lead to degradation of foreign DNA [
41]. In this regard, it has been shown that electroporation of eukaryotic cells with SB transposase and transposon in minicircle format leads to 20-fold higher DNA transfer efficiency compared to conventional plasmids, and to profoundly reduced cellular toxicity in human cells by up to 50% [
42]. Of note, our concept using minicircle Sleeping Beauty transposons goes beyond a recent concept using biodegradable poly(β-amino ester)-based nanoparticles electrostatically complexed with polyglutamatic acid-modified anti murine CD3ε-(Fab)2 for targeted delivery of full length plasmids encoding PiggyBac transposase and chimeric antigen receptors (CARs) transposon to murine CD3ε -positive cells. In that respect, disappointing ex vivo transfection efficiencies in murine T cells might be related to the above mentioned defense mechanisms [
43]. Yet, this study and our own results clearly indicate the feasibility of transposon systems to achieve long-lasting gene expression. In particular, clonogenic survival assays of chromosomally stable HCT116
p53−/−/PSCA colorectal cancer cells and chromosomally instable H4
PSCA glioma cells demonstrated significant decreased cell survival in response to targeted transposition of TP53 in vitro using our PSCA-specific hybrid polyplexes. While the small amount of surviving glioma cells clones could not be propagated further in cell culture, a notable fraction of colon cancer clones that escaped transgenic p53 showed loss of p53 protein expression and was further passaged in cell culture. Remarkably, in these cells, profound transgenic p53 expression was restored by treatment with the DNA-damaging bleomycin family antibiotic zeocin, which demonstrates efficiency of the transposon system and furthermore suggests that cancer cells with stable karyotype are also a legitimate target for p53 gene therapy.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}