
Combined Alcohol Exposure and KRAS Mutation in Human Pancreatic Ductal Epithelial Cells Induces Proliferation and Alters Subtype Signatures Determined by Multi-Omics Analysis

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Proliferation Assay
2.3. Caspase-3 Apoptosis Assay
2.4. Cell Cycle Analysis
2.5. Acetaldehyde Assay
2.6. Cellular ROS Measurements
2.7. RNA Sequencing (RNA-Seq)
2.8. Peptide Preparation
2.9. TMT Tagging
2.10. LC/MS-MS and Protein Identification
2.11. Bioinformatics and Statistical Analysis
2.11.1. Differential Expression
2.11.2. GO Term Analysis
2.11.3. Multiple Co-Inertia Analysis
2.11.4. Differential CoExpression
2.11.5. TCGA Data
2.12. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.13. Western Blotting
2.14. Chemotherapeutic Agent Treatment and Viability Assay
3. Results
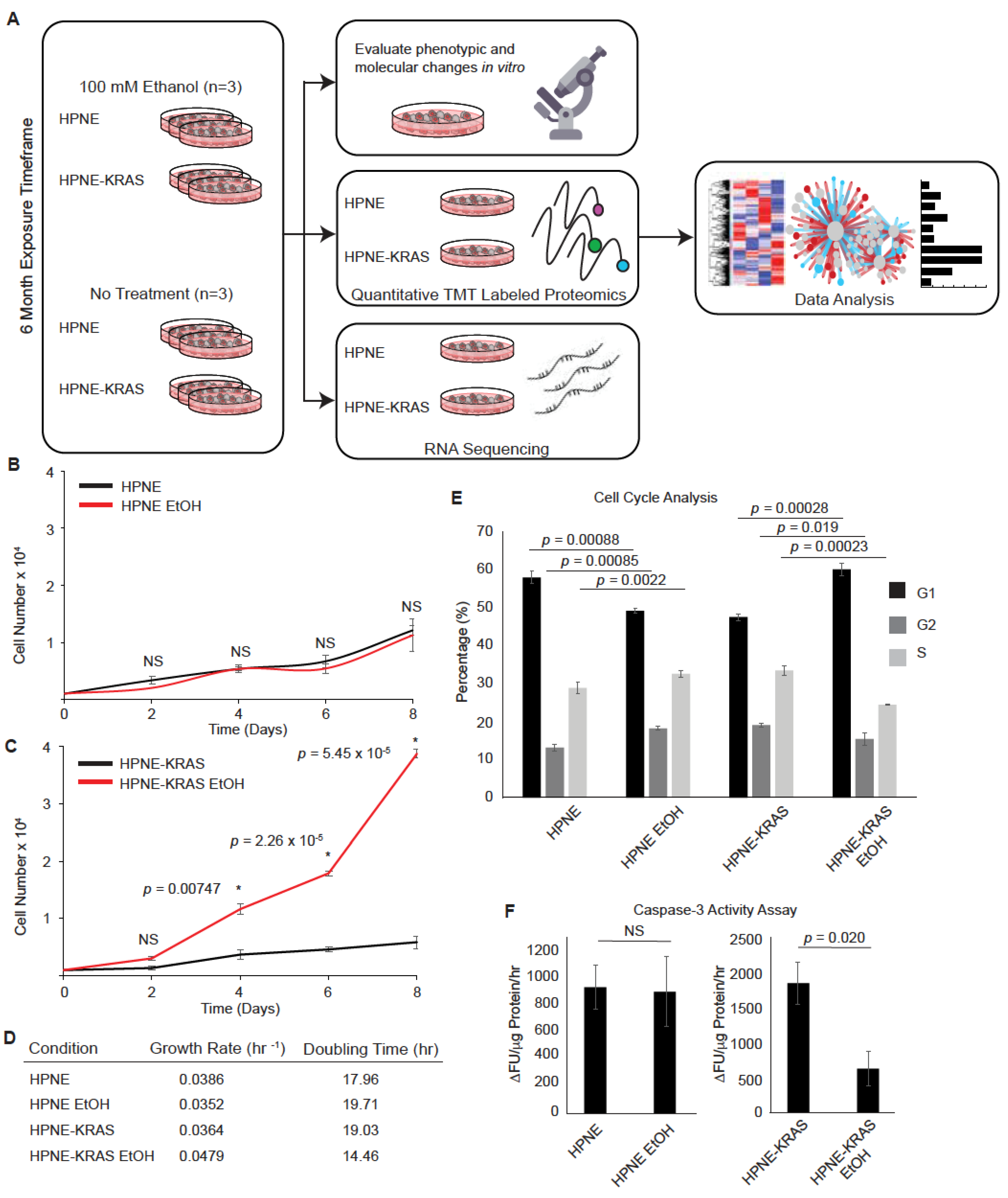
3.1. Chronic Ethanol Treatment Promotes Proliferation in KRAS Mutated HPNE Cells
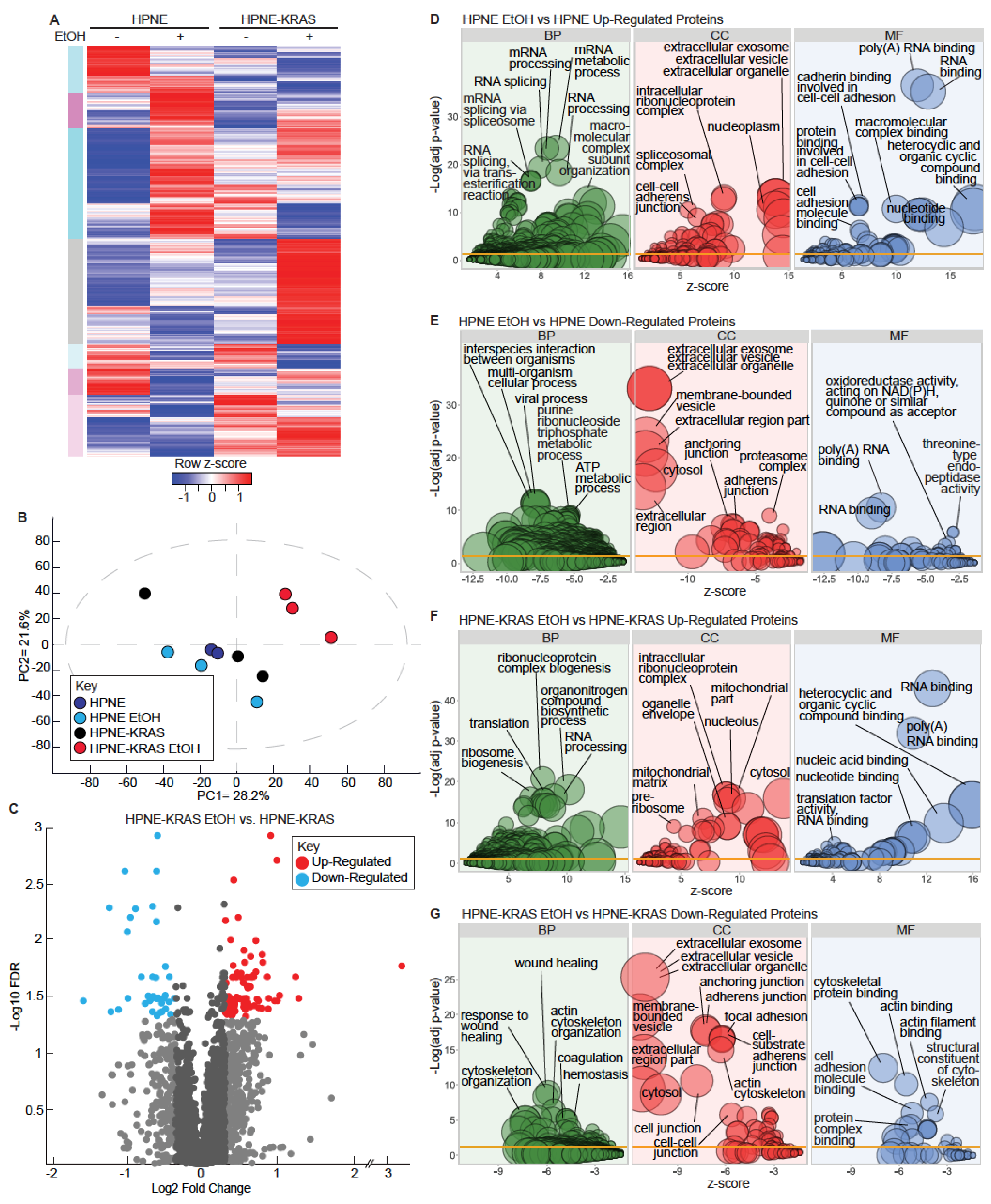
3.2. Mutant KRAS and Ethanol Treatment Increases Ribosomal and RNA Metabolism Gene Expression
3.3. Mutant KRAS and Ethanol Treatment Increases Ribosomal and RNA Processing Proteins
3.4. Ethanol Metabolism Is Marginally Impacted Following Chronic Ethanol Conditioning
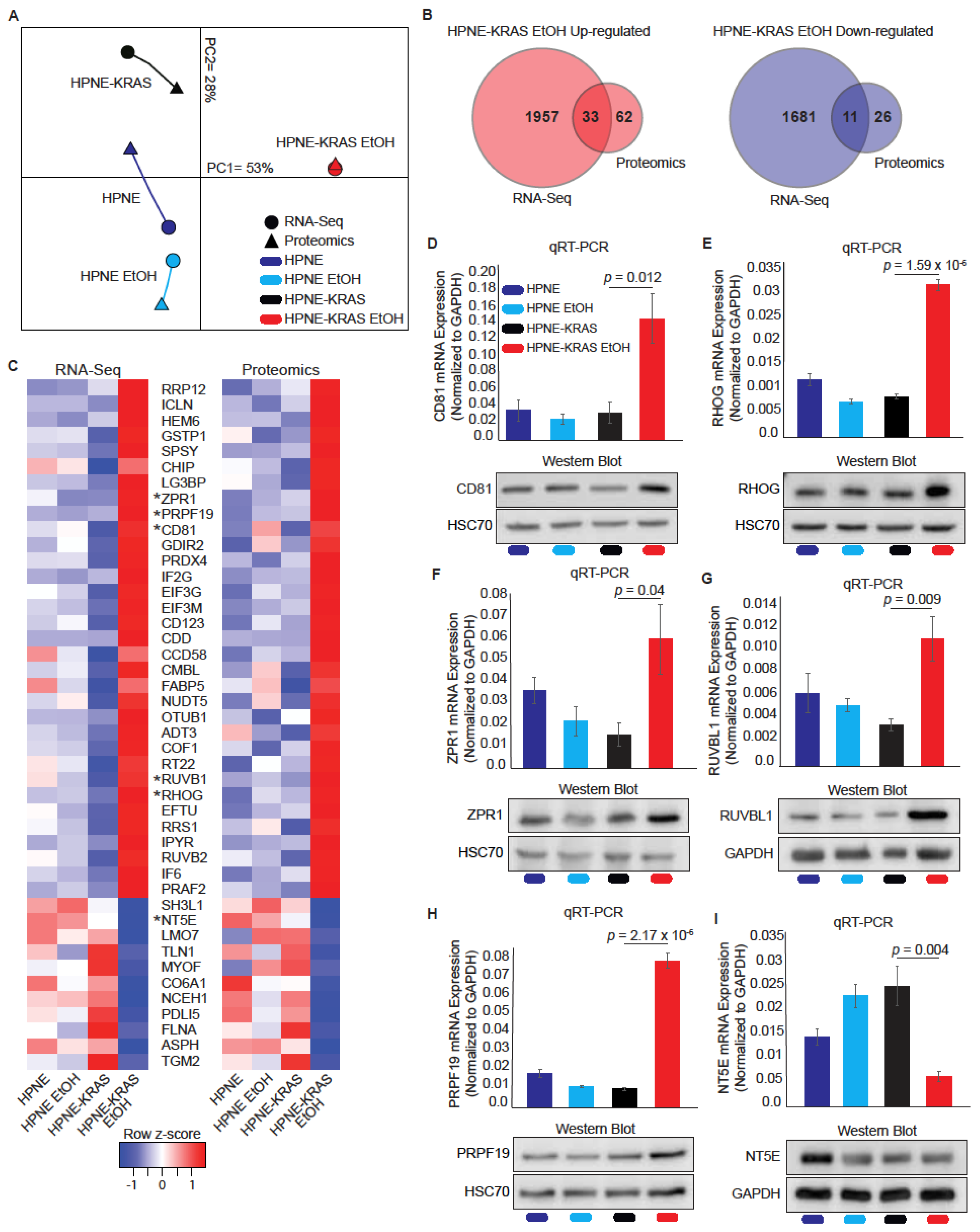
3.5. Mutant KRAS Cells Treated with Ethanol Display Increased Correlation between RNA and Protein Expression through Multi-Omics Comparison
3.6. Differential Co-Expression Analysis Identifies SERPINE1 as a Highly Correlated Protein Unique to EtOH conditioned HPNE-KRAS Cells
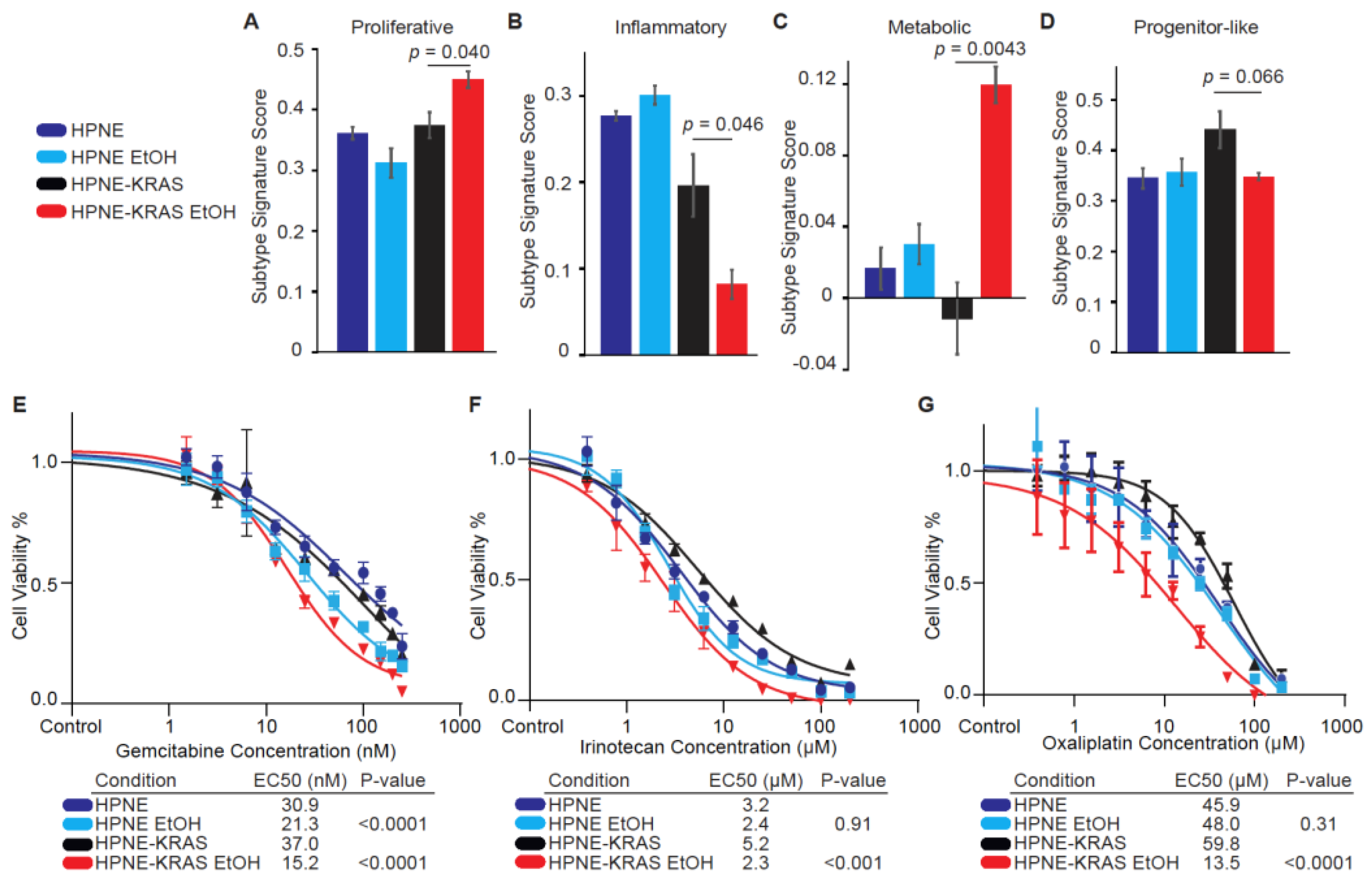
3.7. PDAC Subtypes Are Altered with Ethanol Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Cao, L.; Huang, C.; Cui Zhou, D.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021, 184, 5031–5052.e26. [Google Scholar] [CrossRef]
- Law, H.C.; Lagundzin, D.; Clement, E.J.; Qiao, F.; Wagner, Z.S.; Krieger, K.L.; Costanzo-Garvey, D.; Caffrey, T.C.; Grem, J.L.; DiMaio, D.J.; et al. The Proteomic Landscape of Pancreatic Ductal Adenocarcinoma Liver Metastases Identifies Molecular Subtypes and Associations with Clinical Response. Clin. Cancer Res. 2020, 26, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar]
- Luchini, C.; Paolino, G.; Mattiolo, P.; Piredda, M.L.; Cavaliere, A.; Gaule, M.; Melisi, D.; Salvia, R.; Malleo, G.; Shin, J.I.; et al. KRAS wild-type pancreatic ductal adenocarcinoma: Molecular pathology and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2020, 39, 227. [Google Scholar] [CrossRef]
- Becker, A.E.; Hernandez, Y.G.; Frucht, H.; Lucas, A.L. Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J. Gastroenterol. 2014, 20, 11182–11198. [Google Scholar] [CrossRef]
- Ratna, A.; Mandrekar, P. Alcohol and Cancer: Mechanisms and Therapies. Biomolecules 2017, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longnecker, M.P.; Enger, S.M. Epidemiologic data on alcoholic beverage consumption and risk of cancer. Clin. Chim. Acta 1996, 246, 121–141. [Google Scholar] [CrossRef]
- Duell, E.J. Epidemiology and potential mechanisms of tobacco smoking and heavy alcohol consumption in pancreatic cancer. Mol. Carcinog. 2012, 51, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Wang, F.; Holly, E.A.; Bracci, P.M. Risk of pancreatic cancer by alcohol dose, duration, and pattern of consumption, including binge drinking: A population-based study. Cancer Causes Control 2010, 21, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Michaud, D.S.; Vrieling, A.; Jiao, L.; Mendelsohn, J.B.; Steplowski, E.; Lynch, S.M.; Wactawski-Wende, J.; Arslan, A.A.; Bas Bueno-de-Mesquita, H.; Fuchs, C.S.; et al. Alcohol intake and pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium (PanScan). Cancer Causes Control 2010, 21, 1213–1225. [Google Scholar] [CrossRef]
- Xu, M.; Wang, S.; Ren, Z.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Ke, Z.J.; Shi, X.; Luo, J. Chronic ethanol exposure enhances the aggressiveness of breast cancer: The role of p38gamma. Oncotarget 2016, 7, 3489–3505. [Google Scholar] [CrossRef] [Green Version]
- Testino, G. The burden of cancer attributable to alcohol consumption. Maedica 2011, 6, 313–320. [Google Scholar]
- Asahina, K.; Balog, S.; Hwang, E.; Moon, E.; Wan, E.; Skrypek, K.; Chen, Y.; Fernandez, J.; Romo, J.; Yang, Q.; et al. Moderate alcohol intake promotes pancreatic ductal adenocarcinoma development in mice expressing oncogenic Kras. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G265–G276. [Google Scholar] [CrossRef]
- Yu, W.; Ma, Y.; Roy, S.K.; Srivastava, R.; Shankar, S.; Srivastava, R.K. Ethanol exposure of human pancreatic normal ductal epithelial cells induces EMT phenotype and enhances pancreatic cancer development in KC (Pdx1-Cre and LSL-Kras(G12D)) mice. J. Cell. Mol. Med. 2022, 26, 399–409. [Google Scholar] [CrossRef]
- Campbell, P.M.; Groehler, A.L.; Lee, K.M.; Ouellette, M.M.; Khazak, V.; Der, C.J. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007, 67, 2098–2106. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.F.; Krieger, K.L.; Lagundzin, D.; Li, X.; Cheung, R.S.; Taniguchi, T.; Johnson, K.R.; Bessho, T.; Monteiro, A.N.A.; Woods, N.T. CTDP1 regulates breast cancer survival and DNA repair through BRCT-specific interactions with FANCI. Cell Death Discov. 2019, 5, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagundzin, D.; Hu, W.F.; Law, H.C.H.; Krieger, K.L.; Qiao, F.; Clement, E.J.; Drincic, A.T.; Nedic, O.; Naldrett, M.J.; Alvarez, S.; et al. Delineating the role of FANCA in glucose-stimulated insulin secretion in beta cells through its protein interactome. PLoS ONE 2019, 14, e0220568. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ma, Y.; Shankar, S.; Srivastava, R.K. Chronic ethanol exposure of human pancreatic normal ductal epithelial cells induces cancer stem cell phenotype through SATB2. J. Cell. Mol. Med. 2018, 22, 3920–3928. [Google Scholar] [CrossRef] [PubMed]
- Castilla, R.; Gonzalez, R.; Fouad, D.; Fraga, E.; Muntane, J. Dual effect of ethanol on cell death in primary culture of human and rat hepatocytes. Alcohol Alcohol. 2004, 39, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Gukovskaya, A.S.; Mareninova, O.A.; Odinokova, I.V.; Sung, K.F.; Lugea, A.; Fischer, L.; Wang, Y.L.; Gukovsky, I.; Pandol, S.J. Cell death in pancreatitis: Effects of alcohol. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S10–S13. [Google Scholar] [CrossRef]
- McVicker, B.L.; Tuma, D.J.; Casey, C.A. Effect of ethanol on pro-apoptotic mechanisms in polarized hepatic cells. World J. Gastroenterol. 2007, 13, 4960–4966. [Google Scholar] [CrossRef] [Green Version]
- Dembele, K.; Nguyen, K.H.; Hernandez, T.A.; Nyomba, B.L. Effects of ethanol on pancreatic beta-cell death: Interaction with glucose and fatty acids. Cell Biol. Toxicol. 2009, 25, 141–152. [Google Scholar] [CrossRef]
- Kelkar, S.; Dong, Q.; Xiao, Y.; Joshi-Barve, S.; McClain, C.J.; Barve, S.S. Ethanol enhances activation-induced caspase-3 dependent cell death in T lymphocytes. Alcohol. Clin. Exp. Res. 2002, 26, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, F.D.; Covenas, R. Biochemical Mechanisms Associating Alcohol Use Disorders with Cancers. Cancers 2021, 13, 3548. [Google Scholar] [CrossRef] [PubMed]
- Mameishvili, E.; Serafimidis, I.; Iwaszkiewicz, S.; Lesche, M.; Reinhardt, S.; Bolicke, N.; Buttner, M.; Stellas, D.; Papadimitropoulou, A.; Szabolcs, M.; et al. Aldh1b1 expression defines progenitor cells in the adult pancreas and is required for Kras-induced pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 20679–20688. [Google Scholar] [CrossRef] [Green Version]
- Kubala, M.H.; DeClerck, Y.A. The plasminogen activator inhibitor-1 paradox in cancer: A mechanistic understanding. Cancer Metastasis Rev. 2019, 38, 483–492. [Google Scholar] [CrossRef]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Ollinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Blobner, B.M.; Bellin, M.D.; Beilman, G.J.; Shelton, C.A.; Park, H.J.; Whitcomb, D.C. Gene Expression Profiling of the Pancreas in Patients Undergoing Total Pancreatectomy with Islet Autotransplant Suggests Unique Features of Alcoholic, Idiopathic, and Hereditary Pancreatitis. Pancreas 2020, 49, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Izevbigie, E.B.; Ekunwe, S.I.; Jordan, J.; Howard, C.B. Ethanol modulates the growth of human breast cancer cells in vitro. Exp. Biol. Med. 2002, 227, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Floris, A.; Luo, J.; Frank, J.; Zhou, J.; Orru, S.; Biancolella, M.; Pucci, S.; Orlandi, A.; Campagna, P.; Balzano, A.; et al. Star-related lipid transfer protein 10 (STARD10): A novel key player in alcohol-induced breast cancer progression. J. Exp. Clin. Cancer Res. 2019, 38, 4. [Google Scholar] [CrossRef] [PubMed]
- Charness, M.E.; Safran, R.M.; Perides, G. Ethanol inhibits neural cell-cell adhesion. J. Biol. Chem. 1994, 269, 9304–9309. [Google Scholar] [CrossRef]
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008. [Google Scholar] [CrossRef] [Green Version]
- Elamin, E.; Masclee, A.; Troost, F.; Pieters, H.J.; Keszthelyi, D.; Aleksa, K.; Dekker, J.; Jonkers, D. Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: A combined in vivo and in vitro approach. PLoS ONE 2014, 9, e107421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, T.; Nozu, F.; Imawari, M. Ethanol impairs the assembly and disassembly of actin cytoskeleton and cell adhesion via the RhoA signaling pathway, catenin p120 and E-cadherin in CCK-stimulated pancreatic acini. Biochem. Biophys. Res. Commun. 2011, 405, 558–563. [Google Scholar] [CrossRef]
- Ramanathan, R.; Wilkemeyer, M.F.; Mittal, B.; Perides, G.; Charness, M.E. Alcohol inhibits cell-cell adhesion mediated by human L1. J. Cell Biol. 1996, 133, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013, 5, a012336. [Google Scholar] [CrossRef]
- Minnee, E.; Faller, W.J. Translation initiation and its relevance in colorectal cancer. FEBS J. 2021, 288, 6635–6651. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Zelner, I.; Matlow, J.N.; Natekar, A.; Koren, G. Synthesis of fatty acid ethyl esters in mammalian tissues after ethanol exposure: A systematic review of the literature. Drug Metab. Rev. 2013, 45, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Placencio, V.R.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 in Cancer: Rationale and Insight for Future Therapeutic Testing. Cancer Res. 2015, 75, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Gharibi, A.; La Kim, S.; Molnar, J.; Brambilla, D.; Adamian, Y.; Hoover, M.; Hong, J.; Lin, J.; Wolfenden, L.; Kelber, J.A. ITGA1 is a pre-malignant biomarker that promotes therapy resistance and metastatic potential in pancreatic cancer. Sci. Rep. 2017, 7, 10060. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.L.; Magnus, N.K.C.; Thapar, V.; Morris, R.; Szabolcs, A.; Neyaz, A.; Kulkarni, A.S.; Tai, E.; Chougule, A.; Hillis, A.; et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 26835–26845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Korc, M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol. Cancer Ther. 2012, 11, 2138–2148. [Google Scholar] [CrossRef] [Green Version]
- Lupu-Meiri, M.; Geras-Raaka, E.; Lupu, R.; Shapira, H.; Sandbank, J.; Segal, L.; Gershengorn, M.C.; Oron, Y. Knock-down of plasminogen-activator inhibitor-1 enhances expression of E-cadherin and promotes epithelial differentiation of human pancreatic adenocarcinoma cells. J. Cell. Physiol. 2012, 227, 3621–3628. [Google Scholar] [CrossRef] [Green Version]
- Rossi Sebastiano, M.; Pozzato, C.; Saliakoura, M.; Yang, Z.; Peng, R.W.; Galie, M.; Oberson, K.; Simon, H.U.; Karamitopoulou, E.; Konstantinidou, G. ACSL3-PAI-1 signaling axis mediates tumor-stroma cross-talk promoting pancreatic cancer progression. Sci. Adv. 2020, 6, eabb9200. [Google Scholar] [CrossRef]
- Liu, W.J.; Zhou, L.; Liang, Z.Y.; Zhou, W.X.; You, L.; Zhang, T.P.; Zhao, Y.P. Plasminogen Activator Inhibitor 1 as a Poor Prognostic Indicator in Resectable Pancreatic Ductal Adenocarcinoma. Chin. Med. J. 2018, 131, 2947–2952. [Google Scholar] [CrossRef]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef] [Green Version]
- De Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Marciniak, B.; Kontek, R. Irinotecan-Still an Important Player in Cancer Chemotherapy: A Comprehensive Overview. Int. J. Mol. Sci. 2020, 21, 4919. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clement, E.J.; Law, H.C.-H.; Qiao, F.; Noe, D.; Trevino, J.G.; Woods, N.T. Combined Alcohol Exposure and KRAS Mutation in Human Pancreatic Ductal Epithelial Cells Induces Proliferation and Alters Subtype Signatures Determined by Multi-Omics Analysis. Cancers 2022, 14, 1968. https://doi.org/10.3390/cancers14081968
Clement EJ, Law HC-H, Qiao F, Noe D, Trevino JG, Woods NT. Combined Alcohol Exposure and KRAS Mutation in Human Pancreatic Ductal Epithelial Cells Induces Proliferation and Alters Subtype Signatures Determined by Multi-Omics Analysis. Cancers. 2022; 14(8):1968. https://doi.org/10.3390/cancers14081968
Chicago/Turabian StyleClement, Emalie J., Henry C.-H. Law, Fangfang Qiao, Dragana Noe, Jose G. Trevino, and Nicholas T. Woods. 2022. "Combined Alcohol Exposure and KRAS Mutation in Human Pancreatic Ductal Epithelial Cells Induces Proliferation and Alters Subtype Signatures Determined by Multi-Omics Analysis" Cancers 14, no. 8: 1968. https://doi.org/10.3390/cancers14081968
APA StyleClement, E. J., Law, H. C.-H., Qiao, F., Noe, D., Trevino, J. G., & Woods, N. T. (2022). Combined Alcohol Exposure and KRAS Mutation in Human Pancreatic Ductal Epithelial Cells Induces Proliferation and Alters Subtype Signatures Determined by Multi-Omics Analysis. Cancers, 14(8), 1968. https://doi.org/10.3390/cancers14081968