
Novel Targeted Therapeutic Strategies for Ewing Sarcoma

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
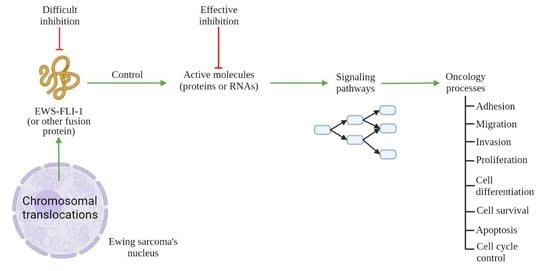
1. Introduction
2. General Consideration: Hallmarks of Cancer
3. Molecular Targets for ES Therapy
3.1. Targeting of ES Pressure on Adhesion, Migration, and Invasion
3.2. Targeting of Ewing Sarcoma Cells with a Focus on Proliferation, Cell Differentiation, and Cell Survival
3.3. Targeting of ES: Induction of Apoptosis and Cell Cycle Arrest
4. Current Clinical Trials
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| CD99 | cluster of differentiation 99 |
| EMT | epithelial to mesenchymal transition |
| ERBB4 | Erb-B2 Receptor Tyrosine Kinase 4 |
| ES | Ewing sarcoma |
| ESFT | Ewing sarcoma family tumors |
| EZH2 | Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| GDF6 | growth and differentiation factor 6 |
| FET | family of genes, Fused in sarcoma, Ewing sarcoma breakpoint region 1, TATA-box binding protein associated factor 15 |
| GLI1 | glioma-associated oncogene 1 |
| IGF-1 | Insulin-like growth factor |
| IGF-1R | insulin-like growth factor 1 receptor |
| LDH | lactate dehydrogenase |
| lncRNAs | long non-coding RNAs |
| MAPK | Mitogen-Activated Protein Kinase |
| MET | mesenchymal to epithelial transition |
| MMP9 | metalloproteinase type 9 |
| MRTFB | of Myocardin-related transcription factor B |
| NAMPT | nicotinamide phosphoribosyl transferase |
| NCAM | neural cell adhesion molecule |
| PAPP-A | Pregnancy-associated plasma protein A |
| PARP | Poly (ADP-ribose) polymerase |
| PDGF | Platelet-derived growth factor |
| PHGDH | 3-phosphoglycerate dehydrogenase |
| PI3K | Phosphoinositide 3-kinases |
| ROCK | Rho-associated coiled-coil kinase |
| RhoA | Ras homolog family member A |
| SHH | Sonic Hedgehog pathway |
| SOX2 | SRY-Box Transcription Factor 2 |
| SRBCT | Small Round Blue Cell Tumors |
| TEAD | TEA domain family member 1 |
| TNF | tumor necrosis factor |
| mTOR | mammalian target of rapamycin |
| TRAIL | TNF-Related Apoptosis Inducing Ligand |
| PNET | Primitive neuroectodermal tumor |
| VEGF-A | Vascular endothelial growth factor A |
| YAP | Yes-associated protein. |
References
- Mer, A.S.; Ba-Alawi, W.; Smirnov, P.; Wang, Y.X.; Brew, B.; Ortmann, J.; Tsao, M.S.; Cescon, D.W.; Goldenberg, A.; Haibe-Kains, B. Integrative Pharmacogenomics Analysis of Patient-Derived Xenografts. Cancer Res. 2019, 79, 4539–4550. [Google Scholar] [CrossRef] [PubMed]
- Esiashvili, N.; Goodman, M.; Marcus, R.B., Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J. Pediatr. Hematol. Oncol. 2008, 30, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.U.; Cheung, M.C.; Min, E.S.; Schneiderbauer, M.M.; Koniaris, L.G.; Scully, S.P. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: An analysis of 1631 cases from the SEER database, 1973–2005. Cancer 2009, 115, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, J.L.; Osuna, D.; Herrero, D.; de Alava, E.; Madoz-Gurpide, J. Advances in Ewing’s sarcoma research: Where are we now and what lies ahead? Cancer Res. 2009, 69, 7140–7150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauer, M.; Ban, J.; Kofler, R.; Walker, B.; Davis, S.; Meltzer, P.; Kovar, H. A molecular function map of Ewing’s sarcoma. PLoS ONE 2009, 4, e5415. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.A.; Fisher, C.; Judson, I.; Thomas, J.M. Soft-tissue sarcomas in adults. N. Engl. J. Med. 2005, 353, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Cabral, A.N.F.; Rocha, R.H.; Amaral, A.; Medeiros, K.B.; Nogueira, P.S.E.; Diniz, L.M. Cutaneous angiosarcoma: Report of three different and typical cases admitted in a unique dermatology clinic. An. Bras. Dermatol. 2017, 92, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Kruse, A.J.; Croce, S.; Kruitwagen, R.F.; Riedl, R.G.; Slangen, B.F.; Van Gorp, T.; Van de Vijver, K.K. Aggressive behavior and poor prognosis of endometrial stromal sarcomas with YWHAE-FAM22 rearrangement indicate the clinical importance to recognize this subset. Int. J. Gynecol. Cancer 2014, 24, 1616–1622. [Google Scholar] [CrossRef]
- Coates, S.J.; Ogunrinade, O.; Lee, H.J.; Desman, G. Epidermotropic metastatic epithelioid sarcoma: A potential diagnostic pitfall. J Cutan Pathol 2014, 41, 672–676. [Google Scholar] [CrossRef]
- Lin, P.P.; Wang, Y.; Lozano, G. Mesenchymal Stem Cells and the Origin of Ewing’s Sarcoma. Sarcoma 2011, 2011, 276463. [Google Scholar] [CrossRef] [Green Version]
- Riggi, N.; Stamenkovic, I. The Biology of Ewing sarcoma. Cancer Lett. 2007, 254, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, E.R.; Lim, J.F.; Tao, W.; Poremba, C.; Chow, C.J.; Kalousek, I.V.; Kovar, H.; MacDonald, T.J.; Sorensen, P.H. The Ewing tumor family of peripheral primitive neuroectodermal tumors expresses human gastrin-releasing peptide. Cancer Res. 1998, 58, 2469–2476. [Google Scholar] [PubMed]
- O’Regan, S.; Diebler, M.F.; Meunier, F.M.; Vyas, S. A Ewing’s sarcoma cell line showing some, but not all, of the traits of a cholinergic neuron. J. Neurochem. 1995, 64, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, M.; Thomsen, C.; Grundevik, P.; Jonasson, E.; Andersson, D.; Runnberg, R.; Dolatabadi, S.; Vannas, C.; Luna Santamariotaa, M.; Fagman, H.; et al. FET family fusion oncoproteins target the SWI/SNF chromatin remodeling complex. EMBO Rep. 2019, 20, e45766. [Google Scholar] [CrossRef] [PubMed]
- Linden, M.; Vannas, C.; Osterlund, T.; Andersson, L.; Osman, A.; Escobar, M.; Fagman, H.; Stahlberg, A.; Aman, P. FET fusion oncoproteins interact with BRD4 and SWI/SNF chromatin remodelling complex subtypes in sarcoma. Mol. Oncol. 2022. [Google Scholar] [CrossRef]
- Trancau, I.O. Chromosomal translocations highlighted in Primitive Neuroectodermal Tumors (PNET) and Ewing sarcoma. J. Med. Life 2014, 7, 44–50. [Google Scholar]
- Van Mater, D.; Wagner, L. Management of recurrent Ewing sarcoma: Challenges and approaches. Onco. Targets Ther. 2019, 12, 2279–2288. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Stolte, B.; Iniguez, A.B.; Dharia, N.V.; Robichaud, A.L.; Conway, A.S.; Morgan, A.M.; Alexe, G.; Schauer, N.J.; Liu, X.; Bird, G.H.; et al. Genome-scale CRISPR-Cas9 screen identifies druggable dependencies in TP53 wild-type Ewing sarcoma. J. Exp. Med. 2018, 215, 2137–2155. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, A.; Ohmura, S.; Orth, M.F.; Knott, M.M.L.; Colombo, M.V.; Arrigoni, C.; Bardinet, V.; Saucier, D.; Wehweck, F.S.; Li, J.; et al. Oncogenic hijacking of a developmental transcription factor evokes vulnerability toward oxidative stress in Ewing sarcoma. Nat. Commun. 2020, 11, 2423. [Google Scholar] [CrossRef] [PubMed]
- Cironi, L.; Riggi, N.; Provero, P.; Wolf, N.; Suva, M.L.; Suva, D.; Kindler, V.; Stamenkovic, I. IGF1 is a common target gene of Ewing’s sarcoma fusion proteins in mesenchymal progenitor cells. PLoS ONE 2008, 3, e2634. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Suva, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suva, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattenden, S.G.; Simon, J.M.; Wali, A.; Jayakody, C.N.; Troutman, J.; McFadden, A.W.; Wooten, J.; Wood, C.C.; Frye, S.V.; Janzen, W.P.; et al. High-throughput small molecule screen identifies inhibitors of aberrant chromatin accessibility. Proc. Natl. Acad. Sci. USA 2016, 113, 3018–3023. [Google Scholar] [CrossRef] [Green Version]
- Welch, D.; Kahen, E.; Fridley, B.; Brohl, A.S.; Cubitt, C.L.; Reed, D.R. Small molecule inhibition of lysine-specific demethylase 1 (LSD1) and histone deacetylase (HDAC) alone and in combination in Ewing sarcoma cell lines. PLoS ONE 2019, 14, e0222228. [Google Scholar] [CrossRef]
- Grohar, P.J.; Woldemichael, G.M.; Griffin, L.B.; Mendoza, A.; Chen, Q.R.; Yeung, C.; Currier, D.G.; Davis, S.; Khanna, C.; Khan, J.; et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J. Natl. Cancer Inst. 2011, 103, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, J.R.; Pioche-Durieu, C.; Ayala, J.; Petit, T.; Girard, H.A.; Malvy, C.P.; Le Cam, E.; Treussart, F.; Arnault, J.C. Plasma hydrogenated cationic detonation nanodiamonds efficiently deliver to human cells in culture functional siRNA targeting the Ewing sarcoma junction oncogene. Biomaterials 2015, 45, 93–98. [Google Scholar] [CrossRef]
- Gauthier, F.; Claveau, S.; Bertrand, J.R.; Vasseur, J.J.; Dupouy, C.; Debart, F. Gymnotic delivery and gene silencing activity of reduction-responsive siRNAs bearing lipophilic disulfide-containing modifications at 2′-position. Bioorg. Med. Chem. 2018, 26, 4635–4643. [Google Scholar] [CrossRef]
- Cervera, S.T.; Rodriguez-Martin, C.; Fernandez-Tabanera, E.; Melero-Fernandez de Mera, R.M.; Morin, M.; Fernandez-Penalver, S.; Iranzo-Martinez, M.; Amhih-Cardenas, J.; Garcia-Garcia, L.; Gonzalez-Gonzalez, L.; et al. Therapeutic Potential of EWSR1-FLI1 Inactivation by CRISPR/Cas9 in Ewing Sarcoma. Cancers 2021, 13, 3783. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heisey, D.A.R.; Lochmann, T.L.; Floros, K.V.; Coon, C.M.; Powell, K.M.; Jacob, S.; Calbert, M.L.; Ghotra, M.S.; Stein, G.T.; Maves, Y.K.; et al. The Ewing Family of Tumors Relies on BCL-2 and BCL-XL to Escape PARP Inhibitor Toxicity. Clin. Cancer Res. 2019, 25, 1664–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palombo, R.; Verdile, V.; Paronetto, M.P. Poison-Exon Inclusion in DHX9 Reduces Its Expression and Sensitizes Ewing Sarcoma Cells to Chemotherapeutic Treatment. Cells 2020, 9, 328. [Google Scholar] [CrossRef] [Green Version]
- Radic-Sarikas, B.; Tsafou, K.P.; Emdal, K.B.; Papamarkou, T.; Huber, K.V.; Mutz, C.; Toretsky, J.A.; Bennett, K.L.; Olsen, J.V.; Brunak, S.; et al. Combinatorial Drug Screening Identifies Ewing Sarcoma-specific Sensitivities. Mol. Cancer Ther. 2017, 16, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Lessnick, S.L.; Ladanyi, M. Molecular pathogenesis of Ewing sarcoma: New therapeutic and transcriptional targets. Annu. Rev. Pathol. 2012, 7, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Spriano, F.; Chung, E.Y.L.; Gaudio, E.; Tarantelli, C.; Cascione, L.; Napoli, S.; Jessen, K.; Carrassa, L.; Priebe, V.; Sartori, G.; et al. The ETS Inhibitors YK-4-279 and TK-216 Are Novel Antilymphoma Agents. Clin. Cancer Res. 2019, 25, 5167–5176. [Google Scholar] [CrossRef]
- Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Ramamoorthy, V.; Aiyer, R.A.; Maywald, R.L.; Buford, A.S.; Doolittle, D.K.; Culotta, K.S.; O’Dorisio, J.E.; Ludwig, J.A. An Oral Formulation of YK-4-279: Preclinical Efficacy and Acquired Resistance Patterns in Ewing Sarcoma. Mol. Cancer Ther. 2015, 14, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Rahim, S.; Minas, T.; Hong, S.H.; Justvig, S.; Celik, H.; Kont, Y.S.; Han, J.; Kallarakal, A.T.; Kong, Y.; Rudek, M.A.; et al. A small molecule inhibitor of ETV1, YK-4-279, prevents prostate cancer growth and metastasis in a mouse xenograft model. PLoS ONE 2014, 9, e114260. [Google Scholar] [CrossRef] [Green Version]
- Rellinger, E.J.; Padmanabhan, C.; Qiao, J.; Appert, A.; Waterson, A.G.; Lindsley, C.W.; Beauchamp, R.D.; Chung, D.H. ML327 induces apoptosis and sensitizes Ewing sarcoma cells to TNF-related apoptosis-inducing ligand. Biochem. Biophys. Res. Commun. 2017, 491, 463–468. [Google Scholar] [CrossRef]
- Zhou, F.; Elzi, D.J.; Jayabal, P.; Ma, X.; Chiu, Y.C.; Chen, Y.; Blackman, B.; Weintraub, S.T.; Houghton, P.J.; Shiio, Y. GDF6-CD99 Signaling Regulates Src and Ewing Sarcoma Growth. Cell. Rep. 2020, 33, 108332. [Google Scholar] [CrossRef] [PubMed]
- Pinca, R.S.; Manara, M.C.; Chiadini, V.; Picci, P.; Zucchini, C.; Scotlandi, K. Targeting ROCK2 rather than ROCK1 inhibits Ewing sarcoma malignancy. Oncol. Rep. 2017, 37, 1387–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehnman, M.; Chaabane, W.; Haglund, F.; Tsagkozis, P. The Tumor Microenvironment of Pediatric Sarcoma: Mesenchymal Mechanisms Regulating Cell Migration and Metastasis. Curr. Oncol. Rep. 2019, 21, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codman, E.A. The classic: Registry of bone sarcoma: Part I.—Twenty-five criteria for establishing the diagnosis of osteogenic sarcoma. Part II.—Thirteen registered cases of “five year cures” analyzed according to these criteria. 1926. Clin. Orthop. Relat. Res. 2009, 467, 2771–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Ware, K.E.; Xu, S.; Gilja, S.; Shetler, S.; Yang, Y.; Wang, X.; Austin, R.G.; Runyambo, D.; Hish, A.J.; et al. E-Cadherin Represses Anchorage-Independent Growth in Sarcomas through Both Signaling and Mechanical Mechanisms. Mol. Cancer Res. 2019, 17, 1391–1402. [Google Scholar] [CrossRef] [Green Version]
- Machado, I.; Navarro, S.; Lopez-Guerrero, J.A.; Alberghini, M.; Picci, P.; Llombart-Bosch, A. Epithelial marker expression does not rule out a diagnosis of Ewing’s sarcoma family of tumours. Virchows Arch. 2011, 459, 409–414. [Google Scholar] [CrossRef]
- Hurtubise, A.; Bernstein, M.L.; Momparler, R.L. Preclinical evaluation of the antineoplastic action of 5-aza-2′-deoxycytidine and different histone deacetylase inhibitors on human Ewing’s sarcoma cells. Cancer Cell Int. 2008, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Sanceau, J.; Truchet, S.; Bauvois, B. Matrix metalloproteinase-9 silencing by RNA interference triggers the migratory-adhesive switch in Ewing’s sarcoma cells. J. Biol. Chem. 2003, 278, 36537–36546. [Google Scholar] [CrossRef] [Green Version]
- Roberto, G.M.; Delsin, L.E.A.; Vieira, G.M.; Silva, M.O.; Hakime, R.G.; Gava, N.F.; Engel, E.E.; Scrideli, C.A.; Tone, L.G.; Brassesco, M.S. ROCK1-PredictedmicroRNAs Dysregulation Contributes to Tumor Progression in Ewing Sarcoma. Pathol. Oncol. Res. 2020, 26, 133–139. [Google Scholar] [CrossRef]
- Gluer, S.; Zense, M.; von Schweinitz, D. Cell adhesion molecules and intermediate filaments on embryonal childhood tumors. Pathol. Res. Pract. 1998, 194, 773–780. [Google Scholar] [CrossRef]
- Hatano, M.; Matsumoto, Y.; Fukushi, J.; Matsunobu, T.; Endo, M.; Okada, S.; Iura, K.; Kamura, S.; Fujiwara, T.; Iida, K.; et al. Cadherin-11 regulates the metastasis of Ewing sarcoma cells to bone. Clin. Exp. Metastasis 2015, 32, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.G.; Jenabi, J.M.; Zhang, J.; Keshelava, N.; Shimada, H.; May, W.A.; Ng, T.; Reynolds, C.P.; Triche, T.J.; Sorensen, P.H. E-cadherin cell-cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res. 2007, 67, 3094–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J.; Kim, Y.; Yoo, Y.H.; Kim, M.S.; Lee, S.H.; Kim, C.G.; Park, K.; Jeoung, D.; Lee, H.; Ko, I.Y.; et al. CD99-Derived Agonist Ligands Inhibit Fibronectin-Induced Activation of beta1 Integrin through the Protein Kinase A/SHP2/Extracellular Signal-Regulated Kinase/PTPN12/Focal Adhesion Kinase Signaling Pathway. Mol. Cell Biol. 2017, 37, e00675-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzetti, G.A.; Laud-Duval, K.; Bellanger, D.; Stern, M.H.; Sastre-Garau, X.; Delattre, O. MiR-30a-5p connects EWS-FLI1 and CD99, two major therapeutic targets in Ewing tumor. Oncogene 2013, 32, 3915–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerisano, V.; Aalto, Y.; Perdichizzi, S.; Bernard, G.; Manara, M.C.; Benini, S.; Cenacchi, G.; Preda, P.; Lattanzi, G.; Nagy, B.; et al. Molecular mechanisms of CD99-induced caspase-independent cell death and cell-cell adhesion in Ewing’s sarcoma cells: Actin and zyxin as key intracellular mediators. Oncogene 2004, 23, 5664–5674. [Google Scholar] [CrossRef] [Green Version]
- Huijbers, E.J.M.; van der Werf, I.M.; Faber, L.D.; Sialino, L.D.; van der Laan, P.; Holland, H.A.; Cimpean, A.M.; Thijssen, V.; van Beijnum, J.R.; Griffioen, A.W. Targeting Tumor Vascular CD99 Inhibits Tumor Growth. Front. Immunol. 2019, 10, 651. [Google Scholar] [CrossRef]
- De Feo, A.; Sciandra, M.; Ferracin, M.; Felicetti, F.; Astolfi, A.; Pignochino, Y.; Picci, P.; Care, A.; Scotlandi, K. Exosomes from CD99-deprived Ewing sarcoma cells reverse tumor malignancy by inhibiting cell migration and promoting neural differentiation. Cell Death Dis. 2019, 10, 471. [Google Scholar] [CrossRef]
- Rocchi, A.; Manara, M.C.; Sciandra, M.; Zambelli, D.; Nardi, F.; Nicoletti, G.; Garofalo, C.; Meschini, S.; Astolfi, A.; Colombo, M.P.; et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J. Clin. Investig. 2010, 120, 668–680. [Google Scholar] [CrossRef]
- Sohn, H.W.; Choi, E.Y.; Kim, S.H.; Lee, I.S.; Chung, D.H.; Sung, U.A.; Hwang, D.H.; Cho, S.S.; Jun, B.H.; Jang, J.J.; et al. Engagement of CD99 induces apoptosis through a calcineurin-independent pathway in Ewing’s sarcoma cells. Am. J. Pathol. 1998, 153, 1937–1945. [Google Scholar] [CrossRef]
- Manara, M.C.; Terracciano, M.; Mancarella, C.; Sciandra, M.; Guerzoni, C.; Pasello, M.; Grilli, A.; Zini, N.; Picci, P.; Colombo, M.P.; et al. CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1R/RAS/Rac1 signaling. Oncotarget 2016, 7, 79925–79942. [Google Scholar] [CrossRef] [Green Version]
- Katschnig, A.M.; Kauer, M.O.; Schwentner, R.; Tomazou, E.M.; Mutz, C.N.; Linder, M.; Sibilia, M.; Alonso, J.; Aryee, D.N.T.; Kovar, H. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017, 36, 5995–6005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS ONE 2009, 4, e7608. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, K.L.; Eisenacher, M.; Braun, Y.; Brachwitz, K.; Wai, D.H.; Dirksen, U.; Lanvers-Kaminsky, C.; Juergens, H.; Herrero, D.; Stegmaier, S.; et al. Microarray analysis of Ewing’s sarcoma family of tumours reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur. J. Cancer 2008, 44, 699–709. [Google Scholar] [CrossRef]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef] [Green Version]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Mullard, M.; Cade, M.; Morice, S.; Dupuy, M.; Danieau, G.; Amiaud, J.; Renault, S.; Lezot, F.; Brion, R.; Thepault, R.A.; et al. Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth. Cancers 2020, 12, 3438. [Google Scholar] [CrossRef]
- Matsumoto, T.; Tabata, K.; Suzuki, T. The GANT61, a GLI inhibitor, induces caspase-independent apoptosis of SK-N-LO cells. Biol. Pharm. Bull. 2014, 37, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Bacelar Sacramento de Araujo, T.; de Oliveira Siquara da Rocha, L.; Torres Andion Vidal, M.; Cerqueira Coelho, P.L.; Galvao Dos Reis, M.; Solano de Freitas Souza, B.; Botelho Pereira Soares, M.; Almeida Pereira, T.; Della Coletta, R.; Pereira Bezerra, D.; et al. GANT61 Reduces Hedgehog Molecule (GLI1) Expression and Promotes Apoptosis in Metastatic Oral Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 6076. [Google Scholar] [CrossRef] [PubMed]
- Karakus, R.; Karakus, E.; Emir, S.; Kacar, A.; Ozyoruk, D. Insulin-like growth factor-1 receptor expression in pediatric tumors: A comparative immunohistochemical study. Turk. J. Med. Sci. 2018, 48, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lian, Y.; Xie, K.; Cai, Y.; Pan, Y.; Zhu, Y. Ropivacaine suppresses tumor biological characteristics of human hepatocellular carcinoma via inhibiting IGF-1R/PI3K/AKT/mTOR signaling axis. Bioengineered 2021, 12, 9162–9173. [Google Scholar] [CrossRef]
- Dobre, M.; Herlea, V.; Vladut, C.; Ciocirlan, M.; Balaban, V.D.; Constantinescu, G.; Diculescu, M.; Milanesi, E. Dysregulation of miRNAs Targeting the IGF-1R Pathway in Pancreatic Ductal Adenocarcinoma. Cells 2021, 10, 1856. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Bai, Y.; Ni, T.; Li, Y.; Cao, R.; Ji, S.; Li, S. MicroRNA1533p suppresses retinoblastoma cell growth and invasion via targeting the IGF1R/Raf/MEK and IGF1R/PI3K/AKT signaling pathways. Int. J. Oncol. 2021, 59, 47. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Y.; Segar, N.; Huang, C.; Zeng, P.; Tan, X.; Mao, L.; Chen, Z.; Haglund, F.; Larsson, O.; et al. Nuclear IGF1R interacts with NuMA and regulates 53BP1dependent DNA doublestrand break repair in colorectal cancer. Oncol. Rep. 2021, 46, 168. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Steinberg, S.M.; Thakar, M.; Counts, D.; Pironis, B.; Parente, C.; Eskenazi, A.; Helman, L.; Wexler, L.H. Insulin-like growth factor type 1 (IGF-1) and IGF binding protein-3 in patients with Ewing sarcoma family of tumors. Cancer 2001, 92, 2941–2947. [Google Scholar] [CrossRef]
- Scotlandi, K.; Manara, M.C.; Serra, M.; Marino, M.T.; Ventura, S.; Garofalo, C.; Alberghini, M.; Magagnoli, G.; Ferrari, S.; Lopez-Guerrero, J.A.; et al. Expression of insulin-like growth factor system components in Ewing’s sarcoma and their association with survival. Eur. J. Cancer 2011, 47, 1258–1266. [Google Scholar] [CrossRef]
- Worrall, C.; Nedelcu, D.; Serly, J.; Suleymanova, N.; Oprea, I.; Girnita, A.; Girnita, L. Novel mechanisms of regulation of IGF-1R action: Functional and therapeutic implications. Pediatr. Endocrinol. Rev. 2013, 10, 473–484. [Google Scholar]
- de Groot, S.; Rottgering, B.; Gelderblom, H.; Pijl, H.; Szuhai, K.; Kroep, J.R. Unraveling the Resistance of IGF-Pathway Inhibition in Ewing Sarcoma. Cancers 2020, 12, 3568. [Google Scholar] [CrossRef]
- Heitzeneder, S.; Sotillo, E.; Shern, J.F.; Sindiri, S.; Xu, P.; Jones, R.; Pollak, M.; Noer, P.R.; Lorette, J.; Fazli, L.; et al. Pregnancy-Associated Plasma Protein-A (PAPP-A) in Ewing Sarcoma: Role in Tumor Growth and Immune Evasion. J. Natl. Cancer Inst. 2019, 111, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, A.; Thiede, M.; Grunewald, T.G.; Alba Rubio, R.; Richter, G.H.; Kirchner, T.; Busch, D.H.; Burdach, S.; Thiel, U. Pappalysin-1 T cell receptor transgenic allo-restricted T cells kill Ewing sarcoma in vitro and in vivo. Oncoimmunology 2017, 6, e1273301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardali, E.; van der Schaft, D.W.; Wiercinska, E.; Gorter, A.; Hogendoorn, P.C.; Griffioen, A.W.; ten Dijke, P. Critical role of endoglin in tumor cell plasticity of Ewing sarcoma and melanoma. Oncogene 2011, 30, 334–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdan, R.; Zhou, Z.; Kleinerman, E.S. Blocking SDF-1alpha/CXCR4 downregulates PDGF-B and inhibits bone marrow-derived pericyte differentiation and tumor vascular expansion in Ewing tumors. Mol. Cancer Ther. 2014, 13, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uren, A.; Merchant, M.S.; Sun, C.J.; Vitolo, M.I.; Sun, Y.; Tsokos, M.; Illei, P.B.; Ladanyi, M.; Passaniti, A.; Mackall, C.; et al. Beta-platelet-derived growth factor receptor mediates motility and growth of Ewing’s sarcoma cells. Oncogene 2003, 22, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Zwerner, J.P.; May, W.A. PDGF-C is an EWS/FLI induced transforming growth factor in Ewing family tumors. Oncogene 2001, 20, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Koppenhafer, S.L.; Goss, K.L.; Terry, W.W.; Gordon, D.J. Inhibition of the ATR-CHK1 Pathway in Ewing Sarcoma Cells Causes DNA Damage and Apoptosis via the CDK2-Mediated Degradation of RRM2. Mol. Cancer Res. 2020, 18, 91–104. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, M.; Luo, Z.; Wen, Z.; Yan, X. The dichotomous role of TGF-beta in controlling liver cancer cell survival and proliferation. J. Genet. Genom. 2020, 47, 497–512. [Google Scholar] [CrossRef]
- Pridgeon, M.G.; Grohar, P.J.; Steensma, M.R.; Williams, B.O. Wnt Signaling in Ewing Sarcoma, Osteosarcoma, and Malignant Peripheral Nerve Sheath Tumors. Curr. Osteoporos. Rep. 2017, 15, 239–246. [Google Scholar] [CrossRef]
- Ross, K.A.; Smyth, N.A.; Murawski, C.D.; Kennedy, J.G. The biology of ewing sarcoma. ISRN Oncol 2013, 2013, 759725. [Google Scholar] [CrossRef] [Green Version]
- Burdach, S.; Plehm, S.; Unland, R.; Dirksen, U.; Borkhardt, A.; Staege, M.S.; Muller-Tidow, C.; Richter, G.H. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle 2009, 8, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Abedalthagafi, M.; Anwar, A.E.; Bui, M.M. Akt and Hippo Pathways in Ewing’s Sarcoma Tumors and Their Prognostic Significance. J. Cancer 2015, 6, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.H.; Lawlor, E.R. BMI-1 suppresses contact inhibition and stabilizes YAP in Ewing sarcoma. Oncogene 2011, 30, 2077–2085. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.M.; Bensard, C.; Wei, P.; Krah, N.M.; Schell, J.C.; Gardiner, J.; Schiffman, J.; Lessnick, S.L.; Rutter, J. EWS/FLI is a Master Regulator of Metabolic Reprogramming in Ewing Sarcoma. Mol. Cancer Res. 2017, 15, 1517–1530. [Google Scholar] [CrossRef] [Green Version]
- Sen, N.; Cross, A.M.; Lorenzi, P.L.; Khan, J.; Gryder, B.E.; Kim, S.; Caplen, N.J. EWS-FLI1 reprograms the metabolism of Ewing sarcoma cells via positive regulation of glutamine import and serine-glycine biosynthesis. Mol. Carcinog. 2018, 57, 1342–1357. [Google Scholar] [CrossRef]
- Issaq, S.H.; Mendoza, A.; Kidner, R.; Rosales, T.I.; Duveau, D.Y.; Heske, C.M.; Rohde, J.M.; Boxer, M.B.; Thomas, C.J.; DeBerardinis, R.J.; et al. EWS-FLI1-regulated Serine Synthesis and Exogenous Serine are Necessary for Ewing Sarcoma Cellular Proliferation and Tumor Growth. Mol. Cancer Ther. 2020, 19, 1520–1529. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.M.; Antolin, I.; Puente-Moncada, N.; Suarez, S.; Gomez-Lobo, M.; Rodriguez, C.; Martin, V. Melatonin Cytotoxicity Is Associated to Warburg Effect Inhibition in Ewing Sarcoma Cells. PLoS ONE 2015, 10, e0135420. [Google Scholar] [CrossRef]
- Nie, X.; Wang, H.; Wei, X.; Li, L.; Xue, T.; Fan, L.; Ma, H.; Xia, Y.; Wang, Y.D.; Chen, W.D. LRP5 promotes gastric cancer via activating canonical Wnt/beta-catenin and glycolysis pathways. Am. J. Pathol. 2021, 192, 503–517. [Google Scholar] [CrossRef]
- Cortese, N.; Capretti, G.; Barbagallo, M.; Rigamonti, A.; Takis, P.G.; Castino, G.F.; Vignali, D.; Maggi, G.; Gavazzi, F.; Ridolfi, C.; et al. Metabolome of Pancreatic Juice Delineates Distinct Clinical Profiles of Pancreatic Cancer and Reveals a Link between Glucose Metabolism and PD-1(+) Cells. Cancer Immunol. Res. 2020, 8, 493–505. [Google Scholar] [CrossRef] [PubMed]
- van der Schaft, D.W.; Hillen, F.; Pauwels, P.; Kirschmann, D.A.; Castermans, K.; Egbrink, M.G.; Tran, M.G.; Sciot, R.; Hauben, E.; Hogendoorn, P.C.; et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. 2005, 65, 11520–11528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, A.; Trucco, M.; Rainusso, N.; Bernardi, R.J.; Shuck, R.; Kurenbekova, L.; Loeb, D.M.; Yustein, J.T. Metabolic modulation of Ewing sarcoma cells inhibits tumor growth and stem cell properties. Oncotarget 2017, 8, 77292–77308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, X.; Wang, G.; Xiao, B.; Ma, Z.; Huo, H.; Li, W. A seven-lncRNA signature for predicting Ewing’s sarcoma. PeerJ 2021, 9, e11599. [Google Scholar] [CrossRef]
- Ma, L.; Sun, X.; Kuai, W.; Hu, J.; Yuan, Y.; Feng, W.; Lu, X. LncRNA SOX2 overlapping transcript acts as a miRNA sponge to promote the proliferation and invasion of Ewing’s sarcoma. Am. J. Transl. Res. 2019, 11, 3841–3849. [Google Scholar]
- Li, H.; Huang, F.; Liu, X.Q.; Liu, H.C.; Dai, M.; Zeng, J. LncRNA TUG1 promotes Ewing’s sarcoma cell proliferation, migration, and invasion via the miR-199a-3p-MSI2 signaling pathway. Neoplasma 2021, 68, 590–601. [Google Scholar] [CrossRef]
- Goss, K.L.; Koppenhafer, S.L.; Harmoney, K.M.; Terry, W.W.; Gordon, D.J. Inhibition of CHK1 sensitizes Ewing sarcoma cells to the ribonucleotide reductase inhibitor gemcitabine. Oncotarget 2017, 8, 87016–87032. [Google Scholar] [CrossRef] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator across a Range of Preclinical Pediatric Tumor Models. Clin. Cancer Res. 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Henssen, A.G.; Reed, C.; Jiang, E.; Garcia, H.D.; von Stebut, J.; MacArthur, I.C.; Hundsdoerfer, P.; Kim, J.H.; de Stanchina, E.; Kuwahara, Y.; et al. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci. Transl. Med. 2017, 9, eaam9078. [Google Scholar] [CrossRef] [Green Version]
- Koppenhafer, S.L.; Goss, K.L.; Terry, W.W.; Gordon, D.J. mTORC1/2 and Protein Translation Regulate Levels of CHK1 and the Sensitivity to CHK1 Inhibitors in Ewing Sarcoma Cells. Mol. Cancer Ther. 2018, 17, 2676–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.C.; Cimprich, K.A. A new mitotic activity comes into focus. Science 2018, 359, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, F.; Tang, T.; Guo, C. The role of PARP1 in the DNA damage response and its application in tumor therapy. Front. Med. 2012, 6, 156–164. [Google Scholar] [CrossRef]
- Vormoor, B.; Curtin, N.J. Poly(ADP-ribose) polymerase inhibitors in Ewing sarcoma. Curr. Opin. Oncol. 2014, 26, 428–433. [Google Scholar] [CrossRef]
- Stewart, E.; Goshorn, R.; Bradley, C.; Griffiths, L.M.; Benavente, C.; Twarog, N.R.; Miller, G.M.; Caufield, W.; Freeman, B.B., 3rd; Bahrami, A.; et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014, 9, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Vormoor, B.; Schlosser, Y.T.; Blair, H.; Sharma, A.; Wilkinson, S.; Newell, D.R.; Curtin, N. Sensitizing Ewing sarcoma to chemo- and radiotherapy by inhibition of the DNA-repair enzymes DNA protein kinase (DNA-PK) and poly-ADP-ribose polymerase (PARP) 1/2. Oncotarget 2017, 8, 113418–113430. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Engert, F.; Schneider, C.; Weibeta, L.M.; Probst, M.; Fulda, S. PARP Inhibitors Sensitize Ewing Sarcoma Cells to Temozolomide-Induced Apoptosis via the Mitochondrial Pathway. Mol. Cancer Ther. 2015, 14, 2818–2830. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.J.; Travers, J.; Pshenichnaya, I.; Kogera, F.A.; Barthorpe, S.; Mironenko, T.; Richardson, L.; Benes, C.H.; Stratton, M.R.; McDermott, U.; et al. Combinations of PARP Inhibitors with Temozolomide Drive PARP1 Trapping and Apoptosis in Ewing’s Sarcoma. PLoS ONE 2015, 10, e0140988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Hampton, O.A.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr. Blood Cancer 2015, 62, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bademci, G.; Abad, C.; Cengiz, F.B.; Seyhan, S.; Incesulu, A.; Guo, S.; Fitoz, S.; Atli, E.I.; Gosstola, N.C.; Demir, S.; et al. Long-range cis-regulatory elements controlling GDF6 expression are essential for ear development. J. Clin. Investig. 2020, 130, 4213–4217. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.A.; Bhargav, D.; Diwan, A.D. Unveiling the bmp13 enigma: Redundant morphogen or crucial regulator? Int. J. Biol. Sci. 2008, 4, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Sonnemann, J.; Dreyer, L.; Hartwig, M.; Palani, C.D.; Hong, L.T.T.; Klier, U.; Broker, B.; Volker, U.; Beck, J.F. Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing’s sarcoma cells. J. Cancer Res. Clin. Oncol. 2007, 133, 847–858. [Google Scholar] [CrossRef]
- Lu, G.; Punj, V.; Chaudhary, P.M. Proteasome inhibitor Bortezomib induces cell cycle arrest and apoptosis in cell lines derived from Ewing’s sarcoma family of tumors and synergizes with TRAIL. Cancer Biol. Ther. 2008, 7, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Lohberger, B.; Bernhart, E.; Stuendl, N.; Glaenzer, D.; Leithner, A.; Rinner, B.; Bauer, R.; Kretschmer, N. Periplocin mediates TRAIL-induced apoptosis and cell cycle arrest in human myxofibrosarcoma cells via the ERK/p38/JNK pathway. Phytomedicine 2020, 76, 153262. [Google Scholar] [CrossRef]
- Du, J.; Wang, Y.; Chen, D.; Ji, G.; Ma, Q.; Liao, S.; Zheng, Y.; Zhang, J.; Hou, Y. BAY61-3606 potentiates the anti-tumor effects of TRAIL against colon cancer through up-regulating DR4 and down-regulating NF-kappaB. Cancer Lett. 2016, 383, 145–153. [Google Scholar] [CrossRef]
- Hanikoglu, F.; Cort, A.; Ozben, H.; Hanikoglu, A.; Ozben, T. Epoxomicin Sensitizes Resistant Osteosarcoma Cells to TRAIL Induced Apoptosis. Anticancer Agents Med. Chem. 2015, 15, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Brown, R.E.; Buryanek, J.; Trent, J.; Ashkenazi, A.; Herbst, R.; Kurzrock, R. Targeting the apoptotic pathway in chondrosarcoma using recombinant human Apo2L/TRAIL (dulanermin), a dual proapoptotic receptor (DR4/DR5) agonist. Mol. Cancer Ther. 2012, 11, 2541–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, R.; Attard, G.; Pacey, S.; Li, L.; Razak, A.; Perrett, R.; Barrett, M.; Judson, I.; Kaye, S.; Fox, N.L.; et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin. Cancer Res. 2007, 13, 6187–6194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, M.; Wahba, A.; Felix, K.; Cabrera, M.; Segura, M.G.; Kundra, V.; Ravoori, M.K.; Stewart, J.; Kleinerman, E.S.; Jensen, V.B.; et al. Bempegaldesleukin (BEMPEG; NKTR-214) efficacy as a single agent and in combination with checkpoint-inhibitor therapy in mouse models of osteosarcoma. Int. J. Cancer 2021, 148, 1928–1937. [Google Scholar] [CrossRef]
- Gamie, Z.; Kapriniotis, K.; Papanikolaou, D.; Haagensen, E.; Da Conceicao Ribeiro, R.; Dalgarno, K.; Krippner-Heidenreich, A.; Gerrand, C.; Tsiridis, E.; Rankin, K.S. TNF-related apoptosis-inducing ligand (TRAIL) for bone sarcoma treatment: Pre-clinical and clinical data. Cancer Lett. 2017, 409, 66–80. [Google Scholar] [CrossRef]
- Henrich, I.C.; Young, R.; Quick, L.; Oliveira, A.M.; Chou, M.M. USP6 Confers Sensitivity to IFN-Mediated Apoptosis through Modulation of TRAIL Signaling in Ewing Sarcoma. Mol. Cancer Res. 2018, 16, 1834–1843. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.J.; Dai, W.; Haldar, S.; Ma, H.; Anderson, V.M.; Overacker, R.D.; Risinger, A.L.; Loesgen, S.; Houghton, P.J.; Cichewicz, R.H.; et al. Altertoxin II, a Highly Effective and Specific Compound against Ewing Sarcoma. Cancers 2021, 13, 6176. [Google Scholar] [CrossRef]
- Kerschner-Morales, S.L.; Kuhne, M.; Becker, S.; Beck, J.F.; Sonnemann, J. Anticancer effects of the PLK4 inhibitors CFI-400945 and centrinone in Ewing’s sarcoma cells. J. Cancer Res. Clin. Oncol. 2020, 146, 2871–2883. [Google Scholar] [CrossRef]
- Ma, Y.; Baltezor, M.; Rajewski, L.; Crow, J.; Samuel, G.; Staggs, V.S.; Chastain, K.M.; Toretsky, J.A.; Weir, S.J.; Godwin, A.K. Targeted inhibition of histone deacetylase leads to suppression of Ewing sarcoma tumor growth through an unappreciated EWS-FLI1/HDAC3/HSP90 signaling axis. J. Mol. Med. (Berl.) 2019, 97, 957–972. [Google Scholar] [CrossRef]
- Flores, G.; Everett, J.H.; Boguslawski, E.A.; Oswald, B.M.; Madaj, Z.B.; Beddows, I.; Dikalov, S.; Adams, M.; Klumpp-Thomas, C.A.; Kitchen-Goosen, S.M.; et al. CDK9 Blockade Exploits Context-dependent Transcriptional Changes to Improve Activity and Limit Toxicity of Mithramycin for Ewing Sarcoma. Mol. Cancer Ther. 2020, 19, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Hwang, E.E.; Guha, R.; O’Neill, A.F.; Melong, N.; Veinotte, C.J.; Conway Saur, A.; Wuerthele, K.; Shen, M.; McKnight, C.; et al. High-throughput Chemical Screening Identifies Focal Adhesion Kinase and Aurora Kinase B Inhibition as a Synergistic Treatment Combination in Ewing Sarcoma. Clin. Cancer Res. 2019, 25, 4552–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Targetable Molecules | Main Pathways | Tumor Effects |
|---|---|---|
| CD99 | IGF-1R and RAS-Rac1 signaling | Induces caspase-independent cell death, endocytosis, cell aggregation, micropinocytosis, cell adhesion, migration, invasion, metastasis, differentiation |
| GDF6 | GDF6 prodomain signaling pathway | Cell proliferation, tumor growth, differentiation, apoptosis |
| E-cadherin | MAPK Pathway | Anchorage-independent growth and spheroid formation, cell-cell adhesion |
| Endoglin | TGFβ signaling | Tumor cell plasticity, patient survival, invasion, anchorage-independent growth, progression of aggressive tumors |
| EZH2 | Epigenetic | Cell differentiation, phenotypic heterogeneity, self-renewal |
| GLI1 | Sonic Hedgehog (SHH) pathway | Cell proliferation, cell cycle control, apoptosis, cell viability, metastasis, invasion, migration, clonogenicity |
| PDGF family members | PDGF pathway | Self-renewal, invasion, chemotherapy resistance, primary tumor growth, metastasis, drug resistance, poor clinical outcome |
| ROCK2 | RhoA-ROCK pathway | Migration, invasion, proliferation, clonogenic capacity, tumor growth |
| YAP proteins | YAP/TAZ pathway, Hippo signaling, WNT/β-catenin signaling | Migration, cell proliferation, metastasis, anchorage-independent colony formation |
| PAPP-A | IGF signaling | Cell proliferation, migration, cell survival, tumor growth, invasion, metastasis |
| PARP family | DNA repair, replication | Apoptosis |
| TRAIL | TRAIL-pathway | Induces caspase-independent cell death, apoptosis |
| ATR/CHK1 | ATR-CHK1 pathway | Cell cycle regulation, cell cycle arrest |
| LDH | aerobic glycolysis | Cell proliferation, apoptosis, tumor growth, cell survival |
| PHGDH | Serine synthesis | Cell proliferation |
| lncRNA SOX2 | WNT/β-catenin signaling | Cell proliferation, invasion, apoptosis, tumor growth |
| lncRNA TUG1 | TUG-miR-145-5p-TRPC6 pathway | Cell proliferation, migration, invasion |
| S.N. | Number | Number of Patients | Disease | Drug/Target | Results |
|---|---|---|---|---|---|
| 1 | NCT04129151 | 18 | Ewing Sarcoma Recurrent | Palbociclib/CDK4 and CDK6 Ganitumab/IGF-1R | Active |
| 2 | NCT02546544 | 16 | Relapsed Ewing Sarcoma Refractory Ewing Sarcoma | Linsitinib/IGF-1R | Disease progression, limited therapeutical effect |
| 3 | NCT00949325 | 24 | Soft Tissue and Bone Sarcoma | Temsirolimus/mTOR Doxorubicin/topoisomerase II | The response rate was 53%, found a correlation between inhibition of mTOR and therapeutical effect 10.1186/s13569-018-0107-9 |
| 4 | NCT00987636 | 907 | Ewing sarcoma | Zoledronic acid/osteoclast apoptosis Busulfan/guanine N7 Treosulfan/guanine N7 Melphalan/guanine N7 | BuMel treatment was more successful than standard chemotherapy -vincristine, dactinomycin, and ifosfamide (VAI) |
| 5 | NCT00618813 | 35 | Ewing Sarcoma | Radiation therapy therapeutic conventional surgery etoposide/topoisomerase II ifosfamide/DNA doxorubicin hydrochloride/topoisomerase II cyclophosphamide/guanine N7 vincristine sulfate/tubulin topotecan hydrochloride/topoisomerase I filgrastim/Granulocyte | No incidence of death was recorded in 37 weeks of treatment |
| 6 | NCT00516295 | 7 | Ewing Sarcoma of Bone Extraosseous Ewing Sarcoma Peripheral Primitive Neuroectodermal Tumor Recurrent Ewing Sarcoma/Peripheral Primitive Neuroectodermal Tumor | Topotecan hydrochloride/topoisomerase I cyclophosphamide/guanine N7 vincristine sulfate/tubulin bevacizumab/VEGF-A | Days of event free survival—442 |
| 7 | NCT00470275 | 10 | Recurrent or Refractory Ewing Sarcoma | Cytarabine/DNA | Lack of efficacy |
| 8 | NCT02657005 | 45 | Relapsed or Refractory Ewing Sarcoma | TK216/EWS-FLI1 | Active |
| 9 | NCT00061893 | 38 | Ewing Sarcoma Family of Tumors | Radiation therapy conventional surgery etoposide/topoisomerase II ifosfamide/DNA doxorubicin hydrochloride/topoisomerase II cyclophosphamide/guanine N7 vincristine sulfate/tubulin topotecan hydrochloride/topoisomerase I filgrastim/granulocyte vinblastine sulfate/tubulin MESNA/urotoxic metabolites | 24-month event free survival was 35%: 71% for the seven with isolated pulmonary metastases, 26% for all others. |
| 10 | NCT02511132 | 22 | Ewing Sarcoma | Vigil/TGF-β Temozolomide/guanine Irinotecan/topoisomerase I | 1 case report of complete response to therapy |
| 11 | NCT01583543 | 12 | Recurrent/Metastatic Ewing’s Sarcoma | Olaparib/PARP | No significant responses or durable disease control was seen |
| 12 | NCT01331135 | 18 | Ewing sarcoma, osteosarcoma, malignant peripheral nerve sheath tumor, rhabdoid tumor, retinoblastoma | Sirolimus/mTOR | The combination of sirolimus with metronomic chemotherapy is well tolerated in children. A phase II trial of this combination is ongoing. |
| 13 | NCT00428272 | 24 | Ewing Sarcoma Osteosarcoma Neuroblastoma Rhabdomyosarcoma | Lexatumumab/TRAIL-2R | The drug seems to mediate some clinical activity in pediatric solid tumors and may work with radiation to enhance antitumor effects. |
| 14 | NCT02306161 | 312 | Metastatic Ewing Sarcoma Metastatic Malignant Neoplasm in the Bone Metastatic Malignant Neoplasm in the Bone Marrow Metastatic Malignant Neoplasm in the Lung Metastatic Peripheral Primitive Neuroectodermal Tumor of Bone Peripheral Primitive Neuroectodermal Tumor of Soft Tissues | Cyclophosphamide/guanine N7 Doxorubicin/topoisomerase II Etoposide/topoisomerase II Ganitumab/IGevent-freeF-1R Ifosfamide/DNA Vincristine/tubulin | Active |
| 15 | NCT04067115 | 45 | Ewing Sarcoma | Trabectedin/guanine N2 Irinotecan/topoisomerase I | Recruiting |
| 16 | NCT00070109 | 50 | Rhabdomyosarcoma Recurrent Childhood Rhabdomyosarcoma Recurrent Childhood Soft Tissue Sarcoma Recurrent Ewing Sarcoma Peripheral Primitive Neuroectodermal Tumor | Trabectedin/guanine N2 | |
| 17 | NCT03600649 | 50 | Ewing Sarcoma Myxoid Liposarcoma Sarcoma, Soft Tissue Desmoplastic Small Round Cell Tumor Extraskeletal Myxoid Chondrosarcoma Angiomatoid Fibrous Histiocytoma Clear Cell Sarcoma Primary Pulmonary Myxoid Sarcoma Myoepithelial Tumor Sclerosing Epithelioid Fibrosarcoma Fibromyxoid Tumor | Cyclophosphamide/guanine N7 Topotecan/topoisomerase I Seclidemstat/LSD1 | Recruiting |
| 18 | NCT03491371 | 56 | Osteosarcoma Ewing sarcoma Chondrosarcoma Soft tissue sarcoma | Methylsulfonic apatinib/VEGFR-2 | No data |
| 19 | NCT04690725 | 29 | Osteosarcoma Ewing sarcoma Chondrosarcoma | TQB3525/PI 3-kinases | Active |
| 20 | NCT01610570 | 8 | Ewing Sarcoma Sarcoma | Mithramycin/EWS-FLI1 | The trial was closed to enrollment, due to inability to safely achieve the desired mithramycin exposure |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fayzullina, D.; Tsibulnikov, S.; Stempen, M.; Schroeder, B.A.; Kumar, N.; Kharwar, R.K.; Acharya, A.; Timashev, P.; Ulasov, I. Novel Targeted Therapeutic Strategies for Ewing Sarcoma. Cancers 2022, 14, 1988. https://doi.org/10.3390/cancers14081988
Fayzullina D, Tsibulnikov S, Stempen M, Schroeder BA, Kumar N, Kharwar RK, Acharya A, Timashev P, Ulasov I. Novel Targeted Therapeutic Strategies for Ewing Sarcoma. Cancers. 2022; 14(8):1988. https://doi.org/10.3390/cancers14081988
Chicago/Turabian StyleFayzullina, Daria, Sergey Tsibulnikov, Mikhail Stempen, Brett A. Schroeder, Naveen Kumar, Rajesh Kumar Kharwar, Arbind Acharya, Peter Timashev, and Ilya Ulasov. 2022. "Novel Targeted Therapeutic Strategies for Ewing Sarcoma" Cancers 14, no. 8: 1988. https://doi.org/10.3390/cancers14081988