
Integrative Clinical and DNA Methylation Analyses in a Population-Based Cohort Identifies CDH17 and LRP2 as Risk Recurrence Factors in Stage II Colon Cancer

 , , , ,
, , , ,  ,
,  ,
,
Abstract
Simple Summary
Abstract

1. Introduction
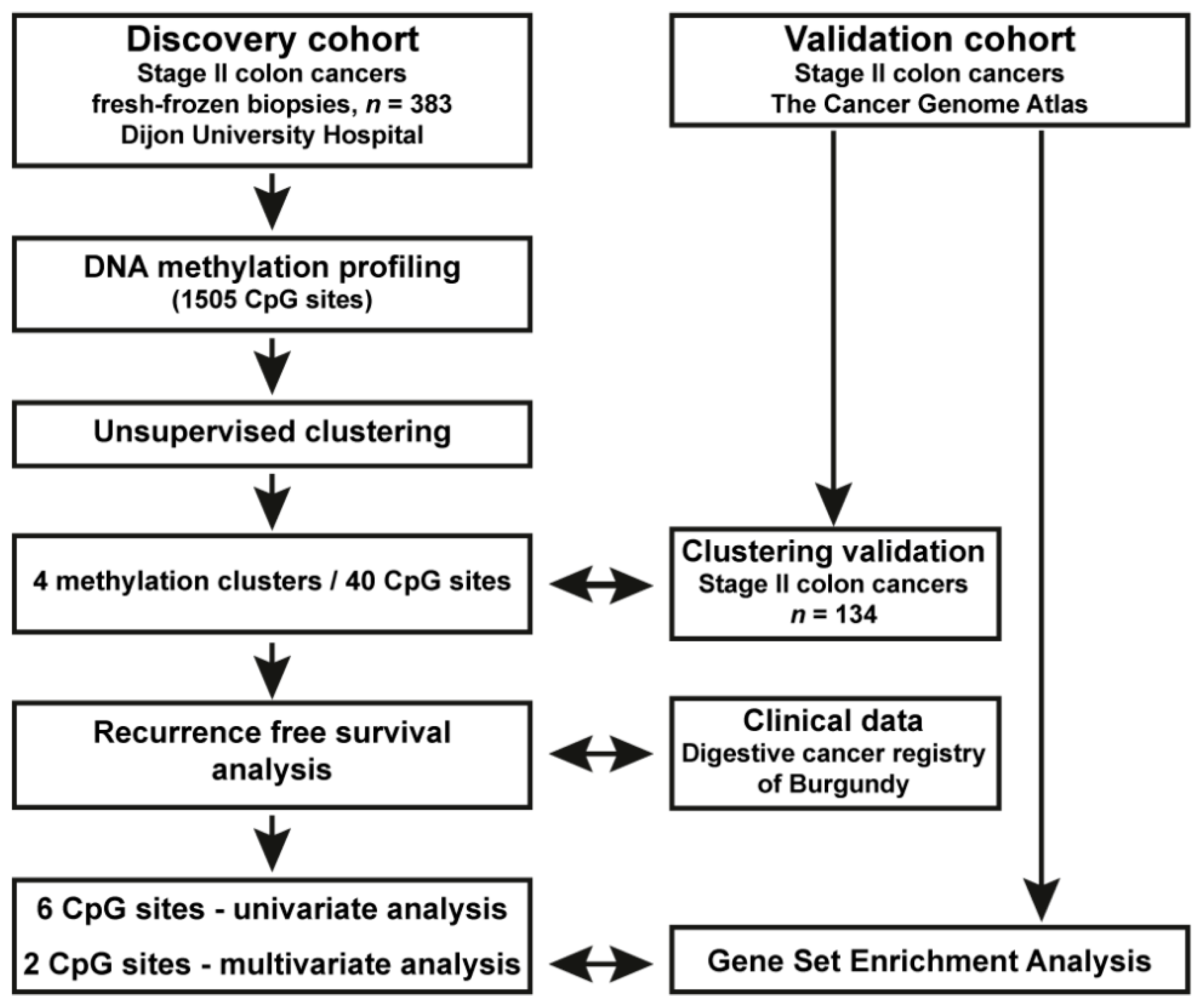
2. Materials and Methods
2.1. Patients, Clinical Data and Tumour Samples
2.2. Characterisation of the CpG Island Methylator Phenotype (CIMP), MSI, Chromosomal Instabilities, Gene Mutations and LINE-1 DNA Methylation Status
2.3. DNA Methylation and Clustering Analyses in the Population-Based Stage II CC Patient Cohort
2.4. Reanalysis of DNA Methylation and RNAseq Data from Stage II CC from TCGA; Validation Cohort
2.5. Statistical Analysis of Clinical Data
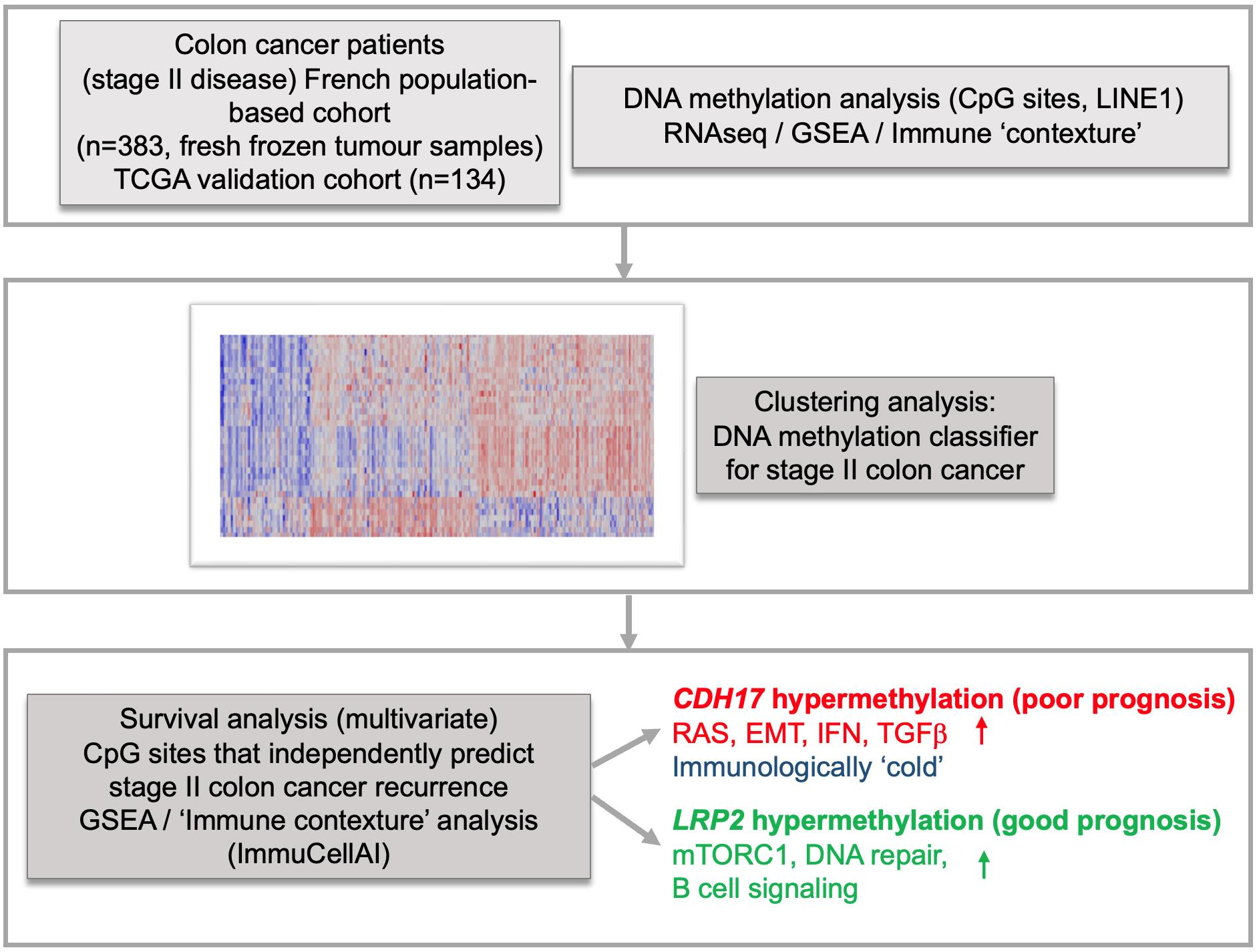
3. Results
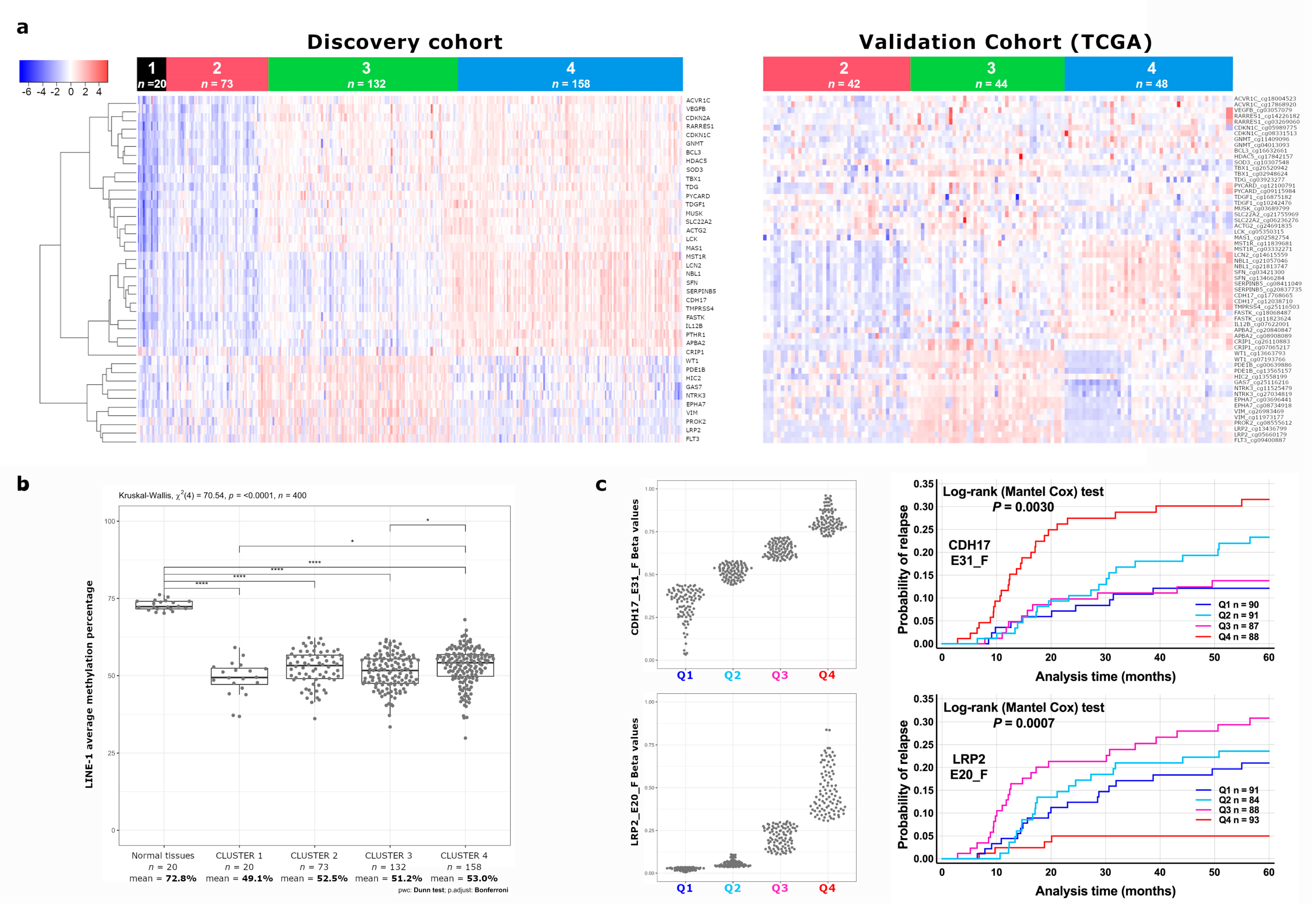
3.1. Identification of a Novel DNA Methylation Classifier in Stage II Colon Cancer
3.2. Identification of CDH17 and LRP2 DNA Methylation Status as Independent Predictive Markers of Disease Recurrence in Stage II CC
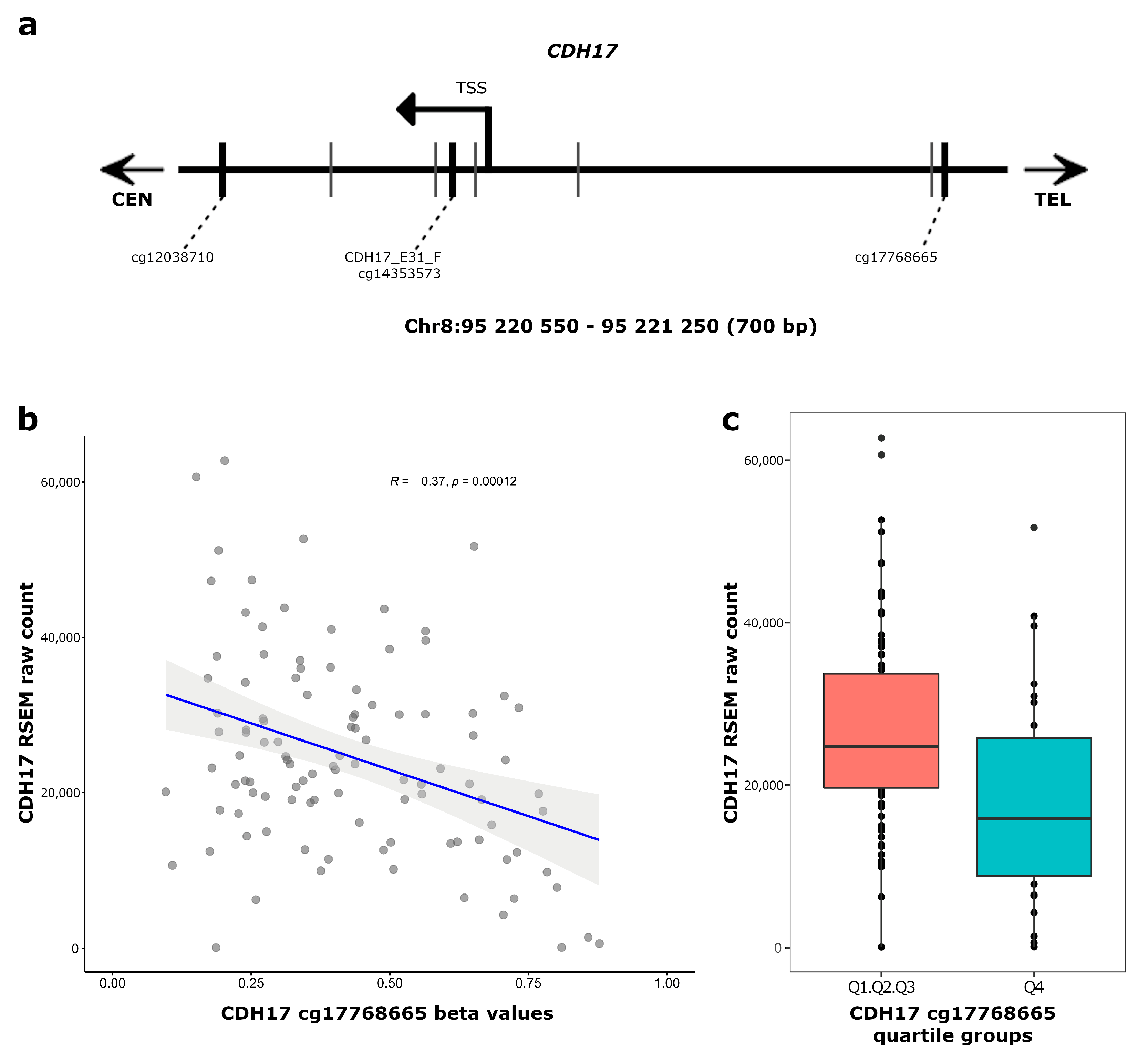
3.3. CDH17 and LRP2 Expression According to DNA Methylation Status in Stage II Colon Cancer
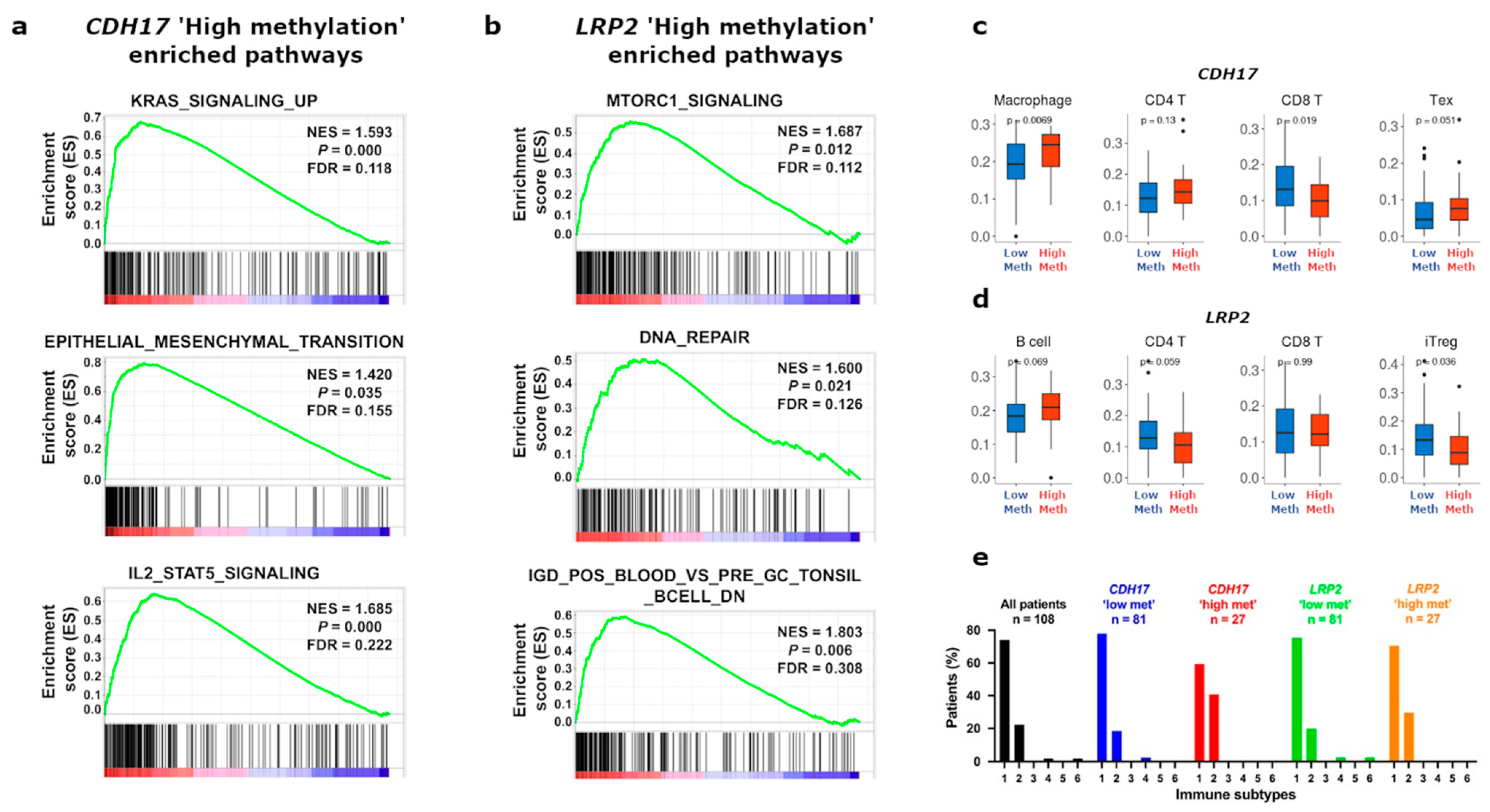
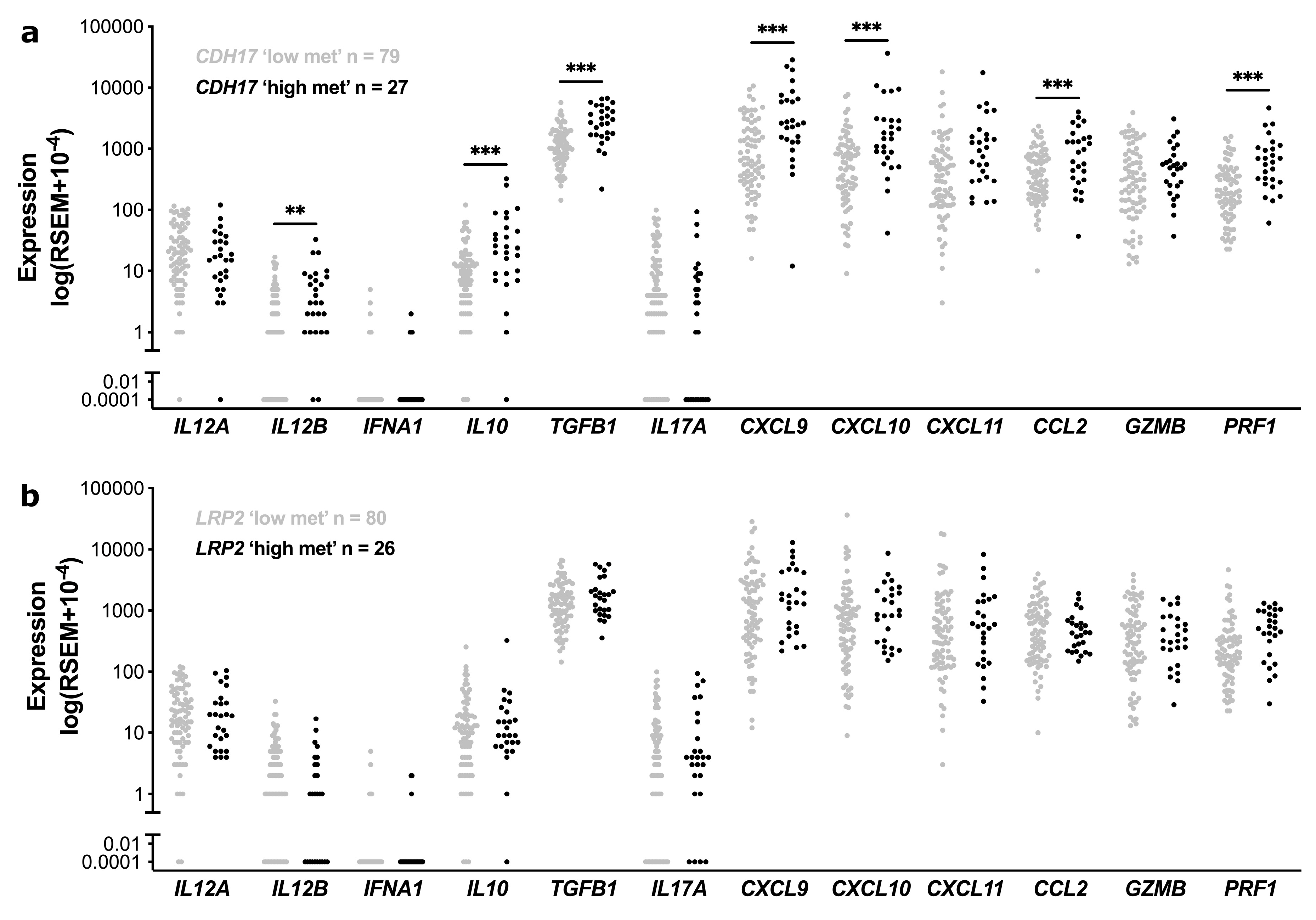
3.4. Functional Enrichment and Immune Cell Subset Analyses in TCGA Stage II CC According to CDH17 and LRP2 Methylation Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.; Bouvier, A.M.; Binquet, C.; Dancourt, V.; Coatmeur, O.; Faivre, J. Are the Recommendations of the French Consensus Conference on the Management of Colon Cancer Followed Up? Eur. J. Cancer Prev. 2006, 15, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Manfredi, S.; Bouvier, A.M.; Lepage, C.; Hatem, C.; Dancourt, V.; Faivre, J. Incidence and Patterns of Recurrence after Resection for Cure of Colonic Cancer in a Well Defined Population. Br. J. Surg. 2006, 93, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Rollot, F.; Chauvenet, M.; Roche, L.; Hamza, S.; Lepage, C.; Faivre, J.; Bouvier, A.M. Long-Term Net Survival in Patients with Colorectal Cancer in France: An Informative Contribution of Recent Methodology. Dis. Colon Rectum 2013, 56, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Quasar Collaborative Group; Gray, R.; Barnwell, J.; McConkey, C.; Hills, R.K.; Williams, N.S.; Kerr, D.J. Adjuvant Chemotherapy Versus Observation in Patients with Colorectal Cancer: A Randomised Study. Lancet 2007, 370, 2020–2029. [Google Scholar] [PubMed]
- Andre, T.; de Gramont, A.; Vernerey, D.; Chibaudel, B.; Bonnetain, F.; Tijeras-Raballand, A.; Scriva, A.; Hickish, T.; Tabernero, J.; van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage Ii to Iii Colon Cancer: Updated 10-Year Survival and Outcomes According to Braf Mutation and Mismatch Repair Status of the Mosaic Study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef]
- Argiles, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised Colon Cancer: Esmo Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From Tumour Heterogeneity to Advances in Precision Treatment of Colorectal Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef]
- Gallois, C.; Taieb, J.; le Corre, D.; le Malicot, K.; Tabernero, J.; Mulot, C.; Seitz, J.F.; Aparicio, T.; Folprecht, G.; Lepage, C.; et al. Prognostic Value of Methylator Phenotype in Stage III Colon Cancer Treated with Oxaliplatin-Based Adjuvant Chemotherapy. Clin. Cancer Res. 2018, 24, 4745–4753. [Google Scholar] [CrossRef]
- Cohen, S.A.; Wu, C.; Yu, M.; Gourgioti, G.; Wirtz, R.; Raptou, G.; Gkakou, C.; Kotoula, V.; Pentheroudakis, G.; Papaxoinis, G.; et al. Evaluation of Cpg Island Methylator Phenotype as a Biomarker in Colorectal Cancer Treated with Adjuvant Oxaliplatin. Clin. Color. Cancer 2016, 15, 164–169. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.; Chapusot, C.; Courcet, E.; Martin, L.; Lepage, C.; Faivre, J.; Piard, F. Why Do Results Conflict Regarding the Prognostic Value of the Methylation Status in Colon Cancers? The Role of the Preservation Method. BMC Cancer 2012, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Sobin, L.H.; Fleming, I.D. Tnm Classification of Malignant Tumors, Fifth Edition (1997). Union Internationale Contre Le Cancer and the American Joint Committee on Cancer. Cancer 1997, 80, 1803–1804. [Google Scholar] [CrossRef]
- Killian, A.; di Fiore, F.; le Pessot, F.; Blanchard, F.; Lamy, A.; Raux, G.; Flaman, J.M.; Paillot, B.; Michel, P.; Sabourin, J.C.; et al. A Simple Method for the Routine Detection of Somatic Quantitative Genetic Alterations in Colorectal Cancer. Gastroenterology 2007, 132, 645–653. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. Factominer: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Miao, Y.R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.Y.; Wang, H.; Guo, A.Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and Its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Jung, G.; Hernandez-Illan, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of Colorectal Cancer: Biomarker and Therapeutic Potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (Msigdb) Hallmark Gene Set Collection. Cell. Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Casal, I.J.; Bartolome, R.A. Beyond N-Cadherin, Relevance of Cadherins 5, 6 and 17 in Cancer Progression and Metastasis. Int. J. Mol. Sci. 2019, 20, 3373. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lei, F.; Tang, Y.; Pan, J.S.; Tong, Q.; Sun, Y.; Sheikh-Hamad, D. Megalin Mediates Plasma Membrane to Mitochondria Cross-Talk and Regulates Mitochondrial Metabolism. Cell. Mol. Life Sci. 2018, 75, 4021–4040. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Christa, A.; Kur, E.; Lioubinski, O.; Bachmann, S.; Willnow, T.E.; Hammes, A. Lrp2 Is an Auxiliary Shh Receptor Required to Condition the Forebrain Ventral Midline for Inductive Signals. Dev. Cell 2012, 22, 268–278. [Google Scholar] [CrossRef]
- Yi, T.; Zhang, Y.; Ng, D.M.; Xi, Y.; Ye, M.; Cen, L.; Li, J.; Fan, X.; Li, Y.; Hu, S.; et al. Regulatory Network Analysis of Mutated Genes Based on Multi-Omics Data Reveals the Exclusive Features in Tumor Immune Microenvironment between Left-Sided and Right-Sided Colon Cancer. Front. Oncol. 2021, 11, 685515. [Google Scholar] [CrossRef]
- Patel, K.; Bhat, F.A.; Patil, S.; Routray, S.; Mohanty, N.; Nair, B.; Sidransky, D.; Ganesh, M.S.; Ray, J.G.; Gowda, H.; et al. Whole-Exome Sequencing Analysis of Oral Squamous Cell Carcinoma Delineated by Tobacco Usage Habits. Front. Oncol. 2021, 11, 660696. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, H.; Zhong, X.; Liu, X.; Cai, Z.; Chen, Y.; He, X.; Lan, P.; Wu, X. Ldl Receptor Related Protein 2 to Promote Colorectal Cancer Metastasis Via Enhancing Gsk3β/Β-Catenin Signaling. J. Clin. Oncol. 2021, 39, e15507. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Yu, L.C.; Yu, I.S.; Jhuang, Y.L.; Huang, W.J.; Yang, C.Y.; Jeng, Y.M. Deletion of Cadherin-17 Enhances Intestinal Permeability and Susceptibility to Intestinal Tumour Formation. J. Pathol. 2018, 246, 289–299. [Google Scholar] [CrossRef]
- Garcia-Martinez, J.M.; Wang, S.; Weishaeupl, C.; Wernitznig, A.; Chetta, P.; Pinto, C.; Ho, J.; Dutcher, D.; Gorman, P.N.; Kroe-Barrett, R.; et al. Selective Tumor Cell Apoptosis and Tumor Regression in Cdh17-Positive Colorectal Cancer Models Using Bi 905711, a Novel Liver-Sparing Trailr2 Agonist. Mol. Cancer Ther. 2021, 20, 96–108. [Google Scholar] [CrossRef]
- Slowicka, K.; Petta, I.; Blancke, G.; Hoste, E.; Dumas, E.; Sze, M.; Vikkula, H.; Radaelli, E.; Haigh, J.J.; Jonckheere, S.; et al. Zeb2 Drives Invasive and Microbiota-Dependent Colon Carcinoma. Nat. Cancer 2020, 1, 620–634. [Google Scholar] [CrossRef]
- Wang, K.; Song, K.; Ma, Z.; Yao, Y.; Liu, C.; Yang, J.; Xiao, H.; Zhang, J.; Zhang, Y.; Zhao, W. Identification of Emt-Related High-Risk Stage Ii Colorectal Cancer and Characterisation of Metastasis-Related Genes. Br. J. Cancer 2020, 123, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Nuttin, L.; Thibaudin, M.; Jacquin, E.; Aucagne, R.; Bon, M.; Revy, S.; Barnestein, R.; Ballot, E.; Truntzer, C.; et al. Mek Inhibition Overcomes Chemoimmunotherapy Resistance by Inducing Cxcl10 in Cancer Cells. Cancer Cell 2022, 40, 136–152.e12. [Google Scholar] [CrossRef] [PubMed]
- Vanhersecke, L.; Brunet, M.; Guegan, J.P.; Rey, C.; Bougouin, A.; Cousin, S.; Moulec, S.L.; Besse, B.; Loriot, Y.; Larroquette, M.; et al. Mature Tertiary Lymphoid Structures Predict Immune Checkpoint Inhibitor Efficacy in Solid Tumors Independently of Pd-L1 Expression. Nat. Cancer 2021, 2, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Melcher, C.; Yu, J.; Duong, V.H.H.; Westphal, K.; Farimany, N.H.S.; Shaverskyi, A.; Zhao, B.; Strowig, T.; Glage, S.; Brand, K.; et al. B Cell-Mediated Regulatory Mechanisms Control Tumor-Promoting Intestinal Inflammation. Cell. Rep. 2022, 40, 111051. [Google Scholar] [CrossRef]
- Lepage, C.; Phelip, J.M.; Cany, L.; Faroux, R.; Manfredi, S.; Ain, J.F.; Pezet, D.; Baconnier, M.; Deguiral, P.; Terrebone, E.; et al. Effect of 5 Years of Imaging and Cea Follow-up to Detect Recurrence of Colorectal Cancer: The Ffcd Prodige 13 Randomised Phase Iii Trial. Dig. Liver Dis. 2015, 47, 529–531. [Google Scholar] [CrossRef]
- Taieb, J.; Benhaim, L.; Puig, P.L.; le Malicot, K.; Emile, J.F.; Geillon, F.; Tougeron, D.; Manfredi, S.; Chauvenet, M.; Taly, V.; et al. Decision for Adjuvant Treatment in Stage Ii Colon Cancer Based on Circulating Tumor DNA:The Circulate-Prodige 70 Trial. Dig. Liver Dis. 2020, 52, 730–733. [Google Scholar] [CrossRef]
- Dumetier, B.; Sauter, C.; Hajmirza, A.; Pernon, B.; Aucagne, R.; Fournier, C.; Row, C.; Guidez, F.; Rossi, C.; Lepage, C.; et al. Repeat Element Activation-Driven Inflammation: Role of NFκB and Implications in Normal Development and Cancer? Biomedicines 2022, 10, 3101. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. | B. | ||||||
|---|---|---|---|---|---|---|---|
| N = 383 | N = 383 | ||||||
| n | % | n | % | ||||
| Sex | MSI status | ||||||
| Women | 169 | 44.1 | MSI | 85 | 22.2 | ||
| Men | 214 | 55.9 | MSS | 298 | 77.8 | ||
| Age class | KRAS codon 12-13 mutational status | ||||||
| ≤64 years old | 87 | 22.7 | Mutated | 116 | 30.3 | ||
| 65–74 years old | 105 | 27.4 | Wild-type | 266 | 69.5 | ||
| ≥75 years old | 191 | 49.9 | Unavailable | 1 | |||
| Tumour site | BRAF codon 600 mutational status | ||||||
| right colon | 172 | 44.9 | Mutated | 54 | 14.1 | ||
| left colon | 141 | 36.8 | Wild-type | 329 | 85.9 | ||
| recto-sigmoïd junction | 65 | 17.0 | PIK3CA codon 542 and 1047 mutational status | ||||
| Unavailable | 5 | Mutated | 70 | 18.3 | |||
| Wild-type | 312 | 81.5 | |||||
| Unavailable | 1 | ||||||
| Staging | CIMP status | ||||||
| T3 | 97 | 25.3 | CIMP-High | 66 | 17.2 | ||
| T4 | 260 | 67.9 | CIMP-Low | 105 | 27.4 | ||
| locoregional extension | 26 | 6.8 | No CIMP | 144 | 37.6 | ||
| Unavailable | 68 | 17.8 | |||||
| Chemotherapy treatment | ERBB2 CNV status | ||||||
| yes | 46 | 12.0 | Amplification | 42 | 11.0 | ||
| no | 337 | 88.0 | No amplification | 274 | 71.5 | ||
| Unavailable | 67 | 17.5 | |||||
| Five-year followup reccurence | TP53 CNV status | ||||||
| no | 315 | 81.8 | Deletion | 80 | 20.9 | ||
| yes | 68 | 17.7 | No Deletion | 236 | 61.6 | ||
| Unavailable | 67 | 17.5 | |||||
| Cluster 2 | Cluster 3 | Cluster 4 | ||||||
| (N = 73) | (N = 132) | (N = 158) | Khi2 Test | |||||
| N | n | % | n | % | n | % | p | |
| Sex | ||||||||
| Men | 214 | 36 | 49.3 | 62 | 47.0 | 105 | 66.5 | 0.002 |
| Women | 169 | 37 | 50.7 | 70 | 53.0 | 53 | 33.5 | |
| Tumour site | ||||||||
| right colon | 172 | 32 | 45.7 | 78 | 59.5 | 52 | 33.1 | <0.001 |
| left colon | 141 | 24 | 34.3 | 40 | 30.5 | 70 | 44.6 | |
| recto-sigmoïd junction | 65 | 14 | 20.0 | 13 | 9.9 | 35 | 22.3 | |
| KRAS | ||||||||
| Mutation | 116 | 30 | 41.1 | 45 | 34.4 | 38 | 24.1 | 0.021 |
| Wild -type | 266 | 43 | 58.9 | 86 | 65.6 | 120 | 75.9 | |
| MSI phenotype | ||||||||
| MSI | 85 | 19 | 26.0 | 47 | 35.6 | 14 | 8.9 | <0.001 |
| MSS | 298 | 54 | 74.0 | 85 | 64.4 | 144 | 91.1 | |
| CIMP phenotype | ||||||||
| CIMP-High | 66 | 11 | 19.3 | 42 | 39.3 | 9 | 6.9 | <0.001 |
| CIMP-Low | 105 | 24 | 42.1 | 36 | 33.6 | 39 | 29.8 | |
| No-CIMP | 144 | 22 | 38.6 | 29 | 27.1 | 83 | 63.4 | |
| BRAF | ||||||||
| Mutation | 54 | 12 | 16.4 | 32 | 24.2 | 8 | 5.1 | <0.001 |
| Wild -type | 329 | 61 | 83.6 | 100 | 75.8 | 150 | 94.9 | |
| MSI/BRAF status | ||||||||
| MSI/BRAF Mutation | 46 | 11 | 15.1 | 28 | 21.2 | 5 | 3.2 | <0.001 |
| Others | 317 | 62 | 84.9 | 104 | 78.8 | 153 | 96.8 | |
| MSI/CIMP/BRAF status | ||||||||
| MSI/CIMP-High/BRAF Mutation | 38 | 9 | 12.3 | 24 | 18.2 | 3 | 1.9 | <0.001 |
| Others | 325 | 64 | 87.7 | 108 | 81.8 | 155 | 98.1 | |
| ERBB2 copy number | ||||||||
| Amplification | 42 | 9 | 15.5 | 12 | 11.3 | 19 | 14.3 | 0.703 |
| Deletion/Neutral | 274 | 49 | 84.5 | 94 | 88.7 | 114 | 85.7 | |
| TP53 copy number | ||||||||
| Deletion | 80 | 9 | 15.5 | 33 | 31.1 | 31 | 23.3 | 0.077 |
| Amplification/Neutral | 236 | 49 | 84.5 | 73 | 68.9 | 102 | 76.7 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tournier, B.; Aucagne, R.; Truntzer, C.; Fournier, C.; Ghiringhelli, F.; Chapusot, C.; Martin, L.; Bouvier, A.M.; Manfredi, S.; Jooste, V.; et al. Integrative Clinical and DNA Methylation Analyses in a Population-Based Cohort Identifies CDH17 and LRP2 as Risk Recurrence Factors in Stage II Colon Cancer. Cancers 2023, 15, 158. https://doi.org/10.3390/cancers15010158
Tournier B, Aucagne R, Truntzer C, Fournier C, Ghiringhelli F, Chapusot C, Martin L, Bouvier AM, Manfredi S, Jooste V, et al. Integrative Clinical and DNA Methylation Analyses in a Population-Based Cohort Identifies CDH17 and LRP2 as Risk Recurrence Factors in Stage II Colon Cancer. Cancers. 2023; 15(1):158. https://doi.org/10.3390/cancers15010158
Chicago/Turabian StyleTournier, Benjamin, Romain Aucagne, Caroline Truntzer, Cyril Fournier, François Ghiringhelli, Caroline Chapusot, Laurent Martin, Anne Marie Bouvier, Sylvain Manfredi, Valérie Jooste, and et al. 2023. "Integrative Clinical and DNA Methylation Analyses in a Population-Based Cohort Identifies CDH17 and LRP2 as Risk Recurrence Factors in Stage II Colon Cancer" Cancers 15, no. 1: 158. https://doi.org/10.3390/cancers15010158
APA StyleTournier, B., Aucagne, R., Truntzer, C., Fournier, C., Ghiringhelli, F., Chapusot, C., Martin, L., Bouvier, A. M., Manfredi, S., Jooste, V., Callanan, M. B., & Lepage, C. (2023). Integrative Clinical and DNA Methylation Analyses in a Population-Based Cohort Identifies CDH17 and LRP2 as Risk Recurrence Factors in Stage II Colon Cancer. Cancers, 15(1), 158. https://doi.org/10.3390/cancers15010158