
Radiotherapeutic Strategies to Overcome Resistance of Breast Cancer Brain Metastases by Considering Immunogenic Aspects of Cancer Stem Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. From the Primary Tumor Site to the Brain—The Metastatic Cascade
1.2. Brain Metastasis Incidence Depending on Breast Cancer Subtype
1.3. Genomic Landscape of BCBM
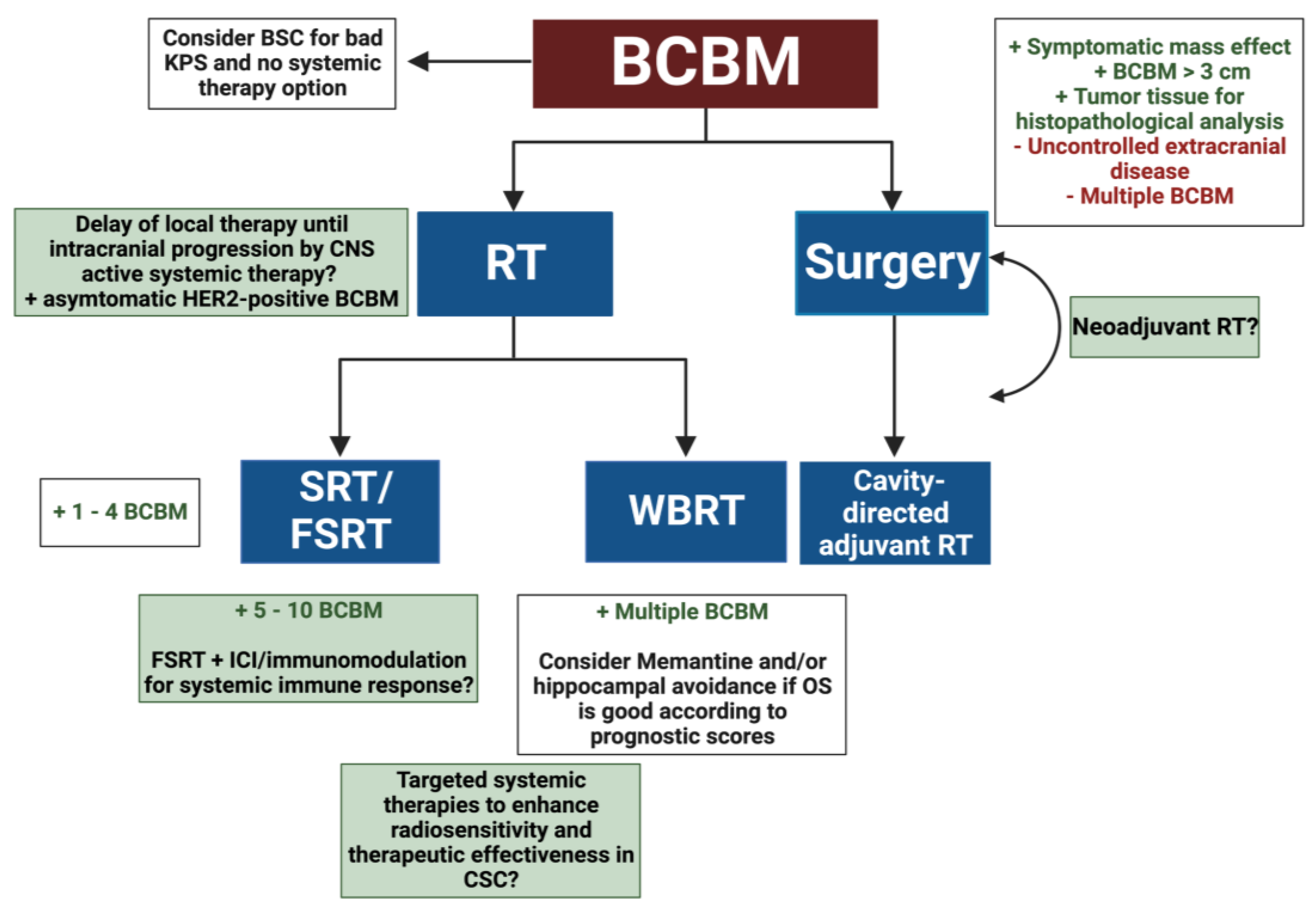
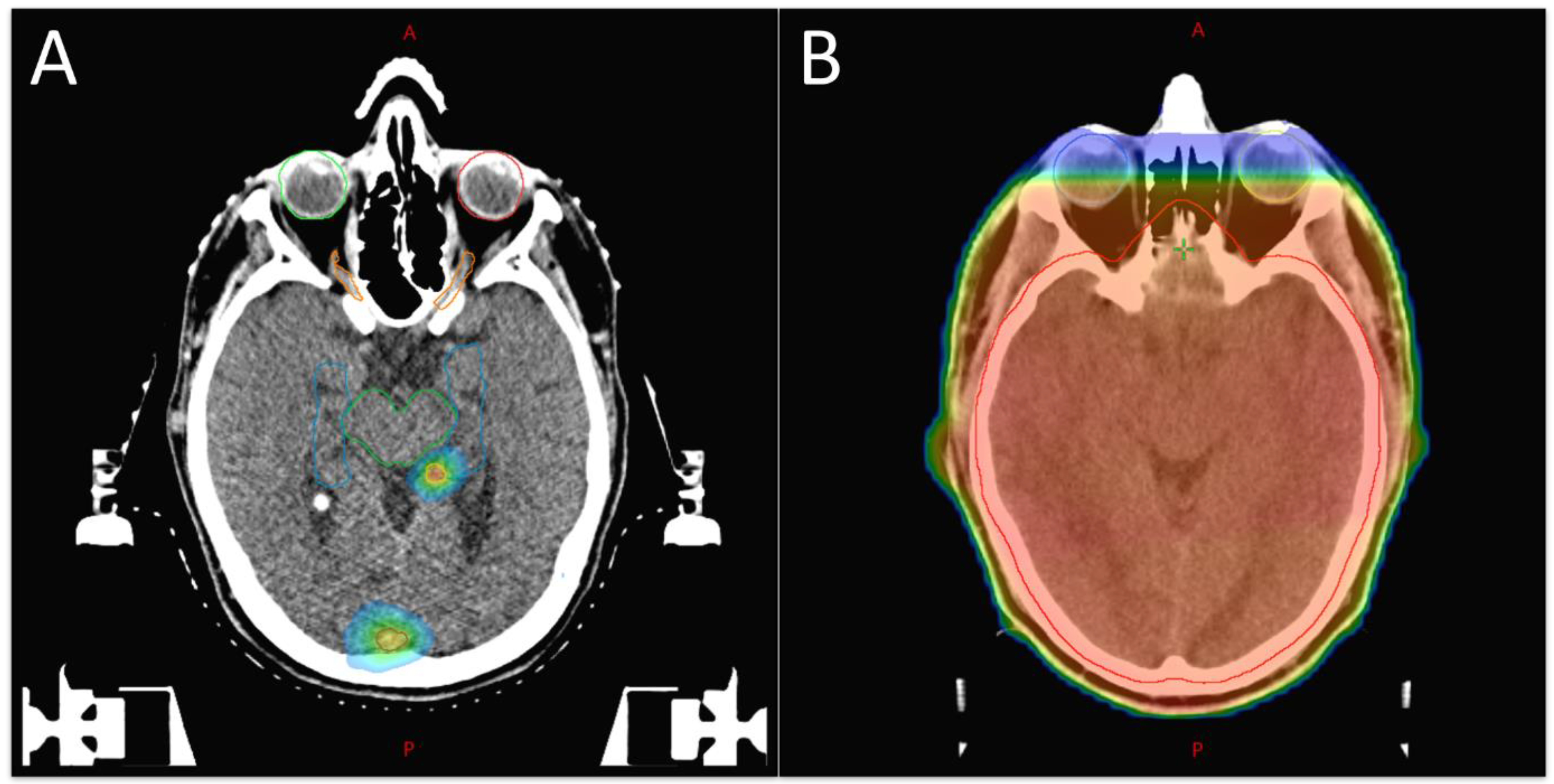
2. Radiotherapeutic Treatment Strategies for BCBM
2.1. Radioresistance in BCBM
2.2. Systemic Therapies of BCBM and RT
3. Mechanisms of Radioresistance and Immune Evasion in CSCs
3.1. Cellular Processes Leading to Radioresistance in CSC
3.2. DNA Repair Mechanisms Contributing to Radioresistance in CSC
3.3. Immune Evasion of CSCs in BCBM
4. New Strategies to Target Radiation Resistance in CSCs
4.1. Targeting DNA Repair in BCBM
4.2. Targeting DNA Repair and Immune Response in BCBM
4.3. Targeting Immune Checkpoints in BCBM
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33, Erratum in CA Cancer J Clin. 2021 71 359. [Google Scholar] [CrossRef]
- Weil, R.J.; Palmieri, D.C.; Bronder, J.L.; Stark, A.M.; Steeg, P.S. Breast Cancer Metastasis to the Central Nervous System. Am. J. Pathol. 2005, 167, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, L.G.; Dawood, S.; Sopik, V.; Haaland, B.; Tan, P.S.; Bhoo-Pathy, N.; Warner, E.; Iqbal, J.; Narod, S.A.; Dent, R. Ten-year survival in women with primary stage IV breast cancer. Breast Cancer Res. Treat. 2016, 160, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Wu, J.; Ding, S.; Goh, C.; Andriani, L.; Lu, S.; Shen, K.; Zhu, L. Subdivision of M1 Stage for De Novo Metastatic Breast Cancer to Better Predict Prognosis and Response to Primary Tumor Surgery. J. Natl. Compr. Cancer Netw. 2019, 17, 1521–1528. [Google Scholar] [CrossRef] [Green Version]
- Paget, S. Distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Campbell, B.K.; Gao, Z.; Corcoran, N.M.; Stylli, S.S.; Hovens, C.M. Molecular Mechanisms Driving the Formation of Brain Metastases. Cancers 2022, 14, 4963. [Google Scholar] [CrossRef]
- Lee, K.-L.; Chen, G.; Chen, T.-Y.; Kuo, Y.-C.; Su, Y.-K. Effects of Cancer Stem Cells in Triple-Negative Breast Cancer and Brain Metastasis: Challenges and Solutions. Cancers 2020, 12, 2122. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Eslami, S.Z.; Cortés-Hernández, L.E.; Thomas, F.; Pantel, K.; Alix-Panabières, C. Functional analysis of circulating tumour cells: The KEY to understand the biology of the metastatic cascade. Br. J. Cancer 2022, 127, 800–810. [Google Scholar] [CrossRef]
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Bryan, S.; Witzel, I.; Borgmann, K.; Oliveira-Ferrer, L. Molecular Mechanisms Associated with Brain Metastases in HER2-Positive and Triple Negative Breast Cancers. Cancers 2021, 13, 4137. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, J.M.; Martin, D.R.; Byrne, M.E. Recent Advances in Delivery through the Blood-Brain Barrier. Curr. Top. Med. Chem. 2014, 14, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef]
- Mittapalli, R.K.; Manda, V.K.; Adkins, C.E.; Geldenhuys, W.J.; Lockman, P.R. Exploiting nutrient transporters at the blood–brain barrier to improve brain distribution of small molecules. Ther. Deliv. 2010, 1, 775–784. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Cai, B.; Zeng, M.; Hao, Y.; Giancotti, F.G.; Fu, B.M. Integrin β4 Signaling Promotes Mammary Tumor Cell Adhesion to Brain Microvascular Endothelium by Inducing ErbB2-Mediated Secretion of VEGF. Ann. Biomed. Eng. 2011, 39, 2223–2241. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef]
- Carvalho, R.; Paredes, J.; Ribeiro, A.S. Impact of breast cancer cells’ secretome on the brain metastatic niche remodeling. Semin. Cancer Biol. 2020, 60, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.F.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Fukuda, K.; Xing, F.; Zhang, Y.; Sharma, S.; Liu, Y.; Chan, M.D.; Zhou, X.; Qasem, S.A.; Pochampally, R.; et al. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J. Biol. Chem. 2015, 290, 9842–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louie, E.; Chen, X.F.; Coomes, A.; Ji, K.; Tsirka, S.; Chen, E.I. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 2013, 32, 4064–4077. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, E.; Ulaner, G.A.; Chin, S.-F. Molecular Classification of Breast Cancer. PET Clin. 2018, 13, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Na Yoon, Y.; Choi, H.S.; Kim, J.-H.; Seol, H.; Lee, J.K.; Seong, M.-K.; Park, I.C.; Kim, K.I.; Kim, H.-A.; et al. Breast cancer stem cells in HER2-negative breast cancer cells contribute to HER2-mediated radioresistance and molecular subtype conversion: Clinical implications for serum HER2 in recurrent HER2-negative breast cancer. Oncotarget 2017, 9, 5811–5822. [Google Scholar] [CrossRef] [Green Version]
- Kuksis, M.; Gao, Y.; Tran, W.; Hoey, C.; Kiss, A.; Komorowski, A.S.; Dhaliwal, A.J.; Sahgal, A.; Das, S.; Chan, K.K.; et al. The incidence of brain metastases among patients with metastatic breast cancer: A systematic review and meta-analysis. Neuro Oncol. 2021, 23, 894–904. [Google Scholar] [CrossRef]
- Koniali, L.; Hadjisavvas, A.; Constantinidou, A.; Christodoulou, K.; Christou, Y.; Demetriou, C.; Panayides, A.S.; Pitris, C.; Pattichis, C.S.; Zamba-Papanicolaou, E.; et al. Risk factors for breast cancer brain metastases: A systematic review. Oncotarget 2020, 11, 650–669. [Google Scholar] [CrossRef] [Green Version]
- Sperduto, P.W.; Berkey, B.; Gaspar, L.E.; Mehta, M.; Curran, W. A New Prognostic Index and Comparison to Three Other Indices for Patients with Brain Metastases: An Analysis of 1960 Patients in the RTOG Database. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 510–514. [Google Scholar] [CrossRef]
- Sperduto, P.W.; Mesko, S.; Li, J.; Cagney, D.; Aizer, A.; Lin, N.U.; Nesbit, E.; Kruser, T.J.; Chan, J.; Braunstein, S.; et al. Beyond an Updated Graded Prognostic Assessment (Breast GPA): A Prognostic Index and Trends in Treatment and Survival in Breast Cancer Brain Metastases from 1985 to Today. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.; Jang, B.-S.; Jeon, S.H.; Kim, J.H.; Kim, Y.J.; Kim, S.H.; Kim, C.-Y.; Han, J.H.; Kim, I.A. Analysis of survival outcomes based on molecular subtypes in breast cancer brain metastases: A single institutional cohort. Breast J. 2018, 24, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Liu, X.; Liao, X.; He, J.; Niu, L. The Clinicopathological features and survival outcomes of patients with different metastatic sites in stage IV breast cancer. BMC Cancer 2019, 19, 1091. [Google Scholar] [CrossRef] [Green Version]
- Ording, A.G.; Heide-Jørgensen, U.; Christiansen, C.F.; Nørgaard, M.; Acquavella, J.; Sørensen, H.T. Site of metastasis and breast cancer mortality: A Danish nationwide registry-based cohort study. Clin. Exp. Metastasis 2017, 34, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, W.; Suijkerbuijk, K.P.M.; van Gils, C.H.; van der Wall, E.; Moelans, C.B.; van Diest, P.J. Receptor Conversion in Distant Breast Cancer Metastases: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2018, 110, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Hulsbergen, A.F.C.; Claes, A.; Kavouridis, V.K.; Ansaripour, A.; Nogarede, C.; Hughes, M.E.; Smith, T.R.; Brastianos, P.K.; Verhoeff, J.J.C.; Lin, N.U.; et al. Subtype switching in breast cancer brain metastases: A multicenter analysis. Neuro Oncol. 2020, 22, 1173–1181. [Google Scholar] [CrossRef]
- Han, C.H.; Brastianos, P.K. Genetic Characterization of Brain Metastases in the Era of Targeted Therapy. Front. Oncol. 2017, 7, 230. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [Green Version]
- Chagpar, A.; Magliocco, A.; Kerviche, A.; Tan, L.; Walley, B.; DeCoteau, J.F. The replication error phenotype is associated with the development of distant metastases in hormonally treated patients with breast carcinoma. Cancer 2004, 100, 913–919. [Google Scholar] [CrossRef]
- Woditschka, S.; Evans, L.; Duchnowska, R.; Reed, L.T.; Palmieri, D.; Qian, Y.; Badve, S.; Sledge, G., Jr.; Gril, B.; Aladjem, M.I.; et al. DNA Double-Strand Break Repair Genes and Oxidative Damage in Brain Metastasis of Breast Cancer. J. Natl. Cancer Inst. 2014, 106, dju145. [Google Scholar] [CrossRef]
- McMullin, R.P.; Wittner, B.S.; Yang, C.; Denton-Schneider, B.R.; Hicks, D.; Singavarapu, R.; Moulis, S.; Lee, J.; Akbari, M.R.; Narod, S.A.; et al. A BRCA1deficient-like signature is enriched in breast cancer brain metastases and predicts DNA damage-induced poly (ADP-ribose) polymerase inhibitor sensitivity. Breast Cancer Res. 2014, 16, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.D.; Zheng, S.; Xiu, J.; Zhou, S.; Khasraw, M.; Brastianos, P.K.; Kesari, S.; Hu, J.; Rudnick, J.; Salacz, M.E.; et al. Profiles of brain metastases: Prioritization of therapeutic targets. Int. J. Cancer 2018, 143, 3019–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyran, M.; Carbuccia, N.; Garnier, S.; Guille, A.; Adelaïde, J.; Finetti, P.; Touzlian, J.; Viens, P.; Tallet, A.; Goncalves, A.; et al. A Comparison of DNA Mutation and Copy Number Profiles of Primary Breast Cancers and Paired Brain Metastases for Identifying Clinically Relevant Genetic Alterations in Brain Metastases. Cancers 2019, 11, 665. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, N.; Varešlija, D.; Keelan, S.; Elangovan, A.; Atkinson, J.M.; Cocchiglia, S.; Bane, F.T.; Singh, V.; Furney, S.; Hu, C.; et al. Mapping molecular subtype specific alterations in breast cancer brain metastases identifies clinically relevant vulnerabilities. Nat. Commun. 2022, 13, 514. [Google Scholar] [CrossRef]
- Kalkanis, S.N.; Kondziolka, D.; Gaspar, L.E.; Burri, S.H.; Asher, A.L.; Cobbs, C.S.; Ammirati, M.; Robinson, P.D.; Andrews, D.W.; Loeffler, J.S.; et al. The role of surgical resection in the management of newly diagnosed brain metastases: A systematic review and evidence-based clinical practice guideline. J. Neurooncol. 2010, 96, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Vecht, C.J.; Haaxma-Reiche, H.; Noordijk, E.M.; Padberg, G.W.; Voormolen, J.H.; Hoekstra, F.H.; Tans, J.T.; Lambooij, N.; Metsaars, J.A.; Wattendorff, A.R.; et al. Treatment of single brain metastasis: Radiotherapy alone or combined with neurosurgery? Ann. Neurol. 1993, 33, 583–590. [Google Scholar] [CrossRef]
- Patchell, R.A.; Tibbs, P.A.; Regine, W.F.; Dempsey, R.J.; Mohiuddin, M.; Kryscio, R.J.; Markesbery, W.R.; Foon, K.A.; Young, B. Postoperative Radiotherapy in the Treatment of Single Metastases to the Brain: A randomized trial. JAMA 1998, 280, 1485–1489. [Google Scholar] [CrossRef]
- Patchell, R.A.; Tibbs, P.A.; Walsh, J.W.; Dempsey, R.J.; Maruyama, Y.; Kryscio, R.J.; Markesbery, W.R.; Macdonald, J.S.; Young, B. A Randomized Trial of Surgery in the Treatment of Single Metastases to the Brain. N. Engl. J. Med. 1990, 322, 494–500. [Google Scholar] [CrossRef]
- Noordijk, E.M.; Vecht, C.J.; Haaxma-Reiche, H.; Padberg, G.W.; Voormolen, J.H.; Hoekstra, F.H.; Tans, J.T.; Lambooij, N.; Metsaars, J.A.; Wattendorff, A.R.; et al. The choice of treatment of single brain metastasis should be based on extracranial tumor activity and age. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 711–717. [Google Scholar] [CrossRef]
- Nahed, B.V.; Alvarez-Breckenridge, C.; Brastianos, P.K.; Shih, H.; Sloan, A.; Ammirati, M.; Kuo, J.S.; Ryken, T.C.; Kalkanis, S.N.; Olson, J.J. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guidelines on the Role of Surgery in the Management of Adults With Metastatic Brain Tumors. Neurosurgery 2019, 84, E152–E155. [Google Scholar] [CrossRef]
- Chao, J.H.; Phillips, R.; Nickson, J.J. Roentgen-ray therapy of cerebral metastases. Cancer 1954, 7, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.N.; Xu, W.; Wong, R.K.; Lloyd, N.; Laperriere, N.; Sahgal, A.; Rakovitch, E.; Chow, E. Whole brain radiotherapy for the treatment of newly diagnosed multiple brain metastases. Cochrane Database Syst. Rev. 2018, 1, CD003869. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Ahluwalia, M.S.; Khan, O.H.; Asher, A.L.; Wefel, J.S.; Gondi, V. Whole-Brain Radiotherapy for Brain Metastases: Evolution or Revolution? J. Clin. Oncol. 2018, 36, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Harth, S.; Abo-Madyan, Y.; Zheng, L.; Siebenlist, K.; Herskind, C.; Wenz, F.; Giordano, F.A. Estimation of intracranial failure risk following hippocampal-sparing whole brain radiotherapy. Radiother. Oncol. 2013, 109, 152–158. [Google Scholar] [CrossRef]
- Khuntia, D.; Brown, P.; Li, J.; Mehta, M.P. Whole-Brain Radiotherapy in the Management of Brain Metastasis. J. Clin. Oncol. 2006, 24, 1295–1304. [Google Scholar] [CrossRef]
- Brown, P.D.; Gondi, V.; Pugh, S.; Tome, W.A.; Wefel, J.S.; Armstrong, T.S.; Bovi, J.A.; Robinson, C.; Konski, A.; Khuntia, D.; et al. Hippocampal Avoidance during Whole-Brain Radiotherapy Plus Memantine for Patients with Brain Metastases: Phase III Trial NRG Oncology CC001. J. Clin. Oncol. 2020, 38, 1019–1029. [Google Scholar] [CrossRef]
- Aoyama, H.; Shirato, H.; Tago, M.; Nakagawa, K.; Toyoda, T.; Hatano, K.; Kenjyo, M.; Oya, N.; Hirota, S.; Shioura, H.; et al. Stereotactic radiosurgery plus whole-brain radiation therapy vs stereotactic radiosurgery alone for treatment of brain metastases: A randomized controlled trial. JAMA 2006, 295, 2483–2491. [Google Scholar] [CrossRef]
- Chang, E.L.; Wefel, J.S.; Hess, K.R.; Allen, P.K.; Lang, F.F.; Kornguth, D.G.; Arbuckle, R.B.; Swint, J.M.; Shiu, A.S.; Maor, M.H.; et al. Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: A randomised controlled trial. Lancet Oncol. 2009, 10, 1037–1044. [Google Scholar] [CrossRef]
- Kocher, M.; Soffietti, R.; Abacioglu, U.; Villà, S.; Fauchon, F.; Baumert, B.G.; Fariselli, L.; Tzuk-Shina, T.; Kortmann, R.-D.; Carrie, C.; et al. Adjuvant Whole-Brain Radiotherapy Versus Observation after Radiosurgery or Surgical Resection of One to Three Cerebral Metastases: Results of the EORTC 22952-26001 Study. J. Clin. Oncol. 2011, 29, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Trifiletti, D.M.; Ruiz-Garcia, H.; Quinones-Hinojosa, A.; Ramakrishna, R.; Sheehan, J.P. The evolution of stereotactic radiosurgery in neurosurgical practice. J. Neurooncol. 2021, 151, 451–459. [Google Scholar] [CrossRef]
- Remick, J.S.; Kowalski, E.; Khairnar, R.; Sun, K.; Morse, E.; Cherng, H.R.; Poirier, Y.; Lamichhane, N.; Becker, S.J.; Chen, S.; et al. A multi-center analysis of single-fraction versus hypofractionated stereotactic radiosurgery for the treatment of brain metastasis. Radiat. Oncol. 2020, 15, 128. [Google Scholar] [CrossRef] [PubMed]
- Loo, M.; Clavier, J.-B.; Khalifa, J.A.; Moyal, E.; Khalifa, J. Dose-Response Effect and Dose-Toxicity in Stereotactic Radiotherapy for Brain Metastases: A Review. Cancers 2021, 13, 6086. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.A.; Cardinale, R.M.; Benedict, S.H.; Kavanagh, B.D.; Zwicker, R.D.; Amir, C.; Broaddus, W.C. Hypofractionated stereotactic radiotherapy as an alternative to radiosurgery for the treatment of patients with brain metastases. Int. J. Radiat. Oncol. Biol. Phys. 2000, 47, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kawabe, T.; Sato, Y.; Higuchi, Y.; Nariai, T.; Watanabe, S.; Kasuya, H. Stereotactic radiosurgery for patients with multiple brain metastases: A case-matched study comparing treatment results for patients with 2–9 versus 10 or more tumors. J. Neurosurg. 2014, 121, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, A.; Ahmed, S.; McAleer, M.F.; Weinberg, J.S.; Li, J.; Brown, P.; Settle, S.; Prabhu, S.S.; Lang, F.F.; Levine, N.; et al. Post-operative stereotactic radiosurgery versus observation for completely resected brain metastases: A single-centre, randomised, controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1040–1048. [Google Scholar] [CrossRef]
- Kayama, T.; Sato, S.; Sakurada, K.; Mizusawa, J.; Nishikawa, R.; Narita, Y.; Sumi, M.; Miyakita, Y.; Kumabe, T.; Sonoda, Y.; et al. Effects of Surgery with Salvage Stereotactic Radiosurgery Versus Surgery with Whole-Brain Radiation Therapy in Patients with One to Four Brain Metastases (JCOG0504): A Phase III, Noninferiority, Randomized Controlled Trial. J. Clin. Oncol. 2018, 36, 3282–3289. [Google Scholar] [CrossRef]
- Cho, E.; Rubinstein, L.; Stevenson, P.; Gooley, T.; Philips, M.; Halasz, L.M.; Gensheimer, M.F.; Linden, H.M.; Rockhill, J.K.; Gadi, V.K. The use of stereotactic radiosurgery for brain metastases from breast cancer: Who benefits most? Breast Cancer Res. Treat. 2015, 149, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Vogelbaum, M.A.; Brown, P.D.; Messersmith, H.; Brastianos, P.K.; Burri, S.; Cahill, D.; Dunn, I.F.; Gaspar, L.E.; Gatson, N.T.N.; Gondi, V.; et al. Treatment for Brain Metastases: ASCO-SNO-ASTRO Guideline. J. Clin. Oncol. 2022, 40, 492–516. [Google Scholar] [CrossRef]
- Donker, M.; van Tienhoven, G.; Straver, M.E.; Meijnen, P.; van de Velde, C.J.H.; Mansel, R.E.; Cataliotti, L.; Westenberg, A.H.; Klinkenbijl, J.H.G.; Orzalesi, L.; et al. Radiotherapy or surgery of the axilla after a positive sentinel node in breast cancer (EORTC 10981-22023 AMAROS): A randomised, multicentre, open-label, phase 3 non-inferiority trial. Lancet Oncol. 2014, 15, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Fisher, B.; Bryant, J.; Dignam, J.J.; Wickerham, D.L.; Mamounas, E.P.; Fisher, E.R.; Margolese, R.G.; Nesbitt, L.; Paik, S.; Pisansky, T.M.; et al. Tamoxifen, Radiation Therapy, or Both for Prevention of Ipsilateral Breast Tumor Recurrence after Lumpectomy in Women with Invasive Breast Cancers of One Centimeter or Less. J. Clin. Oncol. 2002, 20, 4141–4149. [Google Scholar] [CrossRef]
- Louis-Sylvestre, C.; Clough, K.; Asselain, B.; Vilcoq, J.R.; Salmon, R.J.; Campana, F.; Fourquet, A. Axillary Treatment in Conservative Management of Operable Breast Cancer: Dissection or Radiotherapy? Results of a Randomized Study with 15 Years of Follow-Up. J. Clin. Oncol. 2004, 22, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Yaromina, A.; Krause, M.; Baumann, M. Individualization of cancer treatment from radiotherapy perspective. Mol. Oncol. 2012, 6, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschrich, S.; Zhang, H.; Zhao, H.; Boulware, D.; Lee, J.-H.; Bloom, G.; Torres-Roca, J.F. Systems Biology Modeling of the Radiation Sensitivity Network: A Biomarker Discovery Platform. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Cai, S.; Bai, L.; Du, Z.; Li, H.; Sun, P.; Cao, J.; Yi, N.; Liu, S.B.; Tang, Z. Integration of immune and hypoxia gene signatures improves the prediction of radiosensitivity in breast cancer. Am. J. Cancer Res. 2022, 12, 1222–1240. [Google Scholar] [PubMed]
- Monteiro, C.; Miarka, L.; Perea-García, M.; Priego, N.; García-Gómez, P.; Álvaro-Espinosa, L.; de Pablos-Aragoneses, A.; Yebra, N.; Retana, D.; Baena, P.; et al. Stratification of radiosensitive brain metastases based on an actionable S100A9/RAGE resistance mechanism. Nat. Med. 2022, 28, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, A.; Lahijanian, Z.; Panet-Raymond, V.; Siegel, P.M.; Petrecca, K.; Maleki, F.; Dankner, M. Radiomics as an emerging tool in the management of brain metastases. Neurooncol. Adv. 2022, 4, vdac141. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Cortés, J.; O’Shaughnessy, J. Challenges in the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer with brain metastases. Cancer Metastasis Rev. 2016, 35, 323–332. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Sharma, S.; Wu, K.; Tyagi, A.; Zhao, D.; Deshpande, R.P.; Watabe, K. Tamoxifen suppresses brain metastasis of estrogen receptor-deficient breast cancer by skewing microglia polarization and enhancing their immune functions. Breast Cancer Res. 2021, 23, 35. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, Y.; Yue, Y.; Pu, S.; Wu, J.; Lv, Y.; Du, D. Tamoxifen attenuates reactive astrocyte-induced brain metastasis and drug resistance through the IL-6/STAT3 signaling pathway. Acta Biochim. Biophys. Sin. 2020, 52, 1299–1305. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Peiris-Pages, M.; Ozsvari, B.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Targeting cancer stem cell propagation with palbociclib, a CDK4/6 inhibitor: Telomerase drives tumor cell heterogeneity. Oncotarget 2017, 8, 9868–9884. [Google Scholar] [CrossRef]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, S.A.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashizume, R.; Zhang, A.; Mueller, S.; Prados, M.D.; Lulla, R.R.; Goldman, S.; Saratsis, A.M.; Mazar, A.P.; Stegh, A.H.; Cheng, S.-Y.; et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol. 2016, 18, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figura, N.B.; Potluri, T.K.; Mohammadi, H.; Oliver, D.E.; Arrington, J.A.; Robinson, T.J.; Etame, A.B.; Tran, N.D.; Liu, J.K.; Soliman, H.; et al. CDK 4/6 inhibitors and stereotactic radiation in the management of hormone receptor positive breast cancer brain metastases. J. Neurooncol. 2019, 144, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Kim, H.-A.; Seong, M.-K.; Seol, H.; Oh, J.S.; Kim, E.-K.; Chang, J.W.; Hwang, S.-G.; Noh, W.C. STAT3-survivin signaling mediates a poor response to radiotherapy in HER2-positive breast cancers. Oncotarget 2016, 7, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Duru, N.; Fan, M.; Candas, D.; Menaa, C.; Liu, H.-C.; Nantajit, D.; Wen, Y.; Xiao, K.; Eldridge, A.; Chromy, B.A.; et al. HER2-Associated Radioresistance of Breast Cancer Stem Cells Isolated from HER2-Negative Breast Cancer Cells. Clin. Cancer Res. 2012, 18, 6634–6647. [Google Scholar] [CrossRef] [Green Version]
- Liang, K.; Ang, K.; Milas, L.; Hunter, N.; Fan, Z. The epidermal growth factor receptor mediates radioresistance. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 246–254. [Google Scholar] [CrossRef]
- Cao, N.; Li, S.; Wang, Z.; Ahmed, K.M.; Degnan, M.E.; Fan, M.; Dynlacht, J.R.; Li, J.J. NF-kappaB-mediated HER2 overexpression in radiation-adaptive resistance. Radiat. Res. 2009, 171, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Pupa, S.M.; Ligorio, F.; Cancila, V.; Franceschini, A.; Tripodo, C.; Vernieri, C.; Castagnoli, L. HER2 Signaling and Breast Cancer Stem Cells: The Bridge behind HER2-Positive Breast Cancer Aggressiveness and Therapy Refractoriness. Cancers 2021, 13, 4778. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Lopez-Bonet, E.; Cuyàs, E.; Viñas, G.; Pernas, S.; Dorca, J.; Menendez, J.A. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): Towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget 2015, 6, 32317–32338. [Google Scholar] [CrossRef]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 Drives Luminal Breast Cancer Stem Cells in the Absence of HER2 Amplification: Implications for Efficacy of Adjuvant Trastuzumab. Cancer Res. 2013, 73, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Zhou, Z.; Chen, X.; Zhao, R.; Yang, Z.; Wei, N.; Ni, Q.; Feng, Y.; Yu, X.; Ma, J.; et al. HER2 reduces breast cancer radiosensitivity by activating focal adhesion kinase in vitro and in vivo. Oncotarget 2016, 7, 45186–45198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Cho, B.J.; Choi, E.J.; Park, J.M.; Kim, D.H.; Kim, I.A. Radiosensitizing effect of lapatinib in human epidermal growth factor receptor 2-positive breast cancer cells. Oncotarget 2016, 7, 79089–79100. [Google Scholar] [CrossRef] [Green Version]
- Mignot, F.; Ajgal, Z.; Xu, H.; Geraud, A.; Chen, J.Y.; Mégnin-Chanet, F.; Kirova, Y. Concurrent administration of anti-HER2 therapy and radiotherapy: Systematic review. Radiother. Oncol. 2017, 124, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Meattini, I.; Livi, L.; Lorito, N.; Becherini, C.; Bacci, M.; Visani, L.; Fozza, A.; Belgioia, L.; Loi, M.; Mangoni, M.; et al. Integrating radiation therapy with targeted treatments for breast cancer: From bench to bedside. Cancer Treat. Rev. 2022, 108, 102417. [Google Scholar] [CrossRef] [PubMed]
- Pivot, X.; Manikhas, A.; Żurawski, B.; Chmielowska, E.; Karaszewska, B.; Allerton, R.; Chan, S.; Fabi, A.; Bidoli, P.; Gori, S.; et al. CEREBEL (EGF111438): A Phase III, Randomized, Open-Label Study of Lapatinib Plus Capecitabine Versus Trastuzumab Plus Capecitabine in Patients with Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2015, 33, 1564–1573. [Google Scholar] [CrossRef]
- Lin, N.U.; Carey, L.A.; Liu, M.C.; Younger, J.; Come, S.E.; Ewend, M.; Harris, G.J.; Bullitt, E.; Van den Abbeele, A.D.; Henson, J.W.; et al. Phase II Trial of Lapatinib for Brain Metastases in Patients with Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. J. Clin. Oncol. 2008, 26, 1993–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.U.; Diéras, V.; Paul, D.; Lossignol, D.; Christodoulou, C.; Stemmler, H.-J.; Roché, H.; Liu, M.C.; Greil, R.; Ciruelos, E.; et al. Multicenter Phase II Study of Lapatinib in Patients with Brain Metastases from HER2-Positive Breast Cancer. Clin. Cancer Res. 2009, 15, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Miller, J.A.; Kotecha, R.; Chao, S.T.; Ahluwalia, M.S.; Peereboom, D.M.; Mohammadi, A.M.; Barnett, G.H.; Murphy, E.S.; Vogelbaum, M.A.; et al. Stereotactic radiosurgery with concurrent HER2-directed therapy is associated with improved objective response for breast cancer brain metastasis. Neuro Oncol. 2019, 21, 659–668. [Google Scholar] [CrossRef]
- Miller, J.A.; Kotecha, R.; Ahluwalia, M.S.; Mohammadi, A.M.; Chao, S.T.; Barnett, G.H.; Murphy, E.S.; Vogelbaum, M.A.; Angelov, L.; Peereboom, D.M.; et al. Overall survival and the response to radiotherapy among molecular subtypes of breast cancer brain metastases trreated with tageted therapies. Cancer 2017, 123, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Parsai, S.; Miller, J.A.; Juloori, A.; Chao, S.T.; Kotecha, R.; Mohammadi, A.M.; Ahluwalia, M.S.; Murphy, E.S.; Barnett, G.H.; Vogelbaum, M.A.; et al. Stereotactic radiosurgery with concurrent lapatinib is associated with improved local control for HER2-positive breast cancer brain metastases. J. Neurosurg. 2019, 132, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Yomo, S.; Hayashi, M.; Cho, N. Impacts of HER2-overexpression and molecular targeting therapy on the efficacy of stereotactic radiosurgery for brain metastases from breast cancer. J. Neurooncol. 2013, 112, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Zhao, Z.; Arooj, S.; Zheng, T.; Liao, G. Lapatinib Plus Local Radiation Therapy for Brain Metastases from HER-2 Positive Breast Cancer Patients and Role of Trastuzumab: A Systematic Review and Meta-Analysis. Front. Oncol. 2020, 10, 576926. [Google Scholar] [CrossRef] [PubMed]
- Le Rhun, E.; Dhermain, F.; Vogin, G.; Reyns, N.; Metellus, P. Radionecrosis after stereotactic radiotherapy for brain metastases. Expert Rev. Neurother. 2016, 16, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Said, B.I.; Chen, H.; Jerzak, K.J.; Warner, E.; Myrehaug, S.; Tseng, C.-L.; Detsky, J.; Husain, Z.; Sahgal, A.; Soliman, H. Trastuzumab emtansine increases the risk of stereotactic radiosurgery-induced radionecrosis in HER2 + breast cancer. J. Neurooncol. 2022, 159, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, P.K.; Cittelly, D.M.; Robin, T.P.; Carlson, J.A.; Stuhr, K.A.; Contreras-Zarate, M.J.; Lai, S.; Ormond, D.R.; Rusthoven, C.G.; Gaspar, L.E.; et al. Combination of Trastuzumab Emtansine and Stereotactic Radiosurgery Results in High Rates of Clinically Significant Radionecrosis and Dysregulation of Aquaporin-4. Clin. Cancer Res. 2019, 25, 3946–3953. [Google Scholar] [CrossRef] [Green Version]
- Mills, M.N.; Walker, C.; Thawani, C.; Naz, A.; Figura, N.B.; Kushchayev, S.; Etame, A.; Yu, H.M.; Robinson, T.J.; Liu, J.; et al. Trastuzumab Emtansine (T-DM1) and stereotactic radiation in the management of HER2+ breast cancer brain metastases. BMC Cancer 2021, 21, 223. [Google Scholar] [CrossRef]
- Park, C.; Buckley, E.D.; Van Swearingen, A.E.D.; Giles, W.; Herndon, J.E.I.; Kirkpatrick, J.P.; Anders, C.K.; Floyd, S.R. Systemic Therapy Type and Timing Effects on Radiation Necrosis Risk in HER2+ Breast Cancer Brain Metastases Patients Treated with Stereotactic Radiosurgery. Front. Oncol. 2022, 12, 854364. [Google Scholar] [CrossRef]
- Garcia-Alvarez, A.; Papakonstantinou, A.; Oliveira, M. Brain Metastases in HER2-Positive Breast Cancer: Current and Novel Treatment Strategies. Cancers 2021, 13, 2927. [Google Scholar] [CrossRef]
- Zhu, K.; Wu, Y.; He, P.; Fan, Y.; Zhong, X.; Zheng, H.; Luo, T. PI3K/AKT/mTOR-Targeted Therapy for Breast Cancer. Cells 2022, 11, 2508. [Google Scholar] [CrossRef]
- Morgan, A.J.; Giannoudis, A.; Palmieri, C. The genomic landscape of breast cancer brain metastases: A systematic review. Lancet Oncol. 2021, 22, e7–e17. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: Progress made and future hopes. NPJ Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, M.P.; Wang, D.; Wang, F.; Kleinberg, L.; Brade, A.; Robins, H.I.; Turaka, A.; Leahy, T.; Medina, D.; Xiong, H.; et al. Veliparib in combination with whole brain radiation therapy in patients with brain metastases: Results of a phase 1 study. J. Neurooncol. 2015, 122, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Corti, C.; Antonarelli, G.; Criscitiello, C.; Lin, N.U.; Carey, L.A.; Cortés, J.; Poortmans, P.; Curigliano, G. Targeting brain metastases in breast cancer. Cancer Treat. Rev. 2022, 103, 102324. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Zhao, L.; Li, J.; Chen, W.; Li, X. miR-30d Blocked Transforming Growth Factor β1–Induced Epithelial-Mesenchymal Transition by Targeting Snail in Ovarian Cancer Cells. Int. J. Gynecol. Cancer 2015, 25, 1574–1581. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Ren, D.; Zhu, X.; Kong, R.; Zhao, Z.; Sheng, J.; Wang, J.; Xu, X.; Liu, J.; Cui, K.; Zhang, X.H.; et al. Targeting Brain-Adaptive Cancer Stem Cells Prohibits Brain Metastatic Colonization of Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 2052–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.-I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.L.; Kuo, Y.C.; Ho, Y.S.; Huang, Y.H. Triple-negative breast cancer: Current understanding and future therapeutic breakthrough targeting cancer stemness. Cancers 2019, 11, 1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation Resistance of Cancer Stem Cells: The 4 R’s of Radiobiology Revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [Green Version]
- Nathansen, J.; Meyer, F.; Müller, L.; Schmitz, M.; Borgmann, K.; Dubrovska, A. Beyond the Double-Strand Breaks: The Role of DNA Repair Proteins in Cancer Stem-Cell Regulation. Cancers 2021, 13, 4818. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Meng, Y.; Dekmezian, C.; Kim, K.; Pajonk, F. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res. 2010, 12, R13. [Google Scholar] [CrossRef] [Green Version]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lin, Q.; Yun, Z. BRCA1 regulates the cancer stem cell fate of breast cancer cells in the context of hypoxia and histone deacetylase inhibitors. Sci. Rep. 2019, 9, 9702. [Google Scholar] [CrossRef] [Green Version]
- Obara, E.A.A.; Aguilar-Morante, D.; Rasmussen, R.D.; Frias, A.; Vitting-Serup, K.; Lim, Y.C.; Elbæk, K.J.; Pedersen, H.; Vardouli, L.; Jensen, K.E.; et al. SPT6-driven error-free DNA repair safeguards genomic stability of glioblastoma cancer stem-like cells. Nat. Commun. 2020, 11, 4709. [Google Scholar] [CrossRef]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A Role for Homologous Recombination and Abnormal Cell-Cycle Progression in Radioresistance of Glioma-Initiating Cells. Mol. Cancer Ther. 2012, 11, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Jiang, T.; Zhang, H.; Gou, X.; Han, C.; Wang, J.; Chen, A.T.; Ma, J.; Liu, J.; Chen, Z.; et al. LRRC31 inhibits DNA repair and sensitizes breast cancer brain metastasis to radiation therapy. Nat. Cell Biol. 2020, 22, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285.e275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, M.; Vidovic, D.; Paine, A.S.; Huynh, T.T.; Coyle, K.M.; Thomas, M.L.; Cruickshank, B.M.; Dean, C.A.; Clements, D.R.; Kim, Y.; et al. Epigenetic Silencing of TAP1 in Aldefluor+ Breast Cancer Stem Cells Contributes to Their Enhanced Immune Evasion. Stem Cells 2018, 36, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balbous, A.; Cortes, U.; Guilloteau, K.; Rivet, P.; Pinel, B.; Duchesne, M.; Godet, J.; Boissonnade, O.; Wager, M.; Bensadoun, R.J.; et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 604. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, M.; Strappazzon, F.; Arisi, I.; Brandi, R.; D’Onofrio, M.; Sambucci, M.; Manic, G.; Vitale, I.; Barilà, D.; Stagni, V. ATM kinase sustains breast cancer stem-like cells by promoting ATG4C expression and autophagy. Oncotarget 2017, 8, 21692–21709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef]
- Mampre, D.; Mehkri, Y.; Rajkumar, S.; Sriram, S.; Hernandez, J.; Lucke-Wold, B.; Chandra, V. Treatment of breast cancer brain metastases: Radiotherapy and emerging preclinical approaches. Diagn. Ther. 2022, 1, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Sun, P.; Xia, L.; He, X.; Xia, Z.; Huang, Y.; Liu, W.; Li, L.; Chen, L. A brain-enriched lncRNA shields cancer cells from immune-mediated killing for metastatic colonization in the brain. Proc. Natl. Acad. Sci. USA 2022, 119, e2200230119. [Google Scholar] [CrossRef] [PubMed]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell–dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [Green Version]
- Sheng, H.; Huang, Y.; Xiao, Y.; Zhu, Z.; Shen, M.; Zhou, P.; Guo, Z.; Wang, J.; Wang, H.; Dai, W.; et al. ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000340. [Google Scholar] [CrossRef]
- Patin, E.C.; Dillon, M.T.; Nenclares, P.; Grove, L.; Soliman, H.; Leslie, I.; Northcote, D.; Bozhanova, G.; Crespo-Rodriguez, E.; Baldock, H.; et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage–response inhibition and immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004306. [Google Scholar] [CrossRef]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- Wang, N.; Yang, Y.; Jin, D.; Zhang, Z.; Shen, K.; Yang, J.; Chen, H.; Zhao, X.; Yang, L.; Lu, H. PARP inhibitor resistance in breast and gynecological cancer: Resistance mechanisms and combination therapy strategies. Front. Pharmacol. 2022, 13, 967633. [Google Scholar] [CrossRef]
- Patel, P.; Sun, L.; Robbins, Y.; Clavijo, P.E.; Friedman, J.; Silvin, C.; Van Waes, C.; Cook, J.; Mitchell, J.; Allen, C. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 2019, 8, e1638207. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, E.J.; Peterson, J.; Brown, P.D.; Sheehan, J.P.; Quiñones-Hinojosa, A.; Zaorsky, N.G.; Trifiletti, D.M. Treatment of brain metastases with stereotactic radiosurgery and immune checkpoint inhibitors: An international meta-analysis of individual patient data. Radiother. Oncol. 2018, 130, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.A.; Kim, Y.; Arrington, J.A.; Kim, S.; DeJesus, M.; Soyano, A.E.; Armaghani, A.J.; Costa, R.L.; Khong, H.T.; Loftus, L.S.; et al. Nivolumab and Stereotactic Radiosurgery for Patients with Breast Cancer Brain Metastases: A Nonrandomized, Open-Label Phase 1b Study. Adv. Radiat. Oncol. 2021, 6, 100798. [Google Scholar] [CrossRef] [PubMed]
- Page, D.B.; Beal, K.; Linch, S.N.; Spinelli, K.J.; Rodine, M.; Halpenny, D.; Modi, S.; Patil, S.; Young, R.J.; Kaley, T.; et al. Brain radiotherapy, tremelimumab-mediated CTLA-4-directed blockade +/− trastuzumab in patients with breast cancer brain metastases. NPJ Breast Cancer 2022, 8, 50. [Google Scholar] [CrossRef]
- Anscher, M.S.; Arora, S.; Weinstock, C.; Amatya, A.; Bandaru, P.; Tang, C.; Girvin, A.T.; Fiero, M.H.; Tang, S.; Lubitz, R.; et al. Association of Radiation Therapy with Risk of Adverse Events in Patients Receiving Immunotherapy: A Pooled Analysis of Trials in the US Food and Drug Administration Database. JAMA Oncol. 2022, 8, 232–240. [Google Scholar] [CrossRef]
- Andring, L.; Squires, B.; Seymour, Z.; Fahim, D.; Jacob, J.; Ye, H.; Marvin, K.; Grills, I. Radionecrosis (RN) in patients with brain metastases treated with stereotactic radiosurgery (SRS) and immunotherapy. Int. J. Neurosci. 2021, 1–8. [Google Scholar] [CrossRef]
- Kiess, A.P.; Wolchok, J.D.; Barker, C.A.; Postow, M.A.; Tabar, V.; Huse, J.T.; Chan, T.A.; Yamada, Y.; Beal, K. Stereotactic Radiosurgery for Melanoma Brain Metastases in Patients Receiving Ipilimumab: Safety Profile and Efficacy of Combined Treatment. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Jiang, W.; Allen, P.; Glitza, I.; Guha, N.; Hwu, P.; Ghia, A.; Phan, J.; Mahajan, A.; Tawbi, H.; et al. Radiation necrosis with stereotactic radiosurgery combined with CTLA-4 blockade and PD-1 inhibition for treatment of intracranial disease in metastatic melanoma. J. Neurooncol. 2017, 133, 595–602. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-Mediated Inhibition of Metastases after Treatment with Local Radiation and CTLA-4 Blockade in a Mouse Model of Breast Cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e1620. [Google Scholar] [CrossRef] [PubMed]
- Pangal, D.J.; Yarovinsky, B.; Cardinal, T.; Cote, D.J.; Ruzevick, J.; Attenello, F.J.; Chang, E.L.; Ye, J.; Neman, J.; Chow, F.; et al. The Abscopal Effect: Systematic Review in Patients with Brain and Spine Metastases. Neurooncol. Adv. 2022, 4, vdac132. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; DeMaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morisada, M.; Clavijo, P.E.; Moore, E.; Sun, L.; Chamberlin, M.; Van Waes, C.; Hodge, J.W.; Mitchell, J.B.; Friedman, J.; Allen, C.T. PD-1 blockade reverses adaptive immune resistance induced by high-dose hypofractionated but not low-dose daily fractionated radiation. OncoImmunology 2018, 7, e1395996. [Google Scholar] [CrossRef] [Green Version]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing Tumor Immunity with Fractionated Radiation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 83, 1306–1310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Niedermann, G. Abscopal Effects with Hypofractionated Schedules Extending into the Effector Phase of the Tumor-Specific T-Cell Response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; Van De Vijver, K.K.; De Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Möckl, A.; Salamero-Boix, A.; Alekseeva, T.; Schäffer, A.; Schulz, M.; Niesel, K.; Maas, R.R.; Groth, M.; Elie, B.T.; et al. Compensatory CSF2-driven macrophage activation promotes adaptive resistance to CSF1R inhibition in breast-to-brain metastasis. Nat. Cancer 2021, 2, 1086–1101. [Google Scholar] [CrossRef]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef]
- Patel, K.R.; Burri, S.H.; Asher, A.L.; Crocker, I.R.; Fraser, R.W.; Zhang, C.; Chen, Z.; Kandula, S.; Zhong, J.; Press, R.H.; et al. Comparing Preoperative with Postoperative Stereotactic Radiosurgery for Resectable Brain Metastases: A Multi-institutional Analysis. Neurosurgery 2016, 79, 279–285. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hintelmann, K.; Petersen, C.; Borgmann, K. Radiotherapeutic Strategies to Overcome Resistance of Breast Cancer Brain Metastases by Considering Immunogenic Aspects of Cancer Stem Cells. Cancers 2023, 15, 211. https://doi.org/10.3390/cancers15010211
Hintelmann K, Petersen C, Borgmann K. Radiotherapeutic Strategies to Overcome Resistance of Breast Cancer Brain Metastases by Considering Immunogenic Aspects of Cancer Stem Cells. Cancers. 2023; 15(1):211. https://doi.org/10.3390/cancers15010211
Chicago/Turabian StyleHintelmann, Katharina, Cordula Petersen, and Kerstin Borgmann. 2023. "Radiotherapeutic Strategies to Overcome Resistance of Breast Cancer Brain Metastases by Considering Immunogenic Aspects of Cancer Stem Cells" Cancers 15, no. 1: 211. https://doi.org/10.3390/cancers15010211
APA StyleHintelmann, K., Petersen, C., & Borgmann, K. (2023). Radiotherapeutic Strategies to Overcome Resistance of Breast Cancer Brain Metastases by Considering Immunogenic Aspects of Cancer Stem Cells. Cancers, 15(1), 211. https://doi.org/10.3390/cancers15010211