
Genomic and Transcriptional Profiling of Chinese Melanoma Patients Enhanced Potentially Druggable Targets: A Multicenter Study
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Background
2. Methods
2.1. Patients Population
2.2. DNA Isolation and Targeted Sequencing
2.3. RNA Sequencing
2.4. Statistical Analysis
3. Results
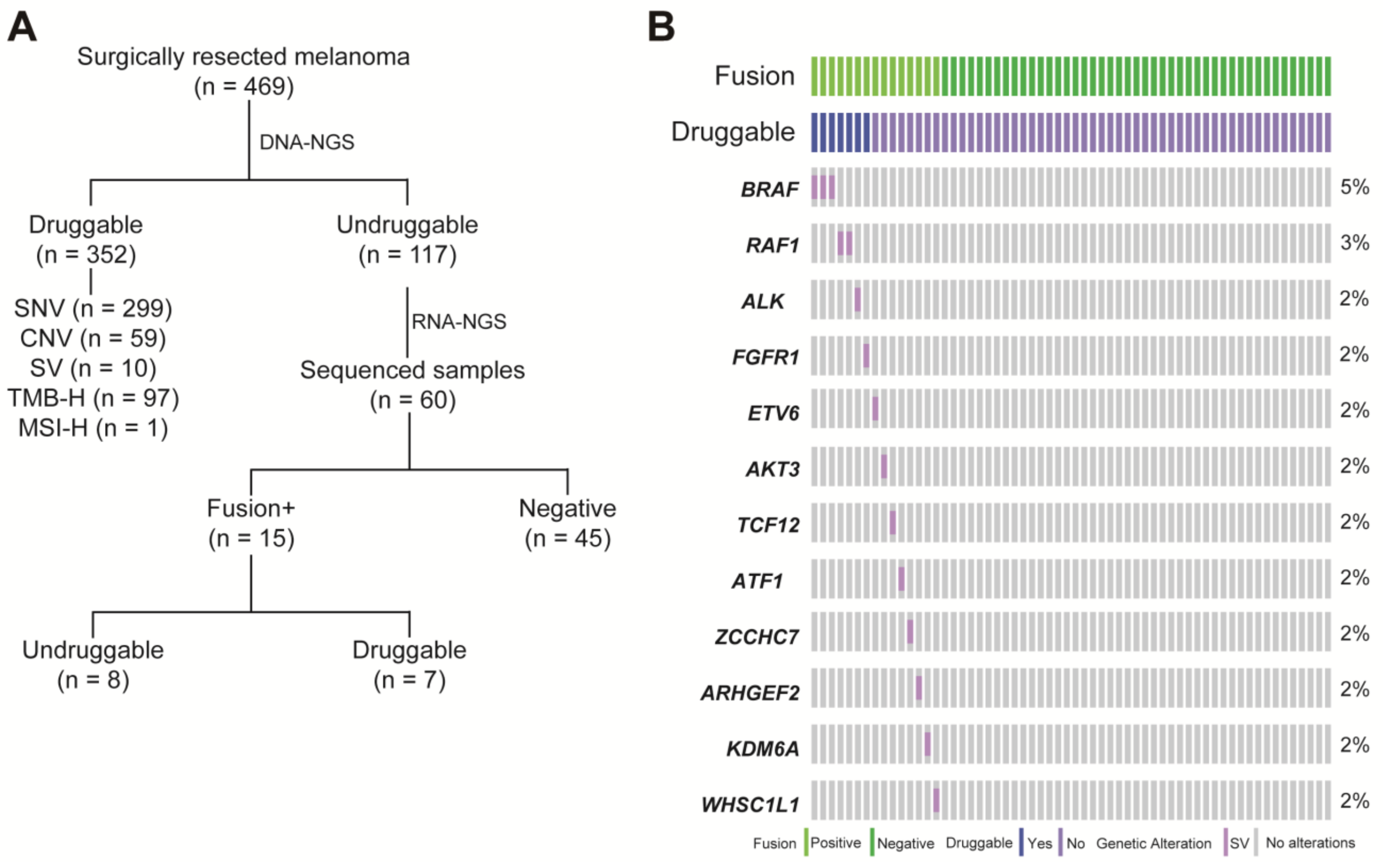
3.1. The Landscape of Somatically Actionable Genes Identified by DNA-NGS
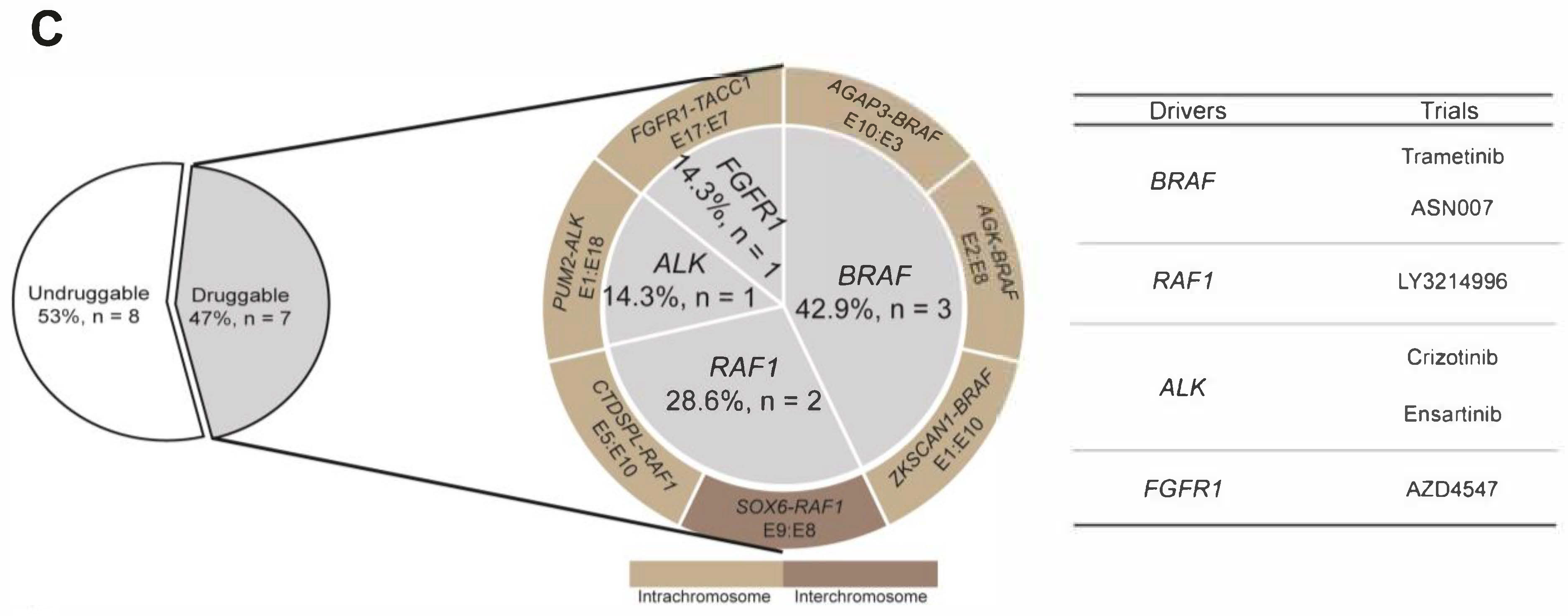
3.2. RNA-NGS Could Identify Actionable Fusions in Undruggable Melanoma Patients
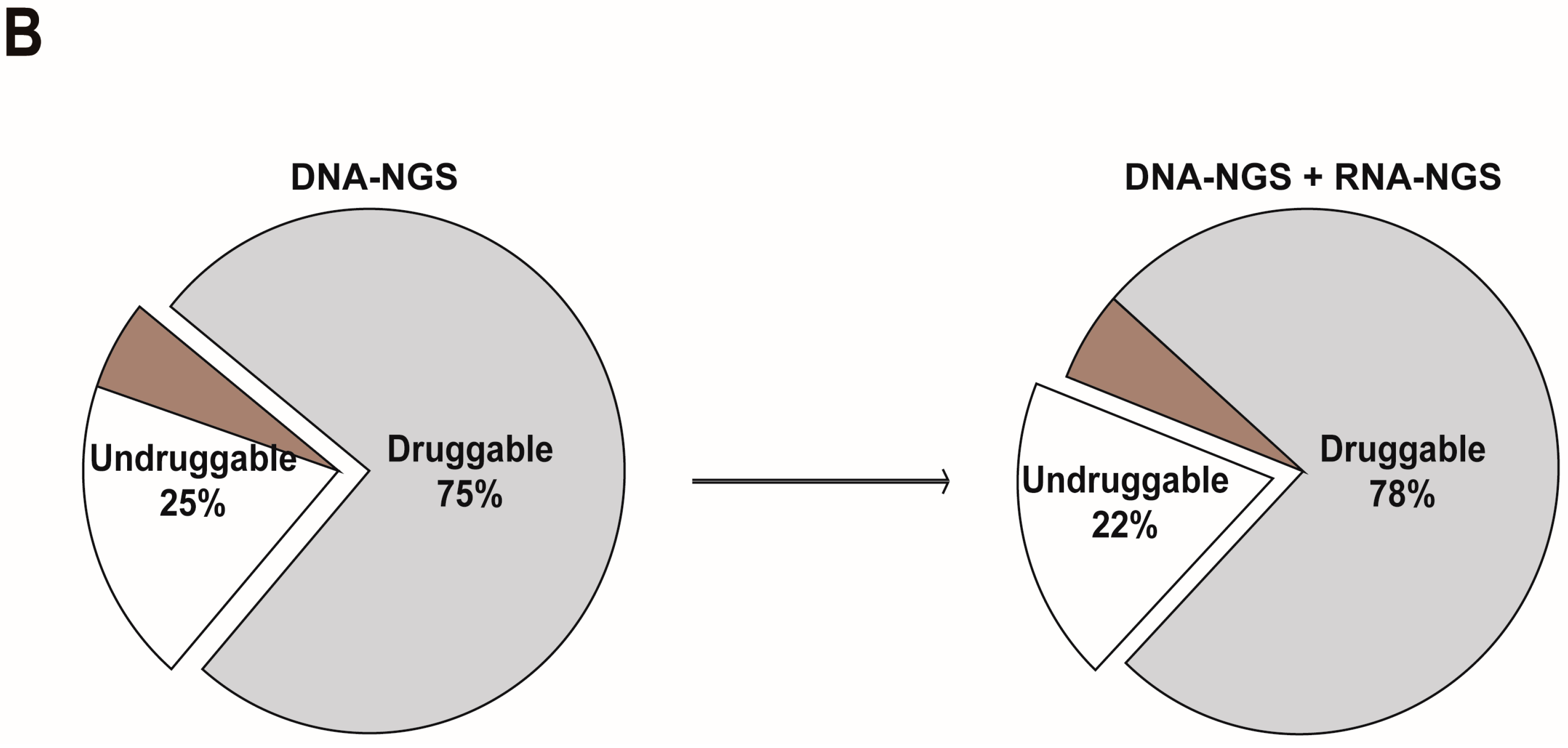
3.3. RNA-NGS Could Significantly Enhance the Proportion of Fusions in Melanoma
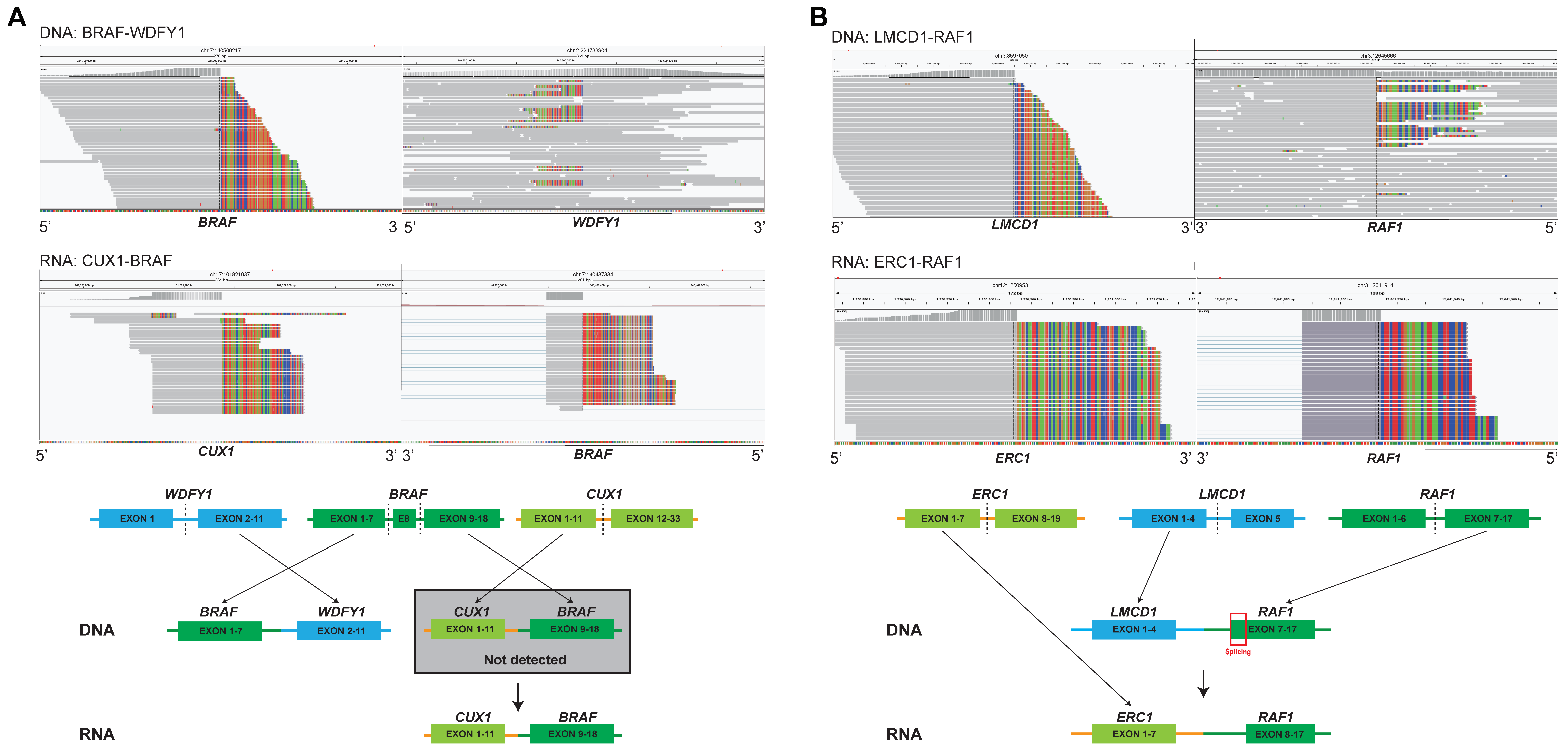
3.4. Unreliability of the Fusion Breakpoint in Predicting Relevant Transcripts Based on DNA-NGS
3.5. Novel Fusions Detected in Melanoma Patients
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| NGS | Next-generation sequencing |
| TKI | Tyrosine kinase inhibitor |
| TMB | Tumor mutation burden |
| MSI | Microsatellite instability |
| NSCLC | Non-small-cell lung cancer |
| FFPE | Formalin-fixed paraffin-embedded |
References
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Manca, A.; Paliogiannis, P.; Colombino, M.; Casula, M.; Lissia, A.; Botti, G.; Caracò, C.; Ascierto, P.A.; Sini, M.C.; Palomba, G.; et al. Mutational concordance between primary and metastatic melanoma: A next-generation sequencing approach. J. Transl. Med. 2019, 17, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Li, J.; Wen, X.; Zhu, B.; Liu, W.; Wang, J.; Jiang, H.; Ding, Y.; Li, D.; Zhang, X. Next-generation sequencing in advanced Chinese melanoma reveals therapeutic targets and prognostic biomarkers for immunotherapy. Sci. Rep. 2022, 12, 9559. [Google Scholar] [CrossRef] [PubMed]
- Afshar, A.R.; Damato, B.E.; Stewart, J.M.; Zablotska, L.B.; Roy, R.; Olshen, A.B.; Joseph, N.M.; Bastian, B.C. Next-Generation Sequencing of Uveal Melanoma for Detection of Genetic Alterations Predicting Metastasis. Transl. Vis. Sci. Technol. 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenbach, R.J.; Lee, J.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Saw, R.P.M.; Howle, J.R.; Spillane, A.J.; Scolyer, R.A.; Kefford, R.F.; et al. Design and Testing of a Custom Melanoma Next Generation Sequencing Panel for Analysis of Circulating Tumor DNA. Cancers 2020, 12, 2228. [Google Scholar] [CrossRef]
- Wen, P.Y.; Stein, A.; van den Bent, M.; De Greve, J.; Wick, A.; de Vos, F.; von Bubnoff, N.; van Linde, M.E.; Lai, A.; Prager, G.W.; et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutant low-grade and high-grade glioma (ROAR): A multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022, 23, 53–64. [Google Scholar] [CrossRef]
- Hutchinson, K.E.; Lipson, D.; Stephens, P.J.; Otto, G.; Lehmann, B.D.; Lyle, P.L.; Vnencak-Jones, C.L.; Ross, J.S.; Pietenpol, J.A.; Sosman, J.A.; et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6696–6702. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.A.; Shah, N.; Montesion, M.; Sharaf, R.; Pavlick, D.C.; Sokol, E.S.; Alexander, B.M.; Venstrom, J.M.; Elvin, J.A.; Ross, J.S.; et al. Melanomas with activating RAF1 fusions: Clinical, histopathologic, and molecular profiles. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2020, 33, 1466–1474. [Google Scholar] [CrossRef] [Green Version]
- Forschner, A.; Forchhammer, S.; Bonzheim, I. NTRK gene fusions in melanoma: Detection, prevalence and potential therapeutic implications. JDDG J. Dtsch. Dermatol. Ges. 2020, 18, 1387–1392. [Google Scholar] [CrossRef]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Kubeček, O.; Kopecký, J. Microsatellite instability in melanoma: A comprehensive review. Melanoma Res. 2016, 26, 545–550. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [CrossRef] [Green Version]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef]
- Ghiasvand, R.; Berge, L.A.M.; Andreassen, B.K.; Stenehjem, J.S.; Heir, T.; Karlstad, Ø.; Juzeniene, A.; Larsen, I.K.; Green, A.C.; Veierød, M.B.; et al. Use of antihypertensive drugs and risk of cutaneous melanoma: A nationwide nested case-control study. Int. J. Epidemiol. 2022. [Google Scholar] [CrossRef]
- Nakamura, K.; Ashida, A.; Kiniwa, Y.; Okuyama, R. Immune status and prognosis of stage II and III primary malignant melanoma: An analysis including 77 cases of acral lentiginous melanoma. J. Dermatol. 2022. [Google Scholar] [CrossRef]
- Bowley, T.Y.; Lagutina, I.V.; Francis, C.; Sivakumar, S.; Selwyn, R.G.; Taylor, E.; Guo, Y.; Fahy, B.N.; Tawfik, B.; Marchetti, D. The RPL/RPS gene signature of melanoma CTCs associates with brain metastasis. Cancer Res. Commun. 2022, 2, 1436–1448. [Google Scholar] [CrossRef]
- Khan, M.; Thompson, J.; Kiiskila, L.; Oboh, O.; Truong, T.; Prentice, A.; Assifi, M.M.; Chung, M.; Wright, G.P. Timing and necessity of staging imaging in clinical stage II cutaneous melanoma: Cost-effectiveness and clinical decision analysis. Am. J. Surg. 2022, 225, 93–98. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, Z.; Liu, J.; Li, L.; Xu, X.; Yao, X.; Dai, Z.; Wang, X.; Yang, S.; Wu, H.; et al. Characterizations of Gene Alterations in Melanoma Patients from Chinese Population. BioMed Res. Int. 2020, 2020, 6096814. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, L.; Lai, Y.; Wang, X.; Han, X.; Liu, S.; Wang, D.; Li, X.; Hu, N.; Kong, Y.; et al. Genetic alteration of Chinese patients with rectal mucosal melanoma. BMC Cancer 2021, 21, 623. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.W.; Jia, D.D.; Zhou, C.C.; Li, T. Mutational profiling of melanomas in patients from the southeast coast of China. Transl. Cancer Res. 2020, 9, 4781–4789. [Google Scholar] [CrossRef] [PubMed]
- Byeon, S.; Cho, H.J.; Jang, K.T.; Kwon, M.; Lee, J.; Lee, J.; Kim, S.T. Molecular profiling of Asian patients with advanced melanoma receiving check-point inhibitor treatment. ESMO Open 2021, 6, 100002. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Choi, J.Y.; Lim, A.R.; Kim, J.W.; Choi, Y.J.; Lee, S.; Sung, J.S.; Chung, H.J.; Jang, B.; Yoon, D.; et al. Genomic Landscape and Clinical Utility in Korean Advanced Pan-Cancer Patients from Prospective Clinical Sequencing: K-MASTER Program. Cancer Discov. 2022, 12, 938–948. [Google Scholar] [CrossRef]
- Bouchè, V.; Aldegheri, G.; Donofrio, C.A.; Fioravanti, A.; Roberts-Thomson, S.; Fox, S.B.; Schettini, F.; Generali, D. BRAF Signaling Inhibition in Glioblastoma: Which Clinical Perspectives? Front. Oncol. 2021, 11, 772052. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Liu, Y.; Dong, L.; Yang, L.; Chen, L.; Liu, K.; Shao, Y.; Ying, J. Potential Unreliability of Uncommon ALK, ROS1, and RET Genomic Breakpoints in Predicting the Efficacy of Targeted Therapy in NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 404–418. [Google Scholar] [CrossRef]
- Couts, K.L.; Bemis, J.; Turner, J.A.; Bagby, S.M.; Murphy, D.; Christiansen, J.; Hintzsche, J.D.; Le, A.; Pitts, T.M.; Wells, K.; et al. ALK Inhibitor Response in Melanomas Expressing EML4-ALK Fusions and Alternate ALK Isoforms. Mol. Cancer Ther. 2018, 17, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.I.; Zhu, V.W.; Nagasaka, M. Catalog of 5’ Fusion Partners in ALK-positive NSCLC Circa 2020. JTO Clin. Res. Rep. 2020, 1, 100015. [Google Scholar] [CrossRef]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, B.; Wang, C.; Zhao, D.; Liu, Z.; Niu, Y.; Wang, X.; Li, W.; Zhu, J.; Tao, H.; et al. Genomic and Transcriptional Profiling of Chinese Melanoma Patients Enhanced Potentially Druggable Targets: A Multicenter Study. Cancers 2023, 15, 283. https://doi.org/10.3390/cancers15010283
Li Y, Wang B, Wang C, Zhao D, Liu Z, Niu Y, Wang X, Li W, Zhu J, Tao H, et al. Genomic and Transcriptional Profiling of Chinese Melanoma Patients Enhanced Potentially Druggable Targets: A Multicenter Study. Cancers. 2023; 15(1):283. https://doi.org/10.3390/cancers15010283
Chicago/Turabian StyleLi, Yue, Baoming Wang, Chunyang Wang, Dandan Zhao, Zhengchuang Liu, Yanling Niu, Xiaojuan Wang, Wei Li, Jianhua Zhu, Houquan Tao, and et al. 2023. "Genomic and Transcriptional Profiling of Chinese Melanoma Patients Enhanced Potentially Druggable Targets: A Multicenter Study" Cancers 15, no. 1: 283. https://doi.org/10.3390/cancers15010283