
Hereditary Colorectal Cancer: State of the Art in Lynch Syndrome

 ,
,  , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. MMR Genes
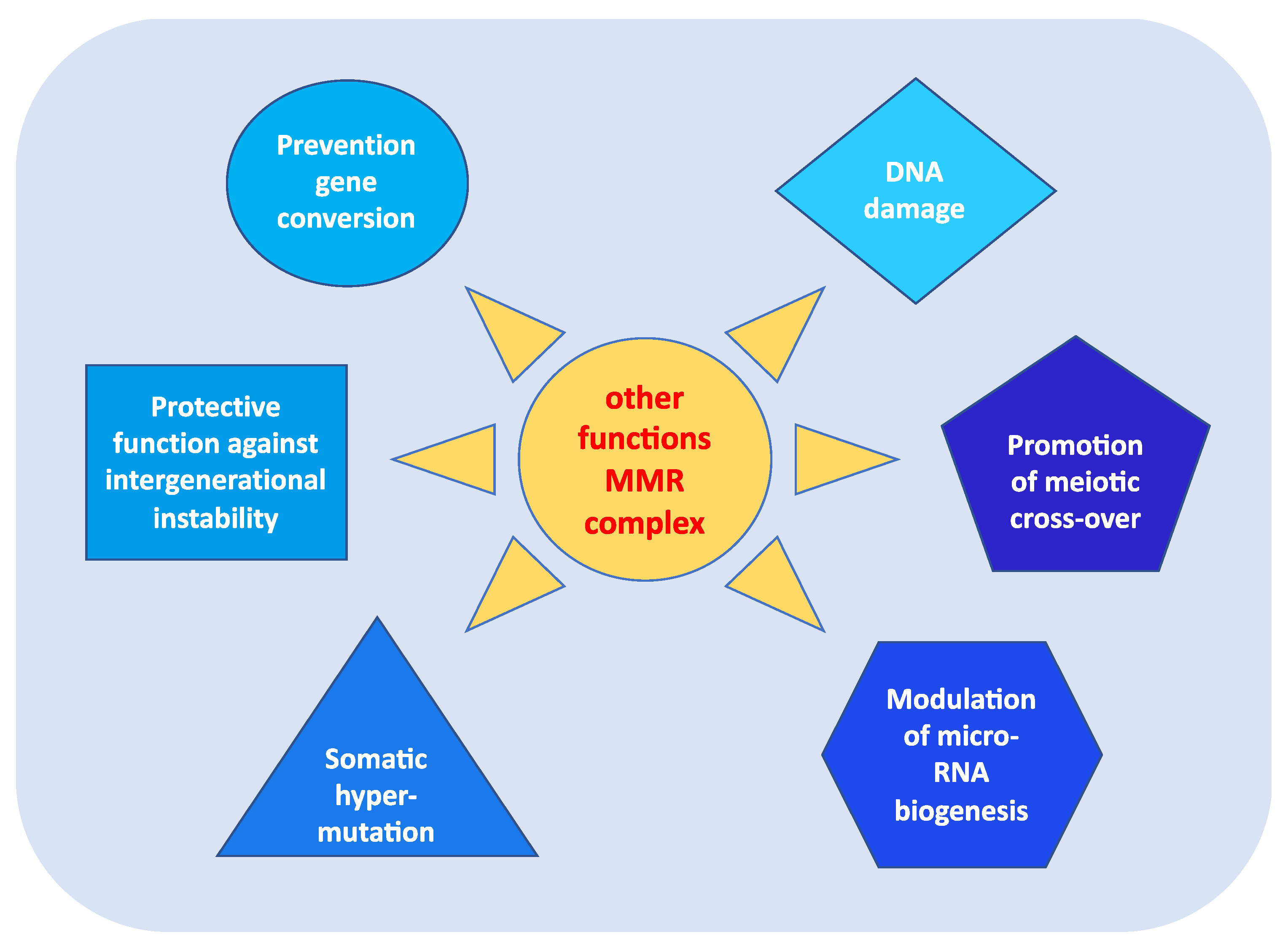
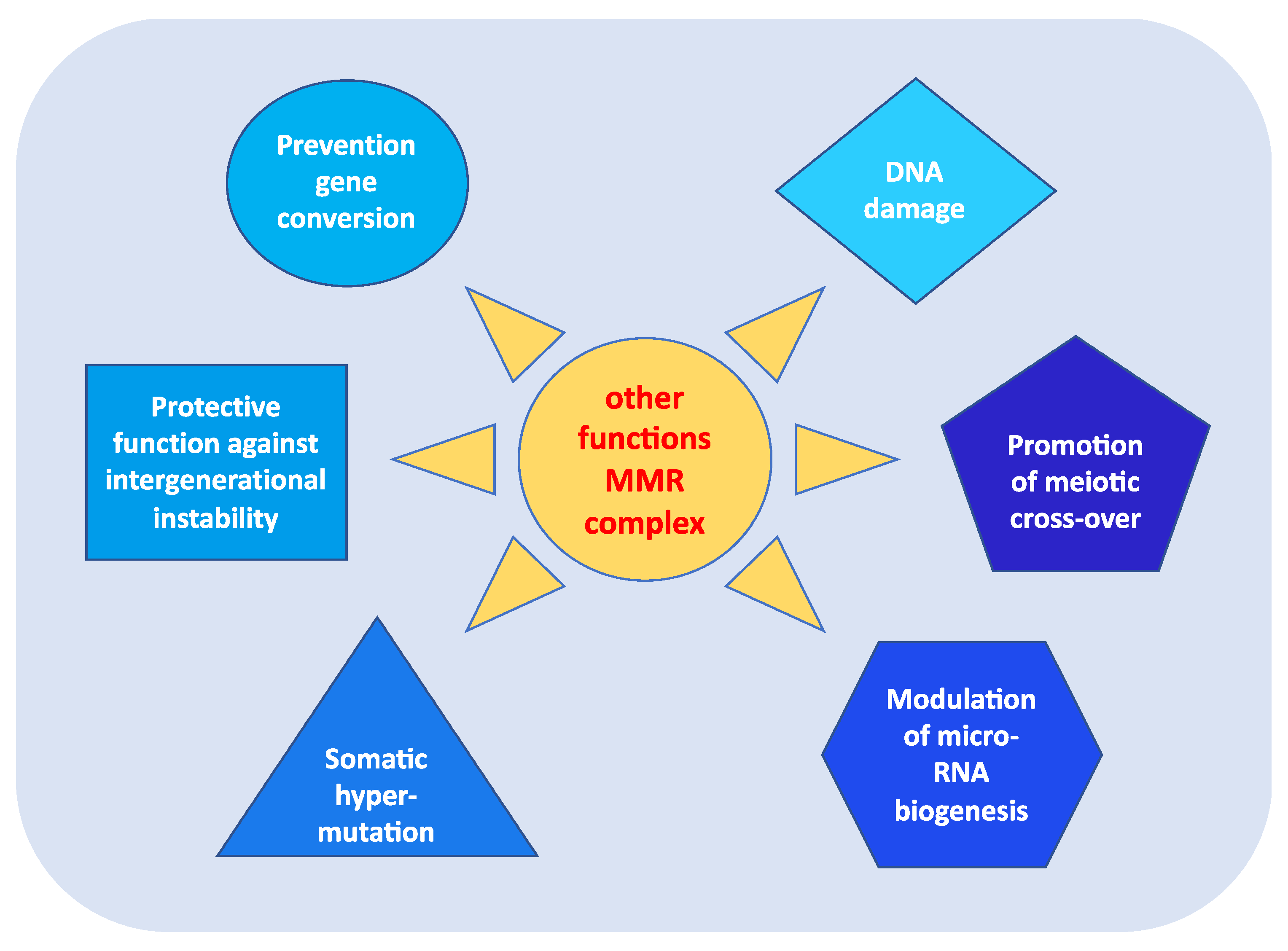
3. Other Functions of MMR Genes
- prevention of reparative recombination (gene conversion) between non-identical sequences [16];
- protection against intergenerational instability resulting from the phenomenon of trinucleotide repeat expansion, which is the basis of the pathogenesis of various neurodegenerative diseases [18];
- the immunoglobulin (Ig) differentiation process based on “somatic hypermutation”, regulated by the MutSα–MutLα complex in combination with two other proteins, AID (activation-induced cytidine deaminase) and Polμ (error-prone DNA polymerase) [19];
- modulation of microRNA (miRNA) biogenesis through the interaction of MMR proteins with the microprocessor complex; in particular, MutLα specifically binds to pri-miRNAs and the Drosha–DGCR8 complex to stimulate the processing of pri-miRNAs into pre-miRNAs in a manner dependent on the ATPase activity of MutLα [20];
- reporting of DNA damage caused by exogenous carcinogens (heterocyclic amines, oxidizing agents, and UV radiation) obtained through a synergistic action between the homologous proteins of p53 (p53, p63, and p73) and the MutSα–MutLα complex. Moreover, in response to exogenous damage, MLH1 interacts with the MRE11 protein, a component of the BRCA1-associated surveillance complex (BASC), and regulates the cell cycle and the apoptotic pathway; indeed, there is a correlation between the MMR system and the G2/M phase of the cell cycle [21].
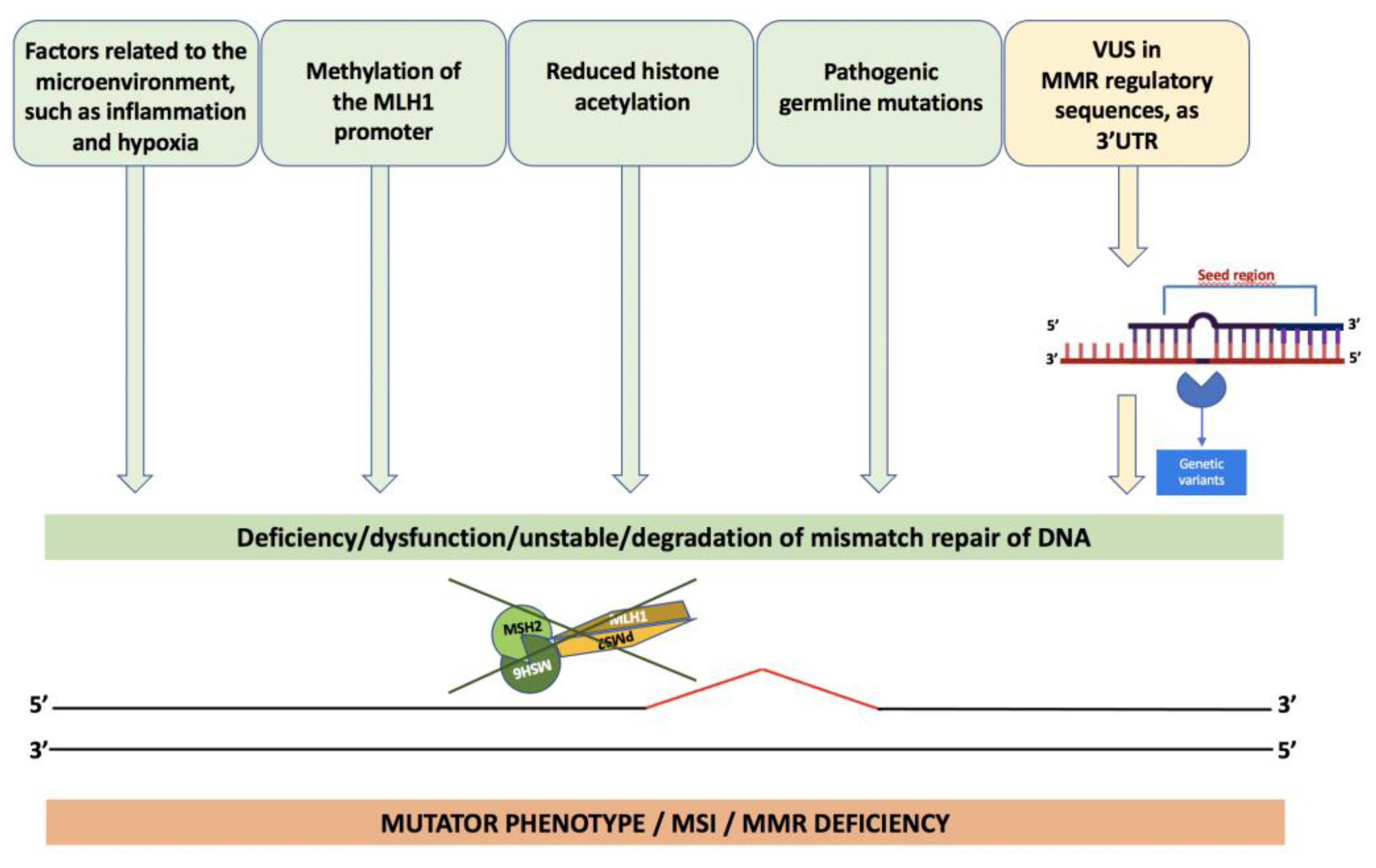
4. The Molecular Alterations of Lynch Syndrome
5. Variants of Uncertain Significance
6. Microsatellite Instability (MSI)
7. K-Endometrium in LS and Loss of MMR Proteins
8. Loss of Expression of MMR Genes
9. Genotype–Phenotype Correlations
10. Lynch-like Syndrome
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abu-Ghazaleh, N.; Kaushik, V.; Gorelik, A.; Jenkins, M.; Macrae, F. Worldwide prevalence of Lynch syndrome in patients with colorectal cancer: Systematic review and meta-analysis. Anesthesia Analg. 2022, 24, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Marina, D.R.; Ugo, P.; Daniela, R.; Valeria, C.; Francesca, D.; Paola, I.; Paola, D. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar]
- Carlomagno, N.; Duraturo, F.; Candida, M.; De Rosa, M.; Varone, V.; Ciancia, G.; Calogero, A.; Santangelo, M.L. Multiple splenic hamartomas and familial adenomatous polyposis: A case report and review of the literature. J. Med Case Rep. 2015, 9, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodaro, C.; Grifasi, C.; Florio, J.; Santangelo, M.L.; Duraturo, F.; De Rosa, M.; Izzo, P.; Renda, A. The role of mutation analysis of the APC gene in the management of FAP patients. A controversial issue. Ann. Ital. di Chir. 2016, 87. [Google Scholar]
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising incidence of early-onset colorectal cancer - a call to action. Nat. Rev. Clin. Oncol. 2021, 18, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Boyle, T.; Keegel, T.; Bull, F.; Heyworth, J.; Fritschi, L. Physical Activity and Risks of Proximal and Distal Colon Cancers: A Systematic Review and Meta-analysis. Gynecol. Oncol. 2012, 104, 1548–1561. [Google Scholar] [CrossRef]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges (Review). Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, G.; Wu, W. Recent advances in Lynch syndrome. Exp. Hematol. Oncol. 2021, 10, 1–8. [Google Scholar] [CrossRef]
- Aune, D.; Chan, D.S.M.; Lau, R.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dietary fibre, whole grains, and risk of colorectal cancer: Systematic review and dose-response meta-analysis of prospective studies. BMJ 2011, 343, d6617. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, P.; Holmm, N.V.; Verkasalom, P.K.; Iliadoum, A.; Kapriom, J.; Koskenvuom, M.; Pukkalam, E.; Skytthem, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- De la Chapelle, A. Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 2004, 4, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.; Kim, D.; Martín-López, J.V.; Lee, R.; Oh, J.; Hanne, J.; Fishel, R.; Lee, J.-B. Dynamic control of strand excision during human DNA mismatch repair. Proc. Natl. Acad. Sci. USA 2016, 113, 3281–3286. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Ijsselsteijn, R.; Jansen, J.G.; de Wind, N. DNA mismatch repair-dependent DNA damage responses and cancer. DNA Repair 2020, 93, 102923. [Google Scholar] [CrossRef] [PubMed]
- Nicholsonm, A.; Hendrixm, M.; Jinks-Robertsonm, S.; Crouse, G.F. Regulation of mitotic homeologous recombination in yeast: Functions of mismatch repair and nucleotide excision repair genes. Genetics 2000, 154, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Long, Y.; Zhou, Y.; Huang, C.; Gu, A.; Wang, X. Common variants in mismatch repair genes associated with increased risk of sperm DNA damage and male infertility. BMC Med. 2012, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Tomé, S.; Holt, I.; Edelmann, W.; Morris, G.E.; Munnich, A.; Pearson, C.; Gourdon, G. MSH2 ATPase Domain Mutation Affects CTG•CAG Repeat Instability in Transgenic Mice. PLoS Genet. 2009, 5, e1000482. [Google Scholar] [CrossRef]
- Zanotti, K.J.; Gearhart, P.J. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair. (Amst.) 2016, 38, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Mao, G.; Pan, X.; Gu, L. Evidence that a mutation in the MLH1 3’-untranslated region confers a mutator phenotype and mismatch repair deficiency in patients with relapsed leukemia. JBC 2008, 283, 3211–3216. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR System. Carcinog 2005, 27, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiricny, J.; Nyström-Lahti, M. Mismatch repair defects in cancer. Curr. Opin. Genet. Dev. 2000, 10, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2015, 38, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Rahman, W.M.; Mecklin, J.-P.; Peltomäki, P. The genetics of HNPCC: Application to diagnosis and screening. Crit. Rev. Oncol. 2006, 58, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Hofstra, R.M.; Spurdle, A.B.; Eccles, D.; Foulkes, W.D.; de Wind, N.; Hoogerbrugge, N.; Hogervorst, F.B. IARC Unclassified Genetic Variants Working Group. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum. Mutat. 2008, 29, 1292–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liccardo, R.; De Rosa, M.; Izzo, P.; Duraturo, F. Novel Implications in Molecular Diagnosis of Lynch Syndrome. Gastroenterol. Res. Pr. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liccardo, R.; Della Ragione, C.; Mitilini, N.; De Rosa, M.; Izzo, P.; Duraturo, F. Novel variants of unknown significance in the PMS2 gene identified in patients with hereditary colon cancer. Cancer Manag. Res. 2019, ume 11, 6719–6725. [Google Scholar] [CrossRef] [Green Version]
- Liccardo, R.; De Rosa, M.; Rossi, G.B.; Carlomagno, N.; Izzo, P.; Duraturo, F. Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome. Int. J. Mol. Sci. 2017, 18, 999. [Google Scholar] [CrossRef] [Green Version]
- Duraturo, F.; Liccardo, R.; Izzo, P. Coexistence of MLH3 germline variants in colon cancer patients belonging to families with Lynch syndrome-associated brain tumors. J. Neuro-Oncol. 2016, 129, 577–578. [Google Scholar] [CrossRef]
- Duraturo, F.; Liccardo, R.; Cavallo, A.; De Rosa, M.; Grosso, M.; Izzo, P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: Probability of synergistic effects. Int. J. Cancer 2010, 129, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Maratt, J.K.; Stoffel, E. Identification of Lynch Syndrome. Gastrointest. Endosc. Clin. N. Am. 2022, 32, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2018, 247, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, R.; De Rosa, M.; Izzo, P.; Duraturo, F. Novel MSH2 splice-site mutation in a young patient with Lynch syndrome. Mol. Med. Rep. 2018, 17, 6942–6946. [Google Scholar] [CrossRef] [Green Version]
- Duraturo, F.; Cavallo, A.; Liccardo, R.; Cudia, B.; De Rosa, M.; Diana, G.; Izzo, P. Contribution of Large Genomic Rearrangements in Italian Lynch Syndrome Patients: Characterization of a Novel Alu-Mediated Deletion. BioMed Res. Int. 2012, 2013, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.M. The 10-Mb paracentric inversion of chromosome arm 2p in activating MSH2 and causing hereditary nonpolyposis colorectal cancer: Re-annotation and mutational mechanisms. Genes Chromosomes Cancer 2008, 47, 543–545. [Google Scholar] [CrossRef]
- Peltomäki, P.; Olkinuora, A.; Nieminen, T.T. Updates in the field of hereditary nonpolyposis colorectal cancer. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 707–720. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Nakken, S.; Tubeuf, H.; Vodak, D.; Ekstrøm, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Martins, A.; Møller, P.; et al. Identification of genetic variants for clinical management of familial colorectal tumors. BMC Med Genet. 2018, 19, 1–19. [Google Scholar] [CrossRef]
- Syngal, S.; Fox, E.A.; Li, C.; Dovidio, M.; Eng, C.; Kolodner, R.D.; Garber, J.E. Interpretation of genetic test results for hereditary nonpolyposis colorectal cancer: Implications for clinical predisposition testing. JAMA 1999, 282, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.J.; Rasmussen, L.J.; Hofstra, R.; Monteiro, A.; Greenblatt, M.S.; de Wind, N. Iarc Unclassified Genetic for the IARC Unclassified Genetic Variants Working Group Assessment of functional effects of unclassified genetic variants. Hum. Mutat. 2008, 29, 1314–1326. [Google Scholar] [CrossRef] [Green Version]
- Aretz, S.; Uhlhaas, S.; Sun, Y. Familial adenomatous polyposis: Aberrant splicing due to missense or silent mutations in the APC gene. Hum. Mutat. 2004, 24, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, A.P.; Suspitsin, E.N.; Kuligina, E.S.; Bizin, I.V.; Frishman, D.; Imyanitov, E.N. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015, 369, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.R.; Syngal, S. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology 2015, 149, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Carr, P.; Alwers, E.; Bienert, S.; Weberpals, J.; Kloor, M.; Brenner, H.; Hoffmeister, M. Lifestyle factors and risk of sporadic colorectal cancer by microsatellite instability status: A systematic review and meta-analyses. Ann. Oncol. 2018, 29, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Cheah, P.L.; Li, J.; Looi, L.M.; Koh, C.C.; Lau, T.P.; Chang, S.W.; Teoh, K.H.; Mun, K.S.; Nazarina, A.R. Screening for microsatellite instability in colorectal carcinoma: Practical utility of immunohistochemistry and PCR with fragment analysis in a diagnostic histopathology setting. J. Pathol. 2019, 41, 91–100. [Google Scholar]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.G.; Gulley, M.L. BRAF mutation testing in colorectal cancer. Arch. Pathol. Lab. Med. 2010, 134, 1225–1228. [Google Scholar] [CrossRef]
- van Lier, M.G.; Wagner, A.; van Leerdam, M.E.; Biermann, K.; Kuipers, E.J.; Steyerberg, E.W.; Dubbink, H.J.; Dinjens, W.N. A review on the molecular diagnostics of Lynch syndrome: A central role for the pathology laboratory. J. Cell. Mol. Med. 2010, 14, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Tafe, L.J.; Riggs, E.R.; Tsongalis, G.J. Lynch Syndrome Presenting as Endometrial Cancer. Clin. Chem. 2014, 60, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.H.; Dinh, M.; Kohlmann, W.; Watson, P.; Green, J.; Syngal, S.; Bandipalliam, P.; Chen, L.-M.; Allen, B.; Conrad, P.; et al. Gynecologic Cancer as a “Sentinel Cancer” for Women With Hereditary Nonpolyposis Colorectal Cancer Syndrome. Obstet. Gynecol. 2005, 105, 569–574. [Google Scholar] [CrossRef]
- Staffa, L.; Echterdiek, F.; Nelius, N.; Benner, A.; Werft, W.; Lahrmann, B.; Grabe, N.; Schneider, M.; Tariverdian, M.; von Knebel Doeberitz, M.; et al. Mismatch Repair-Deficient Crypt Foci in Lynch Syndrome–Molecular Alterations and Association with Clinical Parameters. PLOS ONE 2015, 10, e0121980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, R.K.; Dudley, B.; Karloski, E.; Brand, R.E.; O’Callaghan, N.; Rosty, C.; Buchanan, D.D.; Jenkins, M.A.; Thibodeau, S.N.; French, A.J.; et al. DNA mismatch repair protein deficient non-neoplastic colonic crypts: A novel indicator of Lynch syndrome. Mod. Pathol. 2018, 31, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Hui, P.; Buza, N. Frequent loss of mutation-specific mismatch repair protein expression in nonneoplastic endometrium of Lynch syndrome patients. Mod. Pathol. 2020, 33, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, W.J.; Dierssen, J.; Vasen, H.F.; Wijnen, J.T.; Kenter, G.G.; Meijers-Heijboer, H.; Brocker-Vriends, A.; Stormorken, A.; Moller, P.; Menko, F.; et al. Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J. Pathol. 2000, 192, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Tsunoda, H.; Takano, K.; Oki, A.; Yoshikawa, H. Microsatellite Instability and Immunohistochemical Analysis of MLH1 and MSH2 in Normal Endometrium, Endometrial Hyperplasia and Endometrial Cancer from a Hereditary Nonpolyposis Colorectal Cancer Patient. Jpn. J. Clin. Oncol. 2002, 32, 110–112. [Google Scholar] [CrossRef] [Green Version]
- Berends, M.J.; Hollema, H.; Wu, Y.; van der Sluis, T.; Mensink, R.G.; Hoor, K.A.T.; Sijmons, R.; de Vries, E.; Pras, E.; Mourits, M.J.; et al. MLH1 and MSH2 protein expression as a pre-screening marker in hereditary and non-hereditary endometrial hyperplasia and cancer. Int. J. Cancer 2001, 92, 398–403. [Google Scholar] [CrossRef]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57. [Google Scholar]
- Edwards, R.A.; Witherspoon, M.; Wang, K.; Afrasiabi, K.; Pham, T.; Birnbaumer, L.; Lipkin, S.M. Epigenetic Repression of DNA Mismatch Repair by Inflammation and Hypoxia in Inflammatory Bowel Disease–Associated Colorectal Cancer. Cancer Res. 2009, 69, 6423–6429. [Google Scholar] [CrossRef] [Green Version]
- Kondo, A.; Safaei, R.; Mishima, M.; Niedner, H.; Lin, X.; Howell, S.B. Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 2001, 61. [Google Scholar]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased Expression of the DNA Mismatch Repair Gene Mlh1 under Hypoxic Stress in Mammalian Cells. Mol. Cell. Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531, Correction in Nat. Rev. Genet. 2004, 5, 631. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Bandrés, E.; Cubedo, E.; Agirre, X.; Malumbres, R.; Zárate, R.; Ramirez, N.; Abajo, A.; Navarro, A.; Moreno, I.; Monzó, M.; et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol. Cancer 2006, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, E.K.-O.; Chong, W.W.S.; Jin, H.; Lam, E.K.Y.; Shin, V.Y.; Yu, J.; Poon, T.C.W.; Ng, S.S.M.; Sung, J.J.Y. Differential expression of microRNAs in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut 2009, 58, 1375–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Ferracin, M.; Gafà, R.; Veronese, A.; Spizzo, R.; Pichiorri, F.; Liu, C.-G.; Calin, G.A.; Croce, C.M.; Negrini, M. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Mol. Cancer 2007, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of mismatch repair and genomic stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987. [Google Scholar] [CrossRef] [Green Version]
- Liccardo, R.; Sessa, R.; Trombetti, S.; De Rosa, M.; Izzo, P.; Grosso, M.; Duraturo, F. MiR-137 Targets the 3′ Untranslated Region of MSH2: Potential Implications in Lynch Syndrome-Related Colorectal Cancer. Cancers 2021, 13, 4662. [Google Scholar] [CrossRef]
- Sobocińska, J.; Kolenda, T.; Teresiak, A.; Badziąg-Leśniak, N.; Kopczyńska, M.; Guglas, K.; Przybyła, A.; Filas, V.; Bogajewska-Ryłko, E.; Lamperska, K.; et al. Diagnostics of Mutations in MMR/EPCAM Genes and Their Role in the Treatment and Care of Patients with Lynch Syndrome. Diagnostics 2020, 10, 786. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated With Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [Green Version]
- Baglietto, L.; Lindor, N.M.; Dowty, J.; White, D.M.; Wagner, A.; Gomez-Garcia, E.; Vriends, A.H.J.T.; Cartwright, N.R.; Barnetson, R.A.; Farrington, S.M.; et al. Risks of Lynch Syndrome Cancers for MSH6 Mutation Carriers. Gynecol. Oncol. 2010, 102, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The Clinical Phenotype of Lynch Syndrome Due to Germ-Line PMS2 Mutations. Gastroenterology 2008, 135, 419–428.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.; Chung, D.C. Hereditary colon cancer: Lynch syndrome. Gut Liver 2010, 4, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, Y.M.; Wagner, A.; Morreau, H.; Menko, F.; Stormorken, A.; Quehenberger, F.; Sandkuijl, L.; Møller, P.; Genuardi, M.; Van Houwelingen, H.; et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: Impact on counseling and surveillance. Gastroenterology 2004, 127, 17–25. [Google Scholar] [CrossRef] [Green Version]
- ten Broeke, S.W.; Brohet, R.M.; Tops, C.M.; van der Klift, H.M.; Velthuizen, M.E.; Bernstein, I.; Capellá Munar, G.; Gomez Garcia, E.; Hoogerbrugge, N.; Letteboer, T.G.; et al. Lynch syndrome caused by germline PMS2 mutations: Delineating the cancer risk. J. Clin. Oncol. 2015, 33, 319–325. [Google Scholar] [CrossRef]
- Adam, R.; Spier, I.; Zhao, B.; Kloth, M.; Marquez, J.; Hinrichsen, I.; Kirfel, J.; Tafazzoli, A.; Horpaopan, S.; Uhlhaas, S.; et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am. J. Hum. Genet. 2016, 99, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Syngal, S.; E Brand, R.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [Green Version]
- Liccardo, R.; Lambiase, M.; Nolano, A.; De Rosa, M.; Izzo, P.; Duraturo, F. Significance of rare variants in genes involved in the pathogenesis of Lynch syndrome. Int. J. Mol. Med. 2022, 49, 1–14. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, D.-W.; Lee, H.S.; Bang, S.K.; Seo, S.H.; Park, K.U.; Oh, H.-K.; Kang, S.-B. Universal Screening for Lynch Syndrome Compared with Pedigree-Based Screening: 10-Year Experience in a Tertiary Hospital. Cancer Res. Treat. 2022. [Google Scholar] [CrossRef]
- Crain, P.R.; Zepp, J.M.; Gille, S.; Jenkins, L.; Kauffman, T.L.; Shuster, E.; Goddard, K.A.; Wilfond, B.S.; Hunter, J.E. Identifying patients with Lynch syndrome using a universal tumor screening program in an integrated healthcare system. Hered. Cancer Clin. Pr. 2022, 20, 1–9. [Google Scholar] [CrossRef]
- Liccardo, R.; Nolano, A.; Lambiase, M.; Della Ragione, C.; De Rosa, M.; Izzo, P.; Duraturo, F. MSH2 Overexpression Due to an Unclassified Variant in 3′-Untranslated Region in a Patient with Colon Cancer. Biomedicines 2020, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Geurts-Giele, W.R.; Leenen, C.H.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.; Kuipers, E.J.; Goverde, A.; Ouweland, A.M.V.D.; van Lier, M.G.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Roca, A.; Giner-Calabuig, M.; Murcia, O.; Castillejo, A.; Soto, J.L.; García-Heredia, A.; Jover, R. Lynch-like Syndrome: Potential Mechanisms and Management. Cancers 2022, 14, 1115. [Google Scholar] [CrossRef] [PubMed]
- Golubicki, M.; Díaz-Gay, M.; Bonjoch, L.; Franch-Expósito, S.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Carballido, M.; et al. Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers. Cancers 2021, 13, 1259. [Google Scholar] [CrossRef]
- Magrin, L.; Fanale, D.; Brando, C.; Fiorino, A.; Corsini, L.R.; Sciacchitano, R.; Filorizzo, C.; Dimino, A.; Russo, A.; Bazan, V. POLE, POLD1, and NTHL1: The last but not the least hereditary cancer-predisposing genes. Oncogene 2021, 40, 5893–5901. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Syndrome | Genes | Hereditary | Incidence | Lifetime crc Risk |
|---|---|---|---|---|
| Lynch Syndrome LS | MLH1, MSH2, MSH6, PMS2, EPCAM | AD | 3–5% | 15–90% |
| Familial Adenomatous Polyposis FAP | APC | AD | 1% | Classic forms 100%; Attenuated forms until 70% |
| Mutyh-Associated Polyposis MAP | MUTYH | AR | 1% | 43–99% |
| Peutz-jeghers Syndrome PJS | STK11 | AD | <1% | 39% |
| Juvenile Polyposis JPS | SMAD4, BMPR1A | AD | <1% | 39–69% |
| Bacterial MMR System | Yeast MMR System | Human MMR System | Functions |
|---|---|---|---|
| MutS | MutSα (MSH2/MSH6) MutSβ (MSH2/MSH3) | MutSα (MSH2/MSH6) MutSβ (MSH2/MSH3) | Mismatch recognition |
| MUTL | MutLα (MLH1/PMS1) MutLβ (MLH1/MLH2) MutLγ (MLH1/MLH3) | MutLα (MLH1/PMS2) MutLβ (MLH1/PMS1) MutLγ (MLH1/MLH3) | Match making |
| MutH | PCNA | PCNA | Strand incision |
| RFC | RFC | ||
| MutLα (MLH1/PMS1) MutLγ (MLH1/MLH3) | MutLα (MLH1/PMS2) MutLγ (MLH1/MLH3) | ||
| RecJ | EXO1 | EXO1 | Strand excision (exonuclease) |
| ExoI | |||
| ExoVII | |||
| ExoX | |||
| UvrD | - | - | Strand excision (helicase) |
| DNA polymerase III | DNA polymerase δ | DNA polymerase δ | Repair synthesis |
| Direct Genetic Evidence | Indirect Genetic Evidence |
|---|---|
| Co-segregation with the disease | In silico predictions based on the position and nature of the amminoacid change |
| Co-occurrence with known pathogenic mutations | In vitro functional assays |
| Comparison of allele frequency in cases and controls | Biochemical assays for protein characterization |
| LOH and methylation analysis in vivo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolano, A.; Medugno, A.; Trombetti, S.; Liccardo, R.; De Rosa, M.; Izzo, P.; Duraturo, F. Hereditary Colorectal Cancer: State of the Art in Lynch Syndrome. Cancers 2023, 15, 75. https://doi.org/10.3390/cancers15010075
Nolano A, Medugno A, Trombetti S, Liccardo R, De Rosa M, Izzo P, Duraturo F. Hereditary Colorectal Cancer: State of the Art in Lynch Syndrome. Cancers. 2023; 15(1):75. https://doi.org/10.3390/cancers15010075
Chicago/Turabian StyleNolano, Antonio, Alessia Medugno, Silvia Trombetti, Raffaella Liccardo, Marina De Rosa, Paola Izzo, and Francesca Duraturo. 2023. "Hereditary Colorectal Cancer: State of the Art in Lynch Syndrome" Cancers 15, no. 1: 75. https://doi.org/10.3390/cancers15010075
APA StyleNolano, A., Medugno, A., Trombetti, S., Liccardo, R., De Rosa, M., Izzo, P., & Duraturo, F. (2023). Hereditary Colorectal Cancer: State of the Art in Lynch Syndrome. Cancers, 15(1), 75. https://doi.org/10.3390/cancers15010075