
Adoptive Cell Therapy for T-Cell Malignancies
Abstract
:Simple Summary
Abstract
1. T-Cell Malignancies
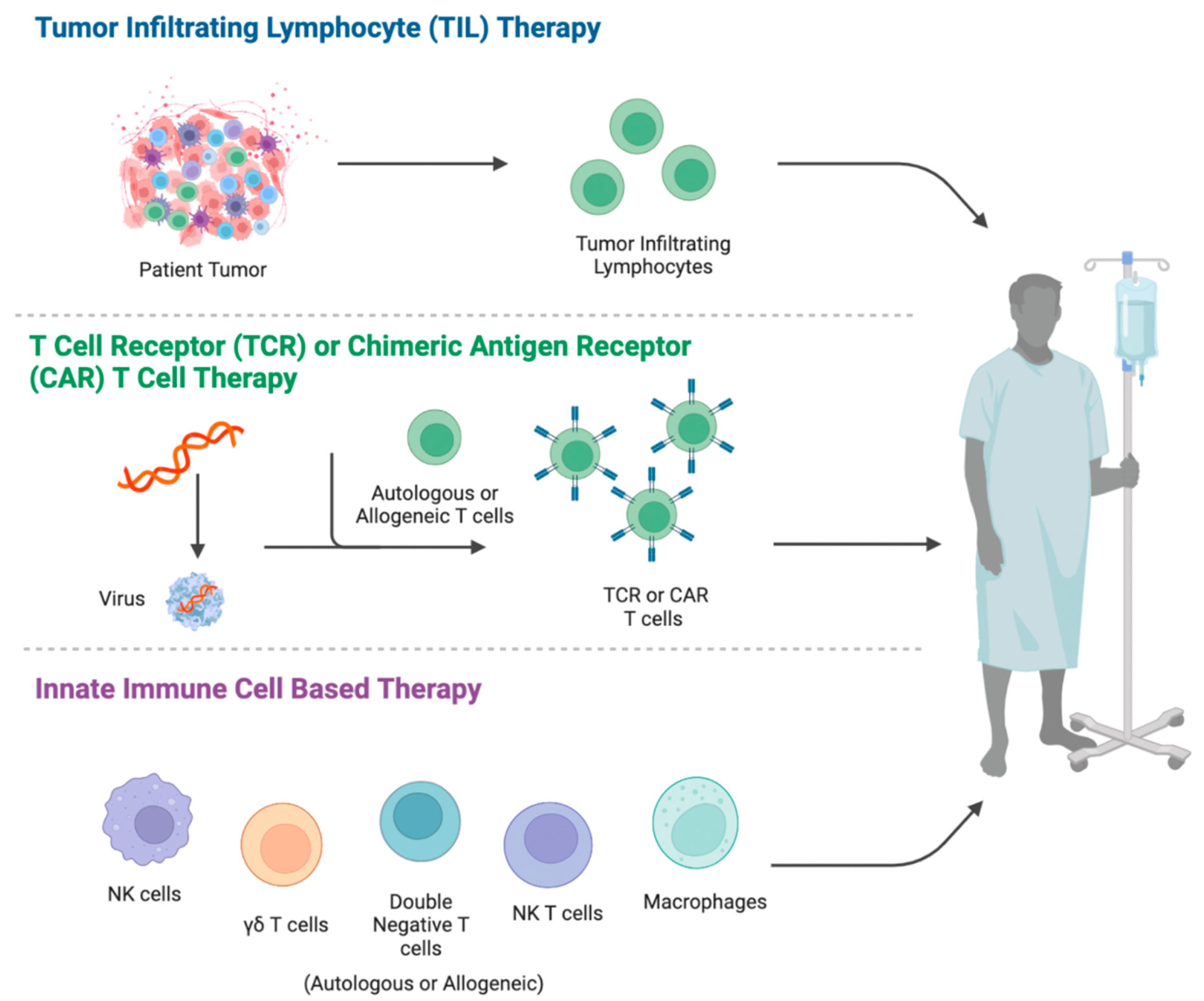
2. Adoptive Cell Therapy
3. Targeting T-Cell Malignancy by CAR-T Therapy
3.1. Challenges
3.2. Solutions
3.3. Clinical Trials
4. Targeting T-Cell Malignancies by Innate Immune Cells
4.1. NK Cells
4.2. γδ T Cells
4.3. DNT Cells
5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cordó, V.; van der Zwet, J.; Canté-Barrett, K.; Pieters, R.; Meijerink, J. T-cell Acute Lymphoblastic Leukemia: A Roadmap to Targeted Therapies. Blood Cancer Discov. 2021, 2, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Pui, C.-H.; Kantarjian, H. Progress and Innovations in the Management of Adult Acute Lymphoblastic Leukemia. JAMA Oncol. 2018, 4, 1413–1420. [Google Scholar] [CrossRef]
- Bellei, M.; Foss, F.M.; Shustov, A.R.; Horwitz, S.M.; Marcheselli, L.; Kim, W.S.; Cabrera, M.E.; Dlouhy, I.; Nagler, A.; Advani, R.H.; et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: A report from the prospective, International T-Cell Project. Haematologica 2018, 103, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- DembiĆ, Z.; Haas, W.; Weiss, S.; McCubrey, J.; Kiefer, H.; von Boehmer, H.; Steinmetz, M. Transfer of specificity by murine α and β T-cell receptor genes. Nature 1986, 320, 232–238. [Google Scholar] [CrossRef]
- Cooper, L.J.N.; Kalos, M.; Lewinsohn, D.A.; Riddell, S.R.; Greenberg, P.D. Transfer of Specificity for Human Immunodeficiency Virus Type 1 into Primary Human T Lymphocytes by Introduction of T-Cell Receptor Genes. J. Virol. 2000, 74, 8207–8212. [Google Scholar] [CrossRef] [Green Version]
- Kessels, H.; Wolkers, M.; Boom, M.D.V.D.; Valk, M.A.V.D.; Schumacher, T. Immunotherapy through TCR gene transfer. Nat. Immunol. 2001, 2, 957–961. [Google Scholar] [CrossRef]
- Clay, T.M.; Custer, M.C.; Sachs, J.; Hwu, P.; Rosenberg, S.A.; Nishimura, M.I. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J. Immunol. 1999, 163, 507–513. [Google Scholar]
- He, Q.; Jiang, X.; Zhou, X.; Weng, J. Targeting cancers through TCR-peptide/MHC interactions. J. Hematol. Oncol. 2019, 12, 139. [Google Scholar] [CrossRef]
- Biernacki, M.A.; Brault, M.; Bleakley, M. T-Cell Receptor–Based Immunotherapy for Hematologic Malignancies. Cancer J. 2019, 25, 179–190. [Google Scholar] [CrossRef]
- Krakow, M.E.F.; Summers, C.; Dahlberg, A.; Bar, M.; Biernacki, M.A.; Cunningham, T.; Vartanian, N.; Hickner, M.; Chaney, C.; Habtetsion, T.; et al. Phase I Study of Adoptive Immunotherapy with HA-1-Specific CD8+ and CD4+ Memory T Cells for Children and Adults with Relapsed Acute Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation (HCT): Trial in Progress. Blood 2020, 136 (Suppl. 1), 45–46. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 1533033819831068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef] [PubMed]
- Kozani, P.S.; Kozani, P.S.; Rahbarizadeh, F. CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res. Ther. 2021, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Scherer, L.D.; Brenner, M.K.; Mamonkin, M. Chimeric Antigen Receptors for T-Cell Malignancies. Front. Oncol. 2019, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, L.C.; Spencer, H.T.; Raikar, S.S. Targeting T cell malignancies using CAR-based immunotherapy: Challenges and potential solutions. J. Hematol. Oncol. 2019, 12, 527. [Google Scholar] [CrossRef] [Green Version]
- Alcantara, M.; Tesio, M.; June, C.H.; Houot, R. CAR T-cells for T-cell malignancies: Challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia 2018, 32, 2307–2315. [Google Scholar] [CrossRef]
- Polgárová, K.; Otáhal, P.; Šálek, C.; Pytlík, R. Chimeric Antigen Receptor Based Cellular Therapy for Treatment Of T-Cell Malignancies. Front. Oncol. 2022, 12, 876758. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Liu, Y.; Yang, J.; Zhang, X.; Yang, X.; Wang, H.; Wang, L.; Wang, Q.; Jin, D.; Li, J.; et al. Naturally Selected CD7 CAR-T Therapy without Genetic Manipulations for T-ALL/LBL: First-in-human Phase I Clinical Trial. Blood 2022, 140, 321–334. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, D.; Fu, X.; Meng, H.; Nan, F.; Sun, Z.; Yu, H.; Zhang, L.; Li, L.; Li, X.; et al. Autologous Nanobody-Derived Fratricide-Resistant CD7-CAR T-cell Therapy for Patients with Relapsed and Refractory T-cell Acute Lymphoblastic Leukemia/Lymphoma. Clin. Cancer Res. 2022, 28, 2830–2843. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017, 1, 2348–2360. [Google Scholar] [CrossRef] [Green Version]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible Transgene Expression Reduces Fratricide and Permits 4-1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol. Res. 2018, 6, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martínez, D.; Baroni, M.L.; Gutiérrez-Agüera, F.; Roca-Ho, H.; Blanch-Lombarte, O.; González-García, S.; Torrebadell, M.; Junca, J.; Ramírez-Orellana, M.; Velasco-Hernández, T.; et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood 2019, 133, 2291–2304. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, D.; Lu, Y.; Dong, S.-C.; Ma, R.; Tang, W.-Y.; Wu, J.-Q.; Feng, J.-F. A new immunotherapy strategy targeted CD30 in peripheral T-cell lymphomas: CAR-modified T-cell therapy based on CD30 mAb. Cancer Gene Ther. 2021, 29, 167–177. [Google Scholar] [CrossRef]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; Van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Georgiadis, C.; Preece, R.; Mock, U.; Qasim, W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. J. Clin. Investig. 2018, 3, e99442. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef]
- Ma, G.; Shen, J.; Pinz, K.; Wada, M.; Park, J.; Kim, S.; Togano, T.; Tse, W. Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell Rev. Rep. 2019, 15, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Alexey, S.; Li, J.; Jones, T.; Grande, G.; Douthit, L.; Xie, J.; Chen, D.; Wu, X.; Michael, M.; et al. Unique CDR3 epitope targeting by CAR-T cells is a viable approach for treating T-cell malignancies. Leukemia 2019, 33, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, P.; Wawrzyniecka, P.A.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Legut, M.; Cole, D.; Sewell, A.K.; Gritti, G.; et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017, 23, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.S.; Guevara, C.I.; Jin, L.; Mbong, N.; Verlinsky, A.; Hsu, S.J.; Aviña, H.; Karki, S.; Abad, J.D.; Yang, P.; et al. AGS67E, an Anti-CD37 Monomethyl Auristatin E Antibody–Drug Conjugate as a Potential Therapeutic for B/T-Cell Malignancies and AML: A New Role for CD37 in AML. Mol. Cancer Ther. 2015, 14, 1650–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehues, T.; Kapaun, P.; Harms, D.; Burdach, S.; Kramm, C.; Körholz, D.; Janka-Schaub, G.; Göbel, U. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia 1999, 13, 614–617. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.; Hansen-Hagge, T.E.; Drexler, H.G.; Gramatzki, M. Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: Suggestion for classification by immunophenotype and T-cell receptor studies. Leuk. Res. 1998, 23, 19–27. [Google Scholar] [CrossRef]
- Falini, B.; Pileri, S.; Pizzolo, G.; Durkop, H.; Flenghi, L.; Stirpe, F.; Martelli, M.; Stein, H. CD30 (Ki-1) molecule: A new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995, 85, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Mo, F.; Zheng, R.; Ma, R.; Bray, V.C.; van Leeuwen, D.G.; Sritabal-Ramirez, J.; Hu, H.; Wang, S.; Mehta, B.; et al. Feasibility and preclinical efficacy of CD7-unedited CD7 CAR T cells for T cell malignancies. Mol. Ther. 2022, 30, 1343–1351. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Benjamin, R.; Jain, N.; Maus, M.V.; Boissel, N.; Graham, C.; Jozwik, A.; Yallop, D.; Konopleva, M.; Frigault, M.J.; Teshima, T.; et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): A phase 1, dose-escalation trial. Lancet Haematol. 2022, 9, e833–e843. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- D’Orsogna, L.J.A.; Roelen, D.L.; Doxiadis, I.I.N.; Claas, F.H.J. Alloreactivity from human viral specific memory T-cells. Transpl. Immunol. 2010, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Houghtelin, A.; Bollard, C.M. Virus-Specific T Cells for the Immunocompromised Patient. Front. Immunol. 2017, 8, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, C.; Wang, Y.; Reppel, L.; D’Aveni, M.; Campidelli, A.; Decot, V.; Bensoussan, D. Viral-specific T-cell transfer from HSCT donor for the treatment of viral infections or diseases after HSCT. Bone Marrow Transplant. 2017, 53, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Rooney, C.; Leen, A. Moving Successful Virus-specific T-cell Therapy for Hematopoietic Stem Cell Recipients to Late Phase Clinical Trials. Mol. Ther.-Nucleic Acids 2012, 1, e55. [Google Scholar] [CrossRef]
- Leen, A.M.; Tripic, T.; Rooney, C.M. Challenges of T cell therapies for virus-associated diseases after hematopoietic stem cell transplantation. Expert Opin. Biol. Ther. 2010, 10, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2016, 27, 154–157. [Google Scholar] [CrossRef]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.-A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Carosella, E.D.; Rouas-Freiss, N.; Roux, D.T.-L.; Moreau, P.; LeMaoult, J. HLA-G: An Immune Checkpoint Molecule. Adv. Immunol. 2015, 127, 33–144. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, F.; Watanabe, N.; McKenna, M.K.; Hicks, M.J.; Srinivasan, M.; Gomes-Silva, D.; Atilla, E.; Smith, T.; Atilla, P.A.; Ma, R.; et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol. 2020, 39, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sherwood, A.; Fromm, J.R.; Winter, S.S.; Dunsmore, K.P.; Loh, M.L.; Greisman, H.A.; Sabath, D.E.; Wood, B.L.; Robins, H. High-Throughput Sequencing Detects Minimal Residual Disease in Acute T Lymphoblastic Leukemia. Sci. Transl. Med. 2012, 4, 134ra63. [Google Scholar] [CrossRef] [PubMed]
- Grover, N.S.; Ivanova, A.; Moore, D.T.; Cheng, C.J.A.; Babinec, C.; West, J.; Cavallo, T.; Morrison, J.K.; Buchanan, F.B.; Bowers, E.; et al. CD30-Directed CAR-T Cells Co-Expressing CCR4 in Relapsed/Refractory Hodgkin Lymphoma and CD30+ Cutaneous T Cell Lymphoma. Blood 2021, 138, 742. [Google Scholar] [CrossRef]
- Riches, M.L.; Shea, T.C.; Ivanova, A.; Cheng, C.; Laing, S.; Seegars, M.B.; Dotti, G.; Savoldo, B.; Grover, N.S.; Serody, J.S. Infusion of CD30 CAR T Cells Is Safe and Effective As Consolidation Following Autologous Hematopoietic Stem Cell Transplant. Transplant. Cell. Ther. 2021, 27, S65–S66. [Google Scholar] [CrossRef]
- Ying, Z.-T.; Chang, L.-J.; Kuo, H.-H.; Liu, Y.-C.; Song, Y.-Q.; Wang, X.-P.; Liu, W.-P.; Zheng, W.; Xie, Y.; Lin, N.-J.; et al. 415. First-In-Patient Proof of Safety and Efficacy of a 4th Generation Chimeric Antigen Receptor-Modified T Cells for the Treatment of Relapsed or Refractory CD30 Positive Lymphomas. Mol. Ther. 2015, 23, S164. [Google Scholar] [CrossRef]
- Quach, D.H.; Ramos, C.A.; Lulla, P.D.; Sharma, S.; Ganesh, H.R.; Hadidi, Y.F.; Thakkar, S.G.; Becerra-Dominguez, L.; Mehta, B.; Perconti, S.; et al. Safety and Efficacy of Off-the-Shelf CD30.CAR-Modified Epstein-Barr Virus-Specific T Cells in Patients with CD30-Positive Lymphoma. Blood 2021, 138 (Suppl. 1), 1763. [Google Scholar] [CrossRef]
- Feng, J.; Xu, H.; Cinquina, A.; Wu, Z.; Zhang, W.; Sun, L.; Chen, Q.; Tian, L.; Song, L.; Pinz, K.G.; et al. Treatment of aggressive T-cell lymphoma/leukemia with anti-CD4 CAR T cells. Front. Immunol. 2022, 13, 997482. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.; Rouce, R.H.; Smith, T.S.; Yang, L.; Srinivasan, M.; Zhang, H.; Perconti, S.; Mehta, B.; Dakhova, O.; Randall, J.; et al. CD5 CAR T-Cells for Treatment of Patients with Relapsed/Refractory CD5 Expressing T-Cell Lymphoma Demonstrates Safety and Anti-Tumor Activity. Biol. Blood Marrow Transplant. 2020, 26, S237. [Google Scholar] [CrossRef]
- Feng, J.; Xu, H.; Cinquina, A.; Wu, Z.; Chen, Q.; Zhang, P.; Wang, X.; Shan, H.; Xu, L.; Zhang, Q.; et al. Treatment of Aggressive T Cell Lymphoblastic Lymphoma/leukemia Using Anti-CD5 CAR T Cells. Stem Cell Rev. Rep. 2021, 17, 652–661. [Google Scholar] [CrossRef]
- Pan, J.; Tan, Y.; Shan, L.; Deng, B.; Ling, Z.; Song, W.; Feng, X.; Hu, G. Phase I study of donor-derived CD5 CAR T cells in patients with relapsed or refractory T-cell acute lymphoblastic leukemia. J. Clin. Oncol. 2022, 40, 7028. [Google Scholar] [CrossRef]
- Zhao, L.; Pan, J.; Tang, K.; Tan, Y.; Deng, B.; Ling, Z.; Song, W.; Chang, A.H.; Feng, X. Autologous CD7-targeted CAR T-cell therapy for refractory or relapsed T-cell acute lymphoblastic leukemia/lymphoma. J. Clin. Oncol. 2022, 40, 7035. [Google Scholar] [CrossRef]
- Yang, J.; Yang, X.; Liu, Y.; Wang, Q.; Wang, H.; Li, J.; Lu, P. A Novel and Successful Patient or Donor-Derived CD7-Targeted CAR T-Cell Therapy for Relapsed or Refractory T-Cell Lymphoblastic Lymphoma (R/R T-LBL). Blood 2021, 138 (Suppl. 1), 652. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, W.; Li, C.; Du, M.; Zhou, F.; Tang, L.; Wu, J.; Jiang, H.; Wei, Q.; Lu, C.; et al. P1466: CD7 Chimeric Antigen Receptor T Cells for Adult Patients with Refractory and Relapsed T Cell Malignancies. HemaSphere 2022, 6, 1348–1349. [Google Scholar] [CrossRef]
- Pan, J.; Tan, Y.; Wang, G.; Deng, B.; Ling, Z.; Song, W.; Seery, S.; Zhang, Y.; Peng, S.; Xu, J.; et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J. Clin. Oncol. 2021, 39, 3340–3351. [Google Scholar] [CrossRef]
- Ramos, C.A.; Ballard, B.; Zhang, H.; Dakhova, O.; Gee, A.P.; Mei, Z.; Bilgi, M.; Wu, M.-F.; Liu, H.; Grilley, B.; et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor–redirected lymphocytes. J. Clin. Investig. 2017, 127, 3462–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J. T Cells Expressing a Fully-Human Anti-CD30 Chimeric Antigen Receptor for Treating CD30-Expressing Lymphomas. Available online: https://www.clinicaltrials.gov/ (accessed on 25 May 2022).
- Cwynarski, K.; Iacoboni, G.; Tholouli, E.; Menne, T.F.; Irvine, D.A.; Balasubramaniam, N.; Wood, L.; Shang, J.; Zhang, Y.; Basilico, S.; et al. First in Human Study of AUTO4, a TRBC1-Targeting CAR T-Cell Therapy in Relapsed/Refractory TRBC1-Positive Peripheral T-Cell Lymphoma. In Proceedings of the 64th Annual American Society of Hematology (ASH) Meeting and Exposition, New Orleans, LA, USA, 10–13 December 2022. [Google Scholar]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Venstrom, J.M.; Pittari, G.; Gooley, T.A.; Chewning, J.H.; Spellman, S.; Haagenson, M.; Gallagher, M.M.; Malkki, M.; Petersdorf, E.; Dupont, B.; et al. HLA-C–Dependent Prevention of Leukemia Relapse by Donor Activating KIR2DS1. N. Engl. J. Med. 2012, 367, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.J.; Ritz, J. Induction of Tumor Immunity Following Allogeneic Stem Cell Transplantation. Adv. Immunol. 2006, 90, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Impola, U.; Eturpeinen, H.; Ealakulppi, N.; Elinjama, T.; Volin, L.; Eniittyvuopio, R.; Partanen, J.; Ekoskela, S. Donor Haplotype B of NK KIR Receptor Reduces the Relapse Risk in HLA-Identical Sibling Hematopoietic Stem Cell Transplantation of AML Patients. Front. Immunol. 2014, 5, 405. [Google Scholar] [CrossRef] [Green Version]
- Leung, W.; Iyengar, R.; Turner, V.; Lang, P.; Bader, P.; Conn, P.; Niethammer, D.; Handgretinger, R. Determinants of Antileukemia Effects of Allogeneic NK Cells. J. Immunol. 2003, 172, 644–650. [Google Scholar] [CrossRef] [Green Version]
- Duault, C.; Kumar, A.; Khani, A.T.; Lee, S.J.; Yang, L.; Huang, M.; Hurtz, C.; Manning, B.; Ghoda, L.Y.; McDonald, T.; et al. Activated natural killer cells predict poor clinical prognosis in high-risk B- and T-cell acute lymphoblastic leukemia. Blood 2021, 138, 1465–1480. [Google Scholar] [CrossRef] [PubMed]
- Elim, O.; Ejung, M.Y.; Ehwang, Y.K.; Eshin, E.-C. Present and Future of Allogeneic Natural Killer Cell Therapy. Front. Immunol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [Green Version]
- Pinz, K.G.; Yakaboski, E.; Jares, A.; Liu, H.; Firor, A.E.; Chen, K.H.; Wada, M.; Salman, H.; Tse, W.; Hagag, N.; et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget 2017, 8, 112783–112796. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.H.; Wada, M.; Pinz, K.G.; Liu, H.; Lin, K.-W.; Jares, A.; Firor, A.E.; Shuai, X.; Salman, H.; Golightly, M.; et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia 2017, 31, 2151–2160. [Google Scholar] [CrossRef] [Green Version]
- Raikar, S.S.; Fleischer, L.C.; Moot, R.; Fedanov, A.; Paik, N.Y.; Knight, K.A.; Doering, C.B.; Spencer, H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology 2017, 7, e1407898. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Ghobadi, A.; Patel, K.; Park, J.H.; Flinn, I.W.; Shah, P.; Wong, C.; Bickers, C.; Szabo, P.; Wong, L.; et al. Safety and Efficacy of FT596, a First-in-Class, Multi-Antigen Targeted, Off-the-Shelf, iPSC-Derived CD19 CAR NK Cell Therapy in Relapsed/Refractory B-Cell Lymphoma. Blood 2021, 138 (Suppl. 1), 823. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.M.; Becker-Hapak, M.; Cashen, A.F.; Jacobs, M.T.; Wong, P.; Foster, M.; McClain, E.; Desai, S.; Pence, P.; Cooley, S.; et al. Systemic IL-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood 2022, 139, 1177–1183. [Google Scholar] [CrossRef]
- Hoerster, K.; Uhrberg, M.; Wiek, C.; Horn, P.A.; Hanenberg, H.; Heinrichs, S. HLA Class I Knockout Converts Allogeneic Primary NK Cells Into Suitable Effectors for “Off-the-Shelf” Immunotherapy. Front. Immunol. 2021, 11, 586168. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H. Challenges of cancer therapy with natural killer cells. Cytotherapy 2015, 17, 245–249. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef]
- Koepsell, S.A.; Miller, J.S.; McKenna, D.H., Jr. Natural killer cells: A review of manufacturing and clinical utility. Transfusion 2013, 53, 404–410. [Google Scholar] [CrossRef]
- Becker, P.S.A.; Suck, G.; Nowakowska, P.; Ullrich, E.; Seifried, E.; Bader, P.; Tonn, T.; Seidl, C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol. Immunother. 2016, 65, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Luevano, M.; Daryouzeh, M.; Alnabhan, R.; Querol, S.; Khakoo, S.; Madrigal, A.; Saudemont, A. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum. Immunol. 2012, 73, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Cai, L.; Hu, Y.; Wang, H. Cord-Blood Natural Killer Cell-Based Immunotherapy for Cancer. Front. Immunol. 2020, 11, 584099. [Google Scholar] [CrossRef] [PubMed]
- Tarn, Y.; Martinson, J.; Doligosa, K.; Klingernann, H.-G. Ex vivo expansion of the highly cytotoxic human natural killer cell line NK-92 under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy 2003, 5, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Hodge, J.W. The emerging role of off-the-shelf engineered natural killer cells in targeted cancer immunotherapy. Mol. Ther.-Oncolytics 2021, 23, 266–276. [Google Scholar] [CrossRef]
- Klingemann, H.G.; Wong, E.; Maki, G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 1996, 2, 68–75. [Google Scholar]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Hor, P.; Kaufman, D.S. iPSC-Derived Natural Killer Cell Therapies —Expansion and Targeting. Front. Immunol. 2022, 13, 217. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. Ebiomedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Borges, L.; Wallet, M.A.; Bullaughey, C.-L.; Naso, M.F.; Gurung, B.; Keating, S.; Carton, J.M.; Wheeler, J.C.; Campion, L.; Mendonca, M.; et al. Development of Multi-Engineered iPSC-Derived CAR-NK Cells for the Treatment of B-Cell Malignancies. Blood 2021, 138 (Suppl. 1), 1729. [Google Scholar] [CrossRef]
- Shankar, K.; Capitini, C.M.; Saha, K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res. Ther. 2020, 11, 234. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Schneider, D.; Pfeifer, R.; Moeker, N.; Verhoeyen, E.; et al. A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.; Wagner, J.A.; Fehniger, T.A. Human Cytokine-Induced Memory-Like Natural Killer Cells. J. Innate Immun. 2015, 7, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Fehniger, T.A.; Cooper, M.A. Harnessing NK Cell Memory for Cancer Immunotherapy. Trends Immunol. 2016, 37, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Goodman, T.; Lefrançois, L. Expression of the γ-δ T-cell receptor on intestinal CD8+ intraepithelial lymphocytes. Nature 1988, 333, 855–858. [Google Scholar] [CrossRef]
- Deusch, K.; Lüling, F.; Reich, K.; Classen, M.; Wagner, H.; Pfeffer, K. A major fraction of human intraepithelial lymphocytes simultaneously expresses the γ/δ T cell receptor, the CD8 accessory molecule and preferentially uses the Vδ1 gene segment. Eur. J. Immunol. 1991, 21, 1053–1059. [Google Scholar] [CrossRef]
- Kalyan, S.; Kabelitz, D. Defining the nature of human γδ T cells: A biographical sketch of the highly empathetic. Cell. Mol. Immunol. 2012, 10, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Holtmeier, W.; Kabelitz, D. γδ T Cells Link Innate and Adaptive Immune Responses. In Mechanisms of Epithelial Defense; KARGER: Basel, Switzerland, 2005; pp. 151–183. [Google Scholar] [CrossRef]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Tanaka, Y.; Harazaki, M.; Mikami, B.; Minato, N. Recognition mechanism of non-peptide antigens by human T cells. Int. Immunol. 2003, 15, 1301–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; El-Diwany, R.; Schaffert, S.; Adams, E.J.; Garcia, K.C.; Pereira, P.; Chien, Y.-H. Antigen Recognition Determinants of γδ T Cell Receptors. Science 2005, 308, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, G.; Zhang, J.; Wu, X.; Chen, X. The Dual Roles of Human γδ T Cells: Anti-Tumor or Tumor-Promoting. Front. Immunol. 2021, 11, 619954. [Google Scholar] [CrossRef] [PubMed]
- Latha, T.S.; Reddy, M.; Durbaka, P.V.R.; Rachamallu, A.; Pallu, R.; Lomada, D. γδ T Cell-Mediated Immune Responses in Disease and Therapy. Front. Immunol. 2014, 5, 571. [Google Scholar] [CrossRef] [Green Version]
- Correia, D.V.; Lopes, A.C.; Silva-Santos, B. Tumor cell recognition by γδ T lymphocytes. OncoImmunology 2013, 2, e22892. [Google Scholar] [CrossRef] [Green Version]
- Legut, M.; Cole, D.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. γδ T cells: Pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigné, C.-M.; Mönkkönen, H.; Mönkkönen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Simões, A.E.; Di Lorenzo, B.; Silva-Santos, B. Molecular Determinants of Target Cell Recognition by Human γδ T Cells. Front. Immunol. 2018, 9, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, D.; Fogli, M.; Hudspeth, K.; da Silva, M.G.; Mavilio, D.; Silva-Santos, B. Differentiation of human peripheral blood Vδ1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 2011, 118, 992–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuyama, H.; Hagi, T.; Mattarollo, S.R.; Morley, J.; Wang, Q.; Fai-So, H.; Moriyasu, F.; Nieda, M.; Nicol, A.J. Vγ9Vδ2 T cell cytotoxicity against tumor cells is enhanced by monoclonal antibody drugs—Rituximab and trastuzumab. Int. J. Cancer 2008, 122, 2526–2534. [Google Scholar] [CrossRef]
- Couzi, L.; Pitard, V.; Sicard, X.; Garrigue, I.; Hawchar, O.; Merville, P.; Moreau, J.-F.; Déchanet-Merville, J. Antibody-dependent anti-cytomegalovirus activity of human γδ T cells expressing CD16 (FcγRIIIa). Blood 2012, 119, 1418–1427. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Q.; Peng, H.; Cheng, R.; Sun, Z.; Ye, Z. IFN-γ enhances HOS and U2OS cell lines susceptibility to γδ T cell-mediated killing through the Fas/Fas ligand pathway. Int. Immunopharmacol. 2011, 11, 496–503. [Google Scholar] [CrossRef]
- Hao, J.; Wu, X.; Xia, S.; Li, Z.; Wen, T.; Zhao, N.; Wu, Z.; Wang, P.; Zhao, L.; Yin, Z. Current progress in γδ T-cell biology. Cell. Mol. Immunol. 2010, 7, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Brandes, M.; Willimann, K.; Moser, B. Professional Antigen-Presentation Function by Human γδ T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Khan, M.W.A.; Curbishley, S.M.; Chen, H.-C.; Thomas, A.D.; Epircher, H.; Mavilio, D.; Steven, N.M.; Eberl, M.; Emoser, B. Expanded Human Blood-Derived γδT Cells Display Potent Antigen-Presentation Functions. Front. Immunol. 2014, 5, 344. [Google Scholar] [CrossRef] [Green Version]
- Gertner-Dardenne, J.; Castellano, R.; Mamessier, E.; Garbit, S.; Kochbati, E.; Etienne, A.; Charbonnier, A.; Collette, Y.; Vey, N.; Olive, D. Human Vγ9Vδ2 T Cells Specifically Recognize and Kill Acute Myeloid Leukemic Blasts. J. Immunol. 2012, 188, 4701–4708. [Google Scholar] [CrossRef] [Green Version]
- Sicard, H.; Al Saati, T.; Delsol, G.; Fournié, J.-J. Synthetic Phosphoantigens Enhance Human Vγ9Vδ2 T Lymphocytes Killing of Non-Hodgkin’s B Lymphoma. Mol. Med. 2001, 7, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Chargui, J.; Combaret, V.; Scaglione, V.; Iacono, I.; Péri, V.; Valteau-Couanet, D.; Dubrel, M.; Angevin, E.; Puisieux, A.; Romagne, F.; et al. Bromohydrin Pyrophosphate-stimulated Vγ9δ2 T Cells Expanded Ex Vivo from Patients With Poor-Prognosis Neuroblastoma Lyse Autologous Primary Tumor Cells. J. Immunother. 2010, 33, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, F.; Pende, D.; Burgio, V.L.; Castelli, C.; Spada, M.; Venditti, M.; Luciani, F.; Lugini, L.; Federici, C.; Ramoni, C.; et al. Effect Of Human Natural Killer and γδ T Cells on the Growth of Human Autologous Melanoma Xenografts in SCID Mice. Cancer Res. 2004, 64, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, N.L.; Suarez-Cuervo, C.; Gillespie, G.Y.; Markert, J.M.; Nabors, L.B.; Meleth, S.; Lopez, R.D.; Lamb, L.S., Jr. Characterization and immunotherapeutic potential of γδ T-cells in patients with glioblastoma. Neuro-Oncology 2009, 11, 357–367. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infil-trating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Weissinger, F.; Tony, H.P.; Wilhelm, M. Stimulation of gammadelta T cells by aminobisphos-phonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2019, 19, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the Efficiency of Vγ9Vδ2 T-Cell Immunotherapy in Cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef] [Green Version]
- Saura-Esteller, J.; de Jong, M.; King, L.A.; Ensing, E.; Winograd, B.; de Gruijl, T.D.; Parren, P.W.H.I.; van der Vliet, H.J. Gamma Delta T-Cell Based Cancer Immunotherapy: Past-Present-Future. Front. Immunol. 2022, 13, 2649. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, X.; Liang, S.; Luo, H.; Alnaggar, M.; Liu, A.; Yin, Z.; Chen, J.; Niu, L.; Jiang, Y. Irreversible electroporation plus allogenic Vγ9Vδ2 T cells enhances antitumor effect for locally advanced pancreatic cancer patients. Signal Transduct. Target. Ther. 2020, 5, 215. [Google Scholar] [CrossRef]
- Xu, Y.; Xiang, Z.; Alnaggar, M.; Kouakanou, L.; Li, J.; He, J.; Yang, J.; Hu, Y.; Chen, Y.; Lin, L.; et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell. Mol. Immunol. 2020, 18, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.; Willcox, C.R.; Joyce, S.P.; Ladell, K.; Kasatskaya, S.; McLaren, J.; Hunter, S.; Salim, M.; Mohammed, F.; Price, D.; et al. Clonal selection in the human Vδ1 T cell repertoire indicates γδ TCR-dependent adaptive immune surveillance. Nat. Commun. 2017, 8, 14760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.R.; Correia, D.V.; Fernandes-Platzgummer, A.; da Silva, C.L.; da Silva, M.G.; Anjos, D.R.; Silva-Santos, B. Delta One T Cells for Immunotherapy of Chronic Lymphocytic Leukemia: Clinical-Grade Expansion/Differentiation and Preclinical Proof of Concept. Clin. Cancer Res. 2016, 22, 5795–5804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferry, G.M.; Agbuduwe, C.; Forrester, M.; Dunlop, S.; Chester, K.; Fisher, J.; Anderson, J.; Barisa, M. A Simple and Robust Single-Step Method for CAR-Vδ1 γδT Cell Expansion and Transduction for Cancer Immunotherapy. Front. Immunol. 2022, 13, 2318. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, B.; Simões, A.E.; Caiado, F.; Tieppo, P.; Correia, D.V.; Carvalho, T.; da Silva, M.G.; Déchanet-Merville, J.; Schumacher, T.N.; Prinz, I.; et al. Broad Cytotoxic Targeting of Acute Myeloid Leukemia by Polyclonal Delta One T Cells. Cancer Immunol. Res. 2019, 7, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells Expressing Polyclonal Repertoire of Endogenous γδ T-cell Receptors and Introduced CD19-specific Chimeric Antigen Receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, K.P.; Barca, T.; Azameera, A.; Makkouk, A.; Romero, J.M.; Bai, L.; Brodey, M.M.; Kennedy-Wilde, J.; Shao, H.; Papaioannou, S.; et al. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin. Transl. Immunol. 2022, 11, e1373. [Google Scholar] [CrossRef]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Harrer, D.C.; Simon, B.; Fujii, S.-I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Makkouk, A.; Yang, X.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef]
- Went, P.; Agostinelli, C.; Gallamini, A.; Piccaluga, P.P.; Ascani, S.; Sabattini, E.; Bacci, F.; Falini, B.; Motta, T.; Paulli, M.; et al. Marker Expression in Peripheral T-Cell Lymphoma: A Proposed Clinical-Pathologic Prognostic Score. J. Clin. Oncol. 2006, 24, 2472–2479. [Google Scholar] [CrossRef]
- Pui, C.H.; Behm, F.G.; Singh, B.; Schell, M.J.; Williams, D.L.; Rivera, G.K.; Kalwinsky, D.K.; Sandlund, J.T.; Crist, W.M.; Raimondi, S.C. Het-erogeneity of presenting features and their relation to treatment outcome in 120 children with T-cell acute lymphoblastic leu-kemia. Blood 1990, 75, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Wawrzyniecka, P.A.; Ibrahim, L.; Gritti, G.; Pule, M.A.; Maciocia, P.M. Chimeric antigen receptor T cells for gamma–delta T cell malignancies. Leukemia 2022, 36, 577–579. [Google Scholar] [CrossRef]
- Adicet Bio Inc. Adicet Bio Reports Positive Data from Ongoing ADI-001 Phase 1 Trial in Patients with Relapsed or Refractory Aggressive B-Cell Non-Hodgkin’s Lymphoma (NHL). 5 December 2022. Available online: https://investor.adicetbio.com/news-releases/news-release-details/adicet-bio-reports-positive-data-ongoing-adi-001-phase-1-trial (accessed on 18 December 2022).
- Tough, D.F.; Sprent, J. Lifespan of gamma/delta T Cells. J. Exp. Med. 1998, 187, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Burnham, R.E.; Zoine, J.T.; Story, J.Y.; Garimalla, S.N.; Gibson, G.; Rae, A.; Williams, E.; Bixby, L.; Archer, D.; Doering, C.B.; et al. Characterization of Donor Variability for γδ T Cell ex vivo Expansion and Development of an Allogeneic γδ T Cell Immunotherapy. Front. Med. 2020, 7, 588453. [Google Scholar] [CrossRef]
- Fleischer, L.C.; Becker, S.A.; Ryan, R.E.; Fedanov, A.; Doering, C.B.; Spencer, H.T. Non-signaling Chimeric Antigen Receptors Enhance Antigen-Directed Killing by γδ T Cells in Contrast to αβ T Cells. Mol. Ther.-Oncolytics 2020, 18, 149–160. [Google Scholar] [CrossRef]
- Fischer, K.; Voelkl, S.; Heymann, J.; Przybylski, G.K.; Mondal, K.; Laumer, M.; Kunz-Schughart, L.; Schmidt, C.A.; Andreesen, R.; Mackensen, A. Isolation and characterization of human antigen-specific TCRαβ+ CD4−CD8− double-negative regulatory T cells. Blood 2005, 105, 2828–2835. [Google Scholar] [CrossRef]
- Juvet, S.C.; Zhang, L. Double negative regulatory T cells in transplantation and autoimmunity: Recent progress and future directions. J. Mol. Cell Biol. 2012, 4, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Tao, W.; Zhu, S. T lymphocytes in the intestinal mucosa: Defense and tolerance. Cell. Mol. Immunol. 2019, 16, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Martina, M.N.; Noel, S.; Saxena, A.; Bandapalle, S.; Majithia, R.; Jie, C.; Arend, L.J.; Allaf, M.E.; Rabb, H.; Hamad, A.R.A. Double-Negative αβ T Cells Are Early Responders to AKI and Are Found in Human Kidney. J. Am. Soc. Nephrol. 2015, 27, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Newman-Rivera, A.M.; Kurzhagen, J.T.; Rabb, H. TCRαβ+ CD4−/CD8− “double negative” T cells in health and disease—Implications for the kidney. Kidney Int. 2022, 102, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.S.; Zhang, Z.-X.; Chen, W.; Zhang, L. Double-Negative T Regulatory Cells Can Develop Outside the Thymus and Do Not Mature from CD8+T Cell Precursors. J. Immunol. 2006, 177, 2803–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, S.M.; Li, L.; Love, P.E. TCR Signal Strength Influences αβ/γδ Lineage Fate. Immunity 2005, 22, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.C.; Chapman, F.M.; Michael, S.D. The production of alpha/beta and gamma/delta double negative (DN) T-cells and their role in the maintenance of pregnancy. Reprod. Biol. Endocrinol. 2015, 13, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Yang, W.; Degauque, N.; Tian, Y.; Mikita, A.; Zheng, X.X. New differentiation pathway for double-negative regulatory T cells that regulates the magnitude of immune responses. Blood 2006, 109, 4071–4079. [Google Scholar] [CrossRef] [Green Version]
- Grishkan, I.V.; Ntranos, A.; Calabresi, P.; Gocke, A.R. Helper T cells down-regulate CD4 expression upon chronic stimulation giving rise to double-negative T cells. Cell. Immunol. 2013, 284, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Crispín, J.C.; Tsokos, G.C. Human TCR-αβ+ CD4− CD8− T Cells Can Derive from CD8+ T Cells and Display an Inflammatory Effector Phenotype. J. Immunol. 2009, 183, 4675–4681. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, N.; Apostolidis, S.A.; Penaloza-MacMaster, P.; Villa, J.M.M.; Barouch, D.H.; Tsokos, G.C.; Crispín, J.C. Programmed Cell Death 1 and Helios Distinguish TCR-αβ+ Double-Negative (CD4−CD8−) T Cells That Derive from Self-Reactive CD8 T Cells. J. Immunol. 2015, 194, 4207–4214. [Google Scholar] [CrossRef] [Green Version]
- Velikkakam, T.; Gollob, K.J.; Dutra, W.O. Double-negative T cells: Setting the stage for disease control or progression. Immunology 2022, 165, 371–385. [Google Scholar] [CrossRef]
- Wu, Z.; Zheng, Y.; Sheng, J.; Han, Y.; Yang, Y.; Pan, H.; Yao, J. CD3+CD4−CD8− (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front. Immunol. 2022, 13, 816005. [Google Scholar] [CrossRef]
- Liu, Z.; Meng, Q.; Bartek, J., Jr.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. OncoImmunology 2017, 6, e1252894. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Blasi, D.; Boldanova, T.; Mori, L.; Terracciano, L.; Heim, M.H.; De Libero, G. Unique T-Cell Populations Define Immune-Inflamed Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Ly, D.; Wang, S.-S.; Lee, J.B.; Kang, H.; Xu, H.; Yao, J.; Tsao, M.-S.; Liu, W.; Zhang, L. Targeting late-stage non-small cell lung cancer with a combination of DNT cellular therapy and PD-1 checkpoint blockade. J. Exp. Clin. Cancer Res. 2019, 38, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Minden, M.D.; Chen, W.C.; Streck, E.; Chen, B.; Kang, H.; Arruda, A.; Ly, D.; Der, S.D.; Kang, S.; et al. Allogeneic Human Double Negative T Cells as a Novel Immunotherapy for Acute Myeloid Leukemia and Its Underlying Mechanisms. Clin. Cancer Res. 2018, 24, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Merims, S.; Li, X.; Joe, B.; Dokouhaki, P.; Han, M.; Childs, R.W.; Wang, Z.-Y.; Gupta, V.; Minden, M.D.; Zhang, L. Anti-leukemia effect of ex vivo expanded DNT cells from AML patients: A potential novel autologous T-cell adoptive immunotherapy. Leukemia 2011, 25, 1415–1422. [Google Scholar] [CrossRef]
- Chen, B.; Lee, J.B.; Kang, H.; Minden, M.D.; Zhang, L. Targeting chemotherapy-resistant leukemia by combining DNT cellular therapy with conventional chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 88. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.B.; Kang, H.; Fang, L.; D’Souza, C.; Adeyi, O.; Zhang, L. Developing Allogeneic Double-Negative T Cells as a Novel Off-the-Shelf Adoptive Cellular Therapy for Cancer. Clin. Cancer Res. 2019, 25, 2241–2253. [Google Scholar] [CrossRef] [Green Version]
- Vasic, D.; Lee, J.B.; Leung, Y.; Khatri, I.; Na, Y.; Abate-Daga, D.; Zhang, L. Allogeneic double-negative CAR-T cells inhibit tumor growth without off-tumor toxicities. Sci. Immunol. 2022, 7, eabl3642. [Google Scholar] [CrossRef]
- Achita, P.; Dervovic, D.; Ly, D.; Lee, J.B.; Haug, T.; Joe, B.; Hirano, N.; Zhang, L. Infusion of ex-vivo expanded human TCR-αβ+ double-negative regulatory T cells delays onset of xenogeneic graft-versus-host disease. Clin. Exp. Immunol. 2018, 193, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Baolin, T.; Lee, B.J.; Cheng, S.; Yao, W.; Wang, D.; Tu, M.; Xiang, Z.; Geng, L.; Wang, M.; Qiang, P.; et al. Safety and Efficacy of Ex Vivo Expanded Healthy Donor-Derived Double Negative T Cells for the Treatment of AML Relapsed after Allogeneic Stem Cell Transplantation: A First in-Human Phase I/IIa Clinical Trial. Blood 2020, 136, 1–2. [Google Scholar] [CrossRef]
- Patel, J.L.; Smith, L.M.; Anderson, J.; Abromowitch, M.; Campana, D.; Jacobsen, J.; Lones, M.A.; Gross, T.G.; Cairo, M.S.; Perkins, S.L. The immunophenotype of T-lymphoblastic lymphoma in children and adolescents: A Children’s Oncology Group report. Br. J. Haematol. 2012, 159, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.M.; Zinzani, P.L.; Vose, J.M.; Gascoyne, R.D.; Rosen, S.T.; Tobinai, K. Peripheral T-cell lymphoma. Blood 2011, 117, 6756–6767. [Google Scholar] [CrossRef] [PubMed]
- Young, K.J.; DuTemple, B.; Phillips, M.J.; Zhang, L. Inhibition of Graft-Versus-Host Disease by Double-Negative Regulatory T Cells. J. Immunol. 2003, 171, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ford, M.S.; Young, K.J.; Cybulsky, M.; Zhang, L. Role of Double-Negative Regulatory T Cells in Long-Term Cardiac Xenograft Survival. J. Immunol. 2003, 170, 1846–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.-X.; Yang, L.; Young, K.; Zhang, L. Suppression of alloimmune responses in vitro and in vivo by CD3+CD8−CD4−αβ+ regulatory T cells. Transplant. Proc. 2001, 33, 84–85. [Google Scholar] [CrossRef]
- Ye, H.; Chang, Y.; Zhao, X.; Huang, X. Characterization of CD3+CD4−CD8− (double negative) T cells reconstitution in patients following hematopoietic stem-cell transplantation. Transpl. Immunol. 2011, 25, 180–186. [Google Scholar] [CrossRef]
- McIver, Z.; Serio, B.; Dunbar, A.; O’Keefe, C.L.; Powers, J.; Wlodarski, M.; Jin, T.; Sobecks, R.; Bolwell, B.; Maciejewski, J.P. Double-negative regulatory T cells induce allotolerance when expanded after allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2008, 141, 170–178. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, D.; Torrealba, J.R.; Waddell, T.K.; Grant, D.; Zhang, L. Donor Lymphocyte Infusion Induces Long-Term Donor-Specific Cardiac Xenograft Survival through Activation of Recipient Double-Negative Regulatory T Cells. J. Immunol. 2005, 175, 3409–3416. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Diao, J.; Stepkowski, S.M.; Zhang, L. Both infiltrating regulatory T cells and insufficient antigen presentation are involved in long-term cardiac xenograft survival. J. Immunol. 2007, 179, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.-C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Rezaei, R.; Ghaleh, H.E.G.; Farzanehpour, M.; Dorostkar, R.; Ranjbar, R.; Bolandian, M.; Nodooshan, M.M.; Alvanegh, A.G. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther. 2021, 29, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Hübbe, M.L.; Jæhger, D.E.; Andresen, T.L.; Andersen, M.H. Leveraging Endogenous Dendritic Cells to Enhance the Therapeutic Efficacy of Adoptive T-Cell Therapy and Checkpoint Blockade. Front. Immunol. 2020, 11, 578349. [Google Scholar] [CrossRef] [PubMed]
- Capelletti, M.; Liegel, J.; Themeli, M.; Mutis, T.; Stroopinsky, D.; Orr, S.; Torres, D.; Morin, A.; Gunset, G.; Ghiasuddin, H.; et al. Potent Synergy between Combination of Chimeric Antigen Receptor (CAR) Therapy Targeting CD19 in Conjunction with Dendritic Cell (DC)/Tumor Fusion Vaccine in Hematological Malignancies. Biol. Blood Marrow Transplant. 2020, 26, S42–S43. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Challenges | Solutions | Examples of CAR-T Cell Product | Modifications of CAR-T Cell | Limitations | Reference |
|---|---|---|---|---|---|
| Fratricide | CAR-T surface antigen modifications | CD7 CAR-T | CD7 knockout (KO) | Risks of unwanted effects from gene editing | [24] |
| CD7 protein expression blocker | Requires multiple transductions, which may reduce yield | [25] | |||
| Reversible CAR expression | CD5 CAR-T | Tet-off | Side effects from drug administration | [26] | |
| Use of antigens restricted to specific T cell subsets or tumors | CD1a CAR-T | None | Only applicable to some cases | [27] | |
| CD30 CAR-T | [28] | ||||
| CD37 CAR-T | Only applicable to some cases | [29] | |||
| Use of alternative cell types | - | - | Depends on the cell type used | - | |
| Product contamination | Use of allogeneic CAR-T | CD3 CAR-T | CD3 & TCRαβ KO | Requires TCR modification to avoid GvHD | [30] |
| CD7 CAR-T | CD7 & TRAC KO | [31] | |||
| - | - | Requires MHC modification to avoid rejection | - | ||
| Use of alternative cell types | - | - | Depends on the cell type used | - | |
| T-cell aplasia | Safety switch | CD4 CAR-T | Use of drug CAMPATH | Potential lack of long-term efficacy and cancer recurrence | [32] |
| Use of antigens restricted to tumors | CDR3 CAR-T | None | More time-consuming and expensive | [33] | |
| Use of antigens restricted to specific T cell subsets | TRBC1 CAR-T | None | Only applicable to some cases | [34] | |
| Use of short-lived alternative cell types | - | - | Potential lack of long-term efficacy and cancer recurrence | - | |
| Bridge to HSCT | - | - | Only some patients are eligible Risks of complications and mortality involved in HSCT | - |
| Treatment Approach | Source | Cell Modifications (If Specified) | Phase | Study Stage | Actual or Estimated Enrollment | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| CD4 CAR-T | Autologous | I | Active | 20 | NCT03829540 | |
| I | Terminated | 9 | NCT04973527 | |||
| I | Terminated | 4 | NCT04219319 | |||
| Active | 50 | NCT04712864 | ||||
| IL-15 secretion | I | Active Preliminary results reported (n = 3) | 12 | NCT04162340 | ||
| CD5 CAR-T | Autologous | CD5 KO via CRISPR/Cas9 | Early I | Not yet recruiting | 18 | NCT04767308 |
| Autologous or HSCT donor | I | Active Preliminary results reported (n = 5) | 42 | NCT03081910 | ||
| IL-15 secretion | I | Active Preliminary results reported (n = 1) | 20 | NCT04594135 | ||
| HSCT donor or new donor | I | Recruiting | 18 | NCT05487495 | ||
| I | Active Preliminary results reported (n = 5) | 18 | NCT05032599 | |||
| CD7 CAR-T | Autologous | I | Recruiting | 15 | NCT05554575 | |
| CD7 blockade with ER anchor | I | Active Preliminary results reported (n = 5) | 20 | NCT04840875 | ||
| I | Recruiting | 9 | NCT04480788 | |||
| I | Recruiting | 20 | NCT05513612 | |||
| I | Recruiting | 18 | NCT05398614 | |||
| pharmacologic inhibitors ibrutinib and dasatinib in vitro | I | Recruiting | 21 | NCT03690011 | ||
| I/II | Recruiting | 108 | NCT04599556 | |||
| I | Recruiting | 4 | NCT05290155 | |||
| CD7 blockade with tandem CD7 nanobody | I/II | Recruiting | 20 | NCT04762485 | ||
| II | Recruiting | 20 | NCT05059912 | |||
| CD7 blockade with tandem CD7 nanobody | I | Completed Full manuscript published | 8 | NCT04004637 | ||
| Autologous or HSCT donor | I | Completed Full manuscript published | 20 | NCT04572308 | ||
| Autologous or donor | N/A | Active Preliminary results reported (n = 8) | 100 | NCT04916860 | ||
| Autologous or HLA-matched sibling donor | CD7 blocking | I | Active Preliminary results reported (n = 10) | 24 | NCT04823091 | |
| HSCT donor or new donor | IntraBlock technology (CAR with ER retention of CD7) | I | Active | 20 | ChiCTR2000034762 | |
| II | Recruiting | 70 | NCT04689659 | |||
| Allogeneic (healthy donor) | Universal CAR-T | Early I | Recruiting | 30 | NCT04264078 | |
| Universal CAR-T | Early I | Not yet recruiting | 15 | NCT04860817 | ||
| Base edited via CRISPR/Cas9 | I | Recruiting | 10 | NCT05397184 | ||
| TRAC, CD7 and HLA-II KO via CRISPR/Cas9 | N/A | Recruiting | 24 | NCT04620655 | ||
| TRAC and CD7 deleted | I | Not yet recruiting | 48 | NCT05377827 | ||
| CJD7 and TRAC deleted | I/II | Recruiting | 44 | NCT04984356 | ||
| I | Recruiting | 30 | NCT05127135 | |||
| Unspecified | Non gene edited | I | Recruiting | 24 | NCT05212584 | |
| Non gene edited | I | Recruiting | 20 | NCT04934774 | ||
| CD7protein expression blocker | I | Recruiting | 20 | NCT05043571 | ||
| N/A | Recruiting | 100 | NCT04928105 | |||
| CD30 CAR-T | Autologous | I | Recruiting | 50 | NCT04008394 | |
| I | Active | 18 | NCT02663297 | |||
| I | Completed Full manuscript published | 9 | NCT01316146 | |||
| I/II | Active Results reported for HL only | 40 | NCT02690545 | |||
| I | Active Results reported for HL only | 66 | NCT02917083 | |||
| I | Recruiting | 20 | NCT03383965 | |||
| I/IIa | Recruiting | 30 | NCT04653649 | |||
| I | Active | 21 | NCT04526834 | |||
| CCR4 receptor expressed | I | Active Preliminary results reported (n = 12) [55] | 59 | NCT03602157 | ||
| I | Completed Outcome reported | 26 | NCT03049449 | |||
| I | Active Preliminary results reported (n = 12) [56] | 20 | NCT04083495 | |||
| Early I | Not yet recruiting | 9 | NCT05208853 | |||
| CAR infused with iCasp9 | I/II | Unknown Result reported for HL only [57] | 20 | NCT02274584 | ||
| Allogeneic (Healthy donor) | Use EBV-specific T cells | I | Active Preliminary results reported (n = 8) [58] | 18 | NCT04288726 | |
| I | Not yet recruiting | 18 | NCT04952584 | |||
| CD37 CAR-T | Autologous | I | Recruiting | NCT04136275 | ||
| CD70 CAR-T | Allogeneic | TCR and MHC I KO via CRISPR/Cas9 | Recruiting | 45 | NCT04502446 | |
| CD123 CAR-T | Autologous | I | Recruiting | 32 | NCT04318678 | |
| CD147 CAR-T | Autologous | Early I | Not yet recruiting | 12 | NCT05013372 | |
| TRBC1 CAR-T | Autologous | I/II | Active Preliminary results reported (n = 10) | 200 | NCT03590574 | |
| I | Recruiting | 9 | NCT04828174 | |||
| Multi-CAR-T | Autologous or HSCT donor | I/II | Recruiting | 30 | NCT04033302 |
| Treatment Approach | Manufacturing Method | Trial Stage, Status and Design | Number of Patients in Report | Age of Patients | Disease Type and Status | Disease Outcome | Treatment Associated Toxicities | Clinical Trial Identifier | Reference |
|---|---|---|---|---|---|---|---|---|---|
| CD4 CAR-T | - Autologous - 3rd generation CAR - IL-15/IL-15 sushi | - Phase I - Active | 3 | >18 yr | - PTCL & CTCL - Prior transplant or 4–6 standard chemotherapy | - CR = 67% (by 8 months & 15 months) - PR = 33% (by 5 months) | - CRS I–II (100%) - No neurotoxicity or severe infection - CD4+ T cell ablation, recovered by 3 months | NCT04162340 | [59] |
| CD5 CAR-T | - Autologous - 2nd generation CAR | - Phase I - Active - Dose escalation - HSCT bridge | 5 | 62–71 yr | r/r T cell NHL | - CR = 66.7% - n = 3 evaluated | - CRS I–II (60%) - Neurotoxicity II (20%) - Prolonged cytopenia (40%) - Reactivation of CMV and BK virus (20%) - No complete T-cell aplasia | NCT03081910 | [60] |
| - Autologous or HSCT donor - IL-15/IL-15 sushi - HSCT bridge | - Phase I - Active | 1 | 22 yr | r/r T-LBL (CNS relapse) | - Disease remission, underwent HSCT | - CRS I - CD5+ T-cell aplasia, recovered by day 9 - No infection | NCT04594135 | [61] | |
| - HSCT donor | - Phase I - Active | 5 | 1–70 yr eligible | - r/r T-ALL - CD7-negative relapse after CD7 CAR-T cell therapy | - CR = 100% (by 1.8–4.1 months) | - CRS I–II (80%) - GvHD I (20%) - Maculopapular rash II (60%) - EBV infection IV (20%) - CD5+ T cell depleted - Hematological toxicity III–IV (100%) | NCT05032599 | [62] | |
| CD7 CAR-T | - Autologous - CD7 blockade with ER anchor | - Phase I - Active | 5 | 1.9–13 yr | r/r T-ALL & T-LBL | - CR = 80% (by 1 months) | - CRS (60%), III (20%) - Hematological toxicity III–IV, recovered to II by 30 days - No neurotoxicity or infection - Reduced CD7+ T cell count (100%) | NCT04840875 | [63] |
| - Autologous - CD7 blockade with tandem CD7 nanobody | - Phase I - Completed | 8 | 7–70 yr eligible | r/r T-ALL and T-LBL | - CR = 87.5% (by 3 months) | - CRS I–II (87.5) - No T cell hypoplasia - No neurotoxicity | NCT04004637 | [23] | |
| - Autologous or HSCT donor - Naturally selected CD7 CAR | - Phase I - Completed | 20 | 3–47 yr | r/r T-ALL (n = 14) & T-LBL (n = 6) | - CR = 95% (BM) by day 28 - Underwent consolidative (50%) or salvage (20%) allo-HSCT by 32-311 days | - CRS I–II (90%) - CRS III (5%) - Neurotoxicity I (10%) - Cytopenia (100%) - Sepsis (10%) - CMV reactivation (5%) | NCT04572308 | [22] | |
| - Autologous (n = 7) or donor (n = 1) - 2nd generation CAR | - Phase I - Active | 8 | 14–47 yr | - r/r T-LBL - 2–10 prior lines of therapies | - CR = 87.5% (BM) by day 28 - Underwent allo-HSCT (75%) by 42–56 days | - CRS I–II (87.5%) - CRS III (12.5%) - Neurotoxicity I (12.5%) | NCT04916860 | [64] | |
| - Autologous (n = 5) or HLA-matched sibling donor (n = 5) - CD7 blocking | - Phase I - Active | 10 | 14–70 yr | r/r T-ALL | - MRD-negative remission (n = 6, 100%) | - CRS I–II (70%) - CRS III–IV (10%) - Cytopenia IV (100%) - GvHD I–II (20%) - Multiple infections (60%) - No neurotoxicity | NCT04823091 | [65] | |
| - HSCT donor or new donor - IntraBlock technology (CAR with ER retention of CD7) | - Phase I - Active | 20 | 2–43 yr | - r/r T-ALL - At least 2 prior therapies - prior HSCT (n = 12) | - CR = 90%, 75% by 6.3 months - Underwent HSCT (35%) | - CRS I–II (90%) - CRS III (10%) - Cytopenia III–IV (100%) - Neurotoxicity I–II (15%) - GvHD I-II (60%) - Viral activation I–II (20%) - CD7+ T cell depleted | ChiCTR2000034762 Phase II: NCT04689659 | [66] | |
| CD30 CAR-T | - Autologous | - Phase I - Completed - Dose escalation | 9 | Child, adult, older adult eligible | - r/r HL (n = 7) & ALCL (n = 2) - 3 or more lines of prior chemotherapy | - CR=50% (n = 1) by 9 months | - No CRS - No toxicities or infections observed | NCT01316146 | [67] |
| - Autologous | - Phase I - Completed - Dose escalation | 22 | 18–65 yr | - r/r PTCL, ALCL, enteropathy-associated T-cell lymphoma, extranodal NK-T-cell lymphoma | - CR = 4.8% - PR = 31.8% - SD = 57% - n = 21 evaluated | - CRS (4.5%) - Sepsis (13.6%) - Maculopapular rash (9%) - Anemia (50%) - Neutropenia (100%) | NCT03049449 | [68] | |
| - Autologous | - Phase I - Active - Consolidation post auto-HSCT | 12 | 16–76 yr | - r/r HL (n = 6), ALCL (n = 4), AITL (n = 1), PTCL (n = 1) - 2–3 lines of prior therapy | - PFS = 57% - OS = 77% at 1yr | - CRS I (8.3%) - No ICANS or dose-limiting toxicity (DLT) - Hematological toxicity (50%) | NCT04083495 | [56] | |
| - Autologous - CCR4 receptor expressed | - Phase I - Active - Dose escalation | 12 | 27–75 yr | - r/r HL (n = 10) & CTCL (n = 2) - 2–10 lines of prior therapy | - SD = 50% (n = 1) | - CRS I-II (25%) - No ICANS or DLT | NCT03602157 | [55] | |
| - Allogeneic (Healthy donor, based on best MHC I & II match) - EBV-specific T cells | - Phase I - Active - Dose escalation | 8 | 12–75 yr eligible | - r/r CD30+ lymphoma - 3–5 lines of prior therapy | - CR = 28.5% (n = 2) - PR = 42.9% (n = 3) - n = 7 evaluated | - No CRS, DLT, or GvHD | NCT04288726 | [58] | |
| TRBC1 CAR-T | - Autologous | - Phase I/II - Active - Dose escalation and expansion | 10 | 34–63 yr | - r/r PTCL NOS (n = 5), AITL (n = 4), ALCL (n = 1) - 1–5 lines of prior therapy | - Complete metabolic response = 55.5% (n = 5) - PR = 11.1% (n = 1) - n = 9 evaluated at 1 month | - Cytopenia (anemia & neutropenia) - CRS I–II (30%) - CRS III (10%) - No neurotoxicity/ICANS or DLT - No grade III or higher infections | NCT03590574 | [69] |
| CAR- Conventional T/CD8+ | CAR-NK | CAR-γδ T | CAR-DNT | |
|---|---|---|---|---|
| Risk of GvHD | High due to alloreactive TCRs - Studies investigating genetic modifications (e.g., TCR KO) or non-alloreactive T cells (e.g., virus-specific) | Low - Protective against GvHD activity by targeting recipient APCs | Low | Low - GvHD suppressive activity |
| Risk of Immune Rejection | High due to MHC mismatch - Studies investigating genetic modifications (e.g., MHC I/II KO) or inhibition of T cell & NK cell cytotoxicity (e.g., certain MHC I alleles) | Present - Require lymphodepletion to suppress T cell activity to minimize NK graft rejection (especially when IL-15 supplementation is used) - Studies investigating genetic modifications (multiple KOs/knock-ins) | Unclear | Low - Resistant to rejection |
| Risk of Fratricide | Present - Studies investigating surface antigen KOs - Antigens restricted to specific T cell subsets | None for T-lineage specific antigens | Depends on target antigen - No fratricide for TCR αβ | Depends on target antigen - No fratricide for CD4 or CD8 |
| Lifespan/ Persistence | Longest persistence - Detectable for 6 months to years after therapy | Shorter - Detectable for only 3 weeks; lacks long-term antitumor efficacy - Requires multiple doses, increasing risk of rejection - Studies investigating memory-like NK | Shorter | Shorter |
| Antitumor Cytotoxicity | MHC-dependent - No endogenous killing ability with TCR KO | MHC independent - NK cell receptors - ADCC, potential use in combination with antibody treatment | MHC independent - TCRγδ - NK cell receptors - ADCC | MHC independent - TCRγδ - TCRαβ - NK cell receptors |
| CAR Construct Suitability | Superior cytotoxicity - CAR originally designed for T cells | Inferior cytotoxicity compared to conventional T cells -NK signaling might affect performance (studies investigating NK-specific CAR constructs) | Comparable with conventional CAR-T | Comparable with conventional CAR-T |
| Toxicities/ Side Effects | - Cytokine release syndrome (CRS) - Studies investigating use of safety switch to prevent T-cell aplasia or severe adverse events | - Reduced risk of CRS due to limited cytokine secretion profile - Studies suggesting limited persistence can reduce risk of T-cell aplasia | Limited CRS | Unclear |
| Cost | High due to necessary modifications | Depends on source of cells | Low | Low |
| Sources | Readily available - Peripheral blood (PB) - Umbilical cord blood | - 10% PB, mature phenotype, harder to expand and standardize product - Umbilical cord blood, immature phenotype - NK-92 cell line commonly used, needs to be irradiated before use, reduced proliferative capacity, derived from lymphoma - iPSC | 1–10% of PB T cells | 3–5% of PB T cells |
| Culture and Expansion | Can be expanded to therapeutic numbers | - Poorer expansion (from PB) than conventional T cells - May involve use of feeder cells (risk of contamination, more complicated/costly) | Can be expanded to therapeutic numbers | Can be expanded to therapeutic numbers |
| Transduction Efficiency | High | Lower - May require multiple transductions or cell sorting | Comparable with conventional CAR-T | Comparable with conventional CAR-T |
| Cryopreservation | Can be cryopreserved | More sensitive to freezing/thawing than conventional T cells | Sensitive to freezing/thawing | Maintain viability and antitumor functions |
| Dependence on Cytokine Support | Yes | Yes | Yes |
| Treatment Approach | Disease | Other Therapy | Title | Study Stage | Phase | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| Expanded haploidentical NK cells | r/r T-ALL | Post HSCT Concurrent chemotherapy | Pilot Study of Expanded, Donor Natural Killer Cell Infusions for Refractory Non-B Lineage Hematologic Malignancies and Solid Tumors | Completed | Phase I | NCT00640796 |
| Donor NK cells | Recurrent or Stage III/IV adult T cell leukemia/lymphoma | Post HSCT | Donor NK Cell Infusion for Progression/Recurrence of Underlying Malignant Disorders After HLA-haploidentical HCT–a Phase 1-2 Study | Completed | Phase I/II | NCT00823524 |
| Third party NK cells | Recurrent or r/r adult T cell leukemia/lymphoma Recurrent or r/r primary cutaneous T cell NHL | Monoclonal antibody (Mogamulizumab) | A Pilot Phase I Trial of IL-21 Expanded, Off the Shelf, Third-Party Natural Killer (NK) Cells in Combination With Mogamulizumab in Patients With Cutaneous T-Cell Lymphomas or Adult T-Cell Leukemia/Lymphomas | Recruiting | Phase I | NCT04848064 |
| Expanded, activated haploidentical NK cells | r/r/ T cell lymphoblastic leukemia and lymphoma | Concurrent Salvage Chemotherapy | Salvage Therapy With Chemotherapy and Natural Killer Cells in Relapsed/Refractory Paediatric T Cell Lymphoblastic Leukaemia and Lymphoma (HNJ-NKAES-2012) | Terminated due to low recruitment | NCT01944982 | |
| Expanded, activated haploidentical NK cells | T-ALL | Concurrent Chemotherapy | Pilot Study of Expanded, Activated Haploidentical Natural Killer Cell Infusions for Non-B Lineage Acute Leukaemia and Myelodysplastic Syndrome | Unknown | Phase I | NCT02123836 |
| Treatment Approach | Disease | Other Therapy | Title | Study Stage | Phase | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| Ex vivo expanded allogeneic γδ T cells | r/r PTCL | None | The Safety and Efficacy Assessment of Ex vivo Expanded Allogeneic γδT Cells Immunotherapy in Patients With Relapsed or Refractory Non-Hodgkin’s Lymphoma (NHL) and Peripheral T Cell Lymphomas (PTCL) | Recruiting | Early Phase I | NCT04696705 |
| Ex vivo expanded γδ T cells | ALL | Post HSCT | Phase I Study of Ex Vivo Expanded/Activated Gamma Delta T-cell Infusion Following Haploidentical Hematopoietic Stem Cell Transplantation and Post-transplant Cyclophosphamide | Recruiting | Phase I | NCT03533816 |
| Ex vivo expanded γδ T cells | ALL | Post HSCT | Safety and Efficiency of γδ T Cell Against Hematological Malignancies After Allo-HSCT. A Dose Escalation, Open-label, Phase 1/2 Study. | Recruiting | Phase I/II | NCT04764513 |
| Ex vivo expanded γδ T cells | ALL | Post HSCT Concurrent chemotherapy CD45RA-depleted donor memory T cells | TCRαβ-depleted Progenitor Cell Graft With Additional Memory T-cell DLI, Plus Selected Use of Blinatumomab, in Naive T-cell Depleted Haploidentical Donor Hematopoietc Cell Transplantation for Hematologic Malignancies | Recruiting | Phase II | NCT03849651 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, K.K.-L.; Lee, J.B.; Zhang, L. Adoptive Cell Therapy for T-Cell Malignancies. Cancers 2023, 15, 94. https://doi.org/10.3390/cancers15010094
Fang KK-L, Lee JB, Zhang L. Adoptive Cell Therapy for T-Cell Malignancies. Cancers. 2023; 15(1):94. https://doi.org/10.3390/cancers15010094
Chicago/Turabian StyleFang, Karen Kai-Lin, Jong Bok Lee, and Li Zhang. 2023. "Adoptive Cell Therapy for T-Cell Malignancies" Cancers 15, no. 1: 94. https://doi.org/10.3390/cancers15010094