
The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks?
 ,
,  , , ,
, , ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. HCC Epidemiology
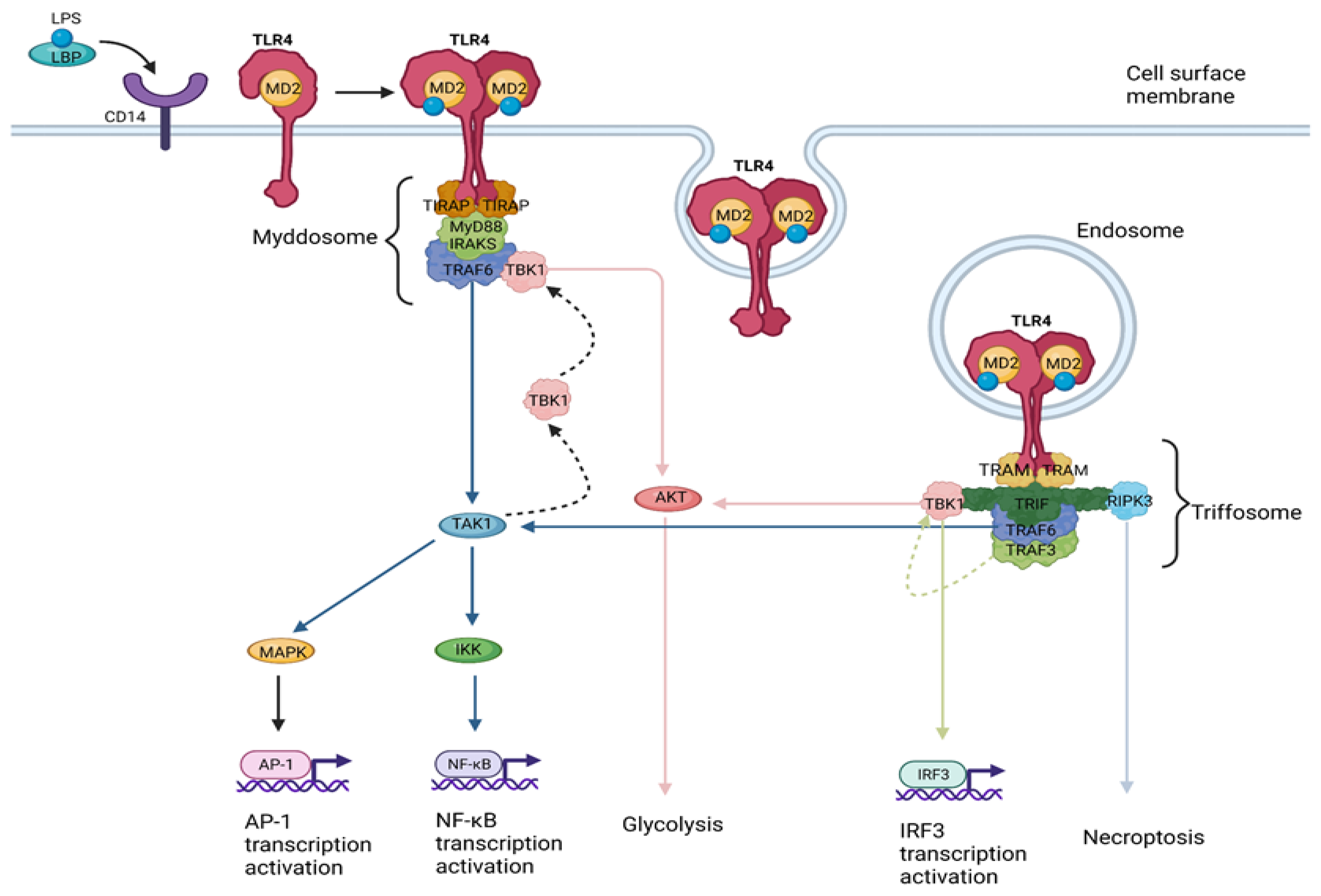
1.2. TLR4 Signaling
- (a)
- The myddosome, a SMOC seeded by TIRAP/MAL, driving the MyD88-dependent pathway which leads to inflammatory cytokine production [22].
- (b)
2. TLR4 Signaling in Liver Disease
3. TLR4 Signaling in HCC
3.1. TLR4 Signaling in HCC Metastasis
3.2. TLR4 Signaling in HCC Drug Resistance
3.3. TLR4 Signaling as Epigenetic Target in HCC
3.4. TLR4 Signaling in HCC Proliferation and Apoptosis
3.5. TLR4 Signaling in HCC Angiogenesis
4. TLR4 Signaling in HCC Immunotherapy
4.1. TLR4 Signaling as a Platform for HCC Vaccines
4.2. TLR4 Signaling as Regulator of HCC Tumor Immune Landscape
4.2.1. Regulation of T and B lymphocytes by TLR4 Signaling
4.2.2. Regulation of DCs by TLR4 Signaling
4.2.3. Regulation of Neutrophils by TLR4 Signaling
4.2.4. Regulation of MDSCs by TLR4 Signaling
4.2.5. Regulation of Macrophages by TLR4 Signaling
4.2.6. Regulation of Fibroblasts by TLR4 Signaling
4.3. Targeting TLR4 Signaling to Enhance PD-1 Blockade
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, C. Innate recognition of microbial-derived signals in immunity and inflammation. Sci. China Life Sci. 2016, 59, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Drexler, S.K.; Foxwell, B.M. The role of Toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef]
- Li, X.; Jiang, S.; Tapping, R.I. Toll-like receptor signaling in cell proliferation and survival. Cytokine 2010, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Avalos, A.M.; Ploegh, H.L. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 2012, 12, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Gioannini, T.L.; Teghanemt, A.; Zhang, D.S.; Coussens, N.P.; Dockstader, W.; Ramaswamy, S.; Weiss, J.P. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. USA 2004, 101, 4186–4191. [Google Scholar] [CrossRef] [PubMed]
- Nomura, F.; Akashi, S.; Sakao, Y.; Sato, S.; Kawai, T.; Matsumoto, M.; Nakanishi, K.; Kimoto, M.; Miyake, K.; Takeda, K.; et al. Cutting edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 2000, 164, 3476–3479. [Google Scholar] [CrossRef]
- Schromm, A.B.; Lien, E.; Henneke, P.; Chow, J.C.; Yoshimura, A.; Heine, H.; Latz, E.; Monks, B.G.; Schwartz, D.A.; Miyake, K.; et al. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: A point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J. Exp. Med. 2001, 194, 79–88. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Verstak, B.; Nagpal, K.; Bottomley, S.P.; Golenbock, D.T.; Hertzog, P.J.; Mansell, A. MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J. Biol. Chem. 2009, 284, 24192–24203. [Google Scholar] [CrossRef]
- Horng, T.; Barton, G.M.; Medzhitov, R. TIRAP: An adapter molecule in the Toll signaling pathway. Nat. Immunol. 2001, 2, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Uematsu, S.; Hoshino, K.; Kaisho, T.; Takeuchi, O.; Takeda, K.; Akira, S. TRAM is specifically involved in the Toll-like receptor 4–mediated MyD88-independent signaling pathway. Nat. Immunol. 2003, 4, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Magupalli, V.G.; Wu, H. SMOCs: Supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 2014, 14, 821–826. [Google Scholar] [CrossRef]
- Kagan, J.C. Signaling Organelles of the Innate Immune System. Cell 2012, 151, 1168–1178. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Bonham, K.S.; Orzalli, M.H.; Hayashi, K.; Wolf, A.I.; Glanemann, C.; Weninger, W.; Iwasaki, A.; Knipe, D.M.; Kagan, J.C. A Promiscuous Lipid-Binding Protein Diversifies the Subcellular Sites of Toll-like Receptor Signal Transduction. Cell 2014, 156, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Henzel, W.J.; Gao, X. IRAK: A Kinase Associated with the Interleukin-1 Receptor. Science 1996, 271, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Zhou, H.; Shan, Y.; Liu, Q.; Li, Q.; Shaw, D.E.; Li, X.; Wu, H. IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly. Mol. Cell 2014, 55, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Gantke, T.; Sriskantharajah, S.; Sadowski, M.; Ley, S.C. IκB kinase regulation of the TPL-2/ERK MAPK pathway. Immunol. Rev. 2012, 246, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Peggie, M.; Plater, L.; Sorcek, R.J.; Young, E.R.R.; Madwed, J.B.; Hough, J.; McIver, E.G.; Cohen, P. Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 2011, 434, 93–104. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; Van Der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Nambu, A.; Jung, J.; Shibata, M.; Aksoylar, H.I.; Lei, J.; Xu, P.; Doan, M.T.; Jiang, H.; MacArthur, M.R.; et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 2019, 20, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Tan, Y.; Kagan, J.C. Innate Immune Signaling Organelles Display Natural and Programmable Signaling Flexibility. Cell 2019, 177, 384–398.e11. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Ozato, K.; Tsujimura, H.; Tamura, T. Toll-like receptor signaling and regulation of cytokine gene expression in the immune system. Biotechniques 2002, 33, S66–S75. [Google Scholar] [CrossRef]
- Oblak, A.; Jerala, R. Toll-like receptor 4 activation in cancer progression and therapy. Clin. Dev. Immunol. 2011, 2011, 609579. [Google Scholar] [CrossRef]
- Soares, J.B.; Pimentel-Nunes, P.; Afonso, L.; Rolanda, C.; Lopes, P.; Roncon-Albuquerque, R.; Gonçalves, N.; Boal-Carvalho, I.; Pardal, F.; Lopes, S.; et al. Increased hepatic expression of TLR2 and TLR4 in the hepatic inflammation-fibrosis-carcinoma sequence. Innate Immun. 2012, 18, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Mann, D.A. Nuclear factor-κB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Real, C.I.; Liu, C.; Baba, H.A.; Gerken, G.; Lu, M.; Broering, R. Hepatic expression of oncogenes Bmi1 and Dkk1 is up-regulated in hepatitis B virus surface antigen–transgenic mice and can be induced by treatment with HBV particles or lipopolysaccharides in vitro. Int. J. Cancer 2017, 141, 354–363. [Google Scholar] [CrossRef]
- Jing, X.; Tian, Z.; Gao, P.; Xiao, H.; Qi, X.; Yu, Y.; Ding, X.; Yang, L.; Zong, L. HBsAg/β2GPI activates the NF-κB pathway via the TLR4/MyD88/IκBα axis in hepatocellular carcinoma. Oncol. Rep. 2018, 40, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Iyer, T.K.; Carpenter, E.; Li, H.; Chang, K.M.; Vonderheide, R.H.; Kaplan, D.E. Dysfunctional B-cell activation in cirrhosis resulting from hepatitis C infection associated with disappearance of CD27-Positive B-cell population. Hepatology 2012, 55, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Uraki, S.; Tameda, M.; Sugimoto, K.; Shiraki, K.; Takei, Y.; Nobori, T.; Ito, M. Substitution in amino acid 70 of hepatitis C virus core protein changes the adipokine profile via toll-like receptor 2/4 signaling. PLoS ONE 2015, 10, e0131346. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.; Naylor, G.; Wickman, G.R.; Rath, N.; Castino, G.; Stevenson, D.; Bryson, S.; Munro, J.; McGarry, L.; Mullin, M.; et al. Defective apoptotic cell contractility provokes sterile inflammation, leading to liver damage and tumour suppression. eLife 2021, 10, e61983. [Google Scholar] [CrossRef]
- Weber, S.N.; Bohner, A.; Dapito, D.H.; Schwabe, R.F.; Lammert, F. TLR4 deficiency protects against hepatic fibrosis and diethylnitrosamine-induced pre-carcinogenic liver injury in fibrotic liver. PLoS ONE 2016, 11, e0158819. [Google Scholar] [CrossRef]
- Nace, G.W.; Huang, H.; Klune, J.R.; Eid, R.E.; Rosborough, B.R.; Korff, S.; Li, S.; Shapiro, R.A.; Stolz, D.B.; Sodhi, C.P.; et al. Cellular-specific role of toll-like receptor 4 in hepatic ischemia-reperfusion injury in mice. Hepatology 2013, 58, 374–387. [Google Scholar] [CrossRef]
- Nadanaka, S.; Hashiguchi, T.; Kitagawa, H. Aberrant glycosaminoglycan biosynthesis by tumor suppressor EXTL2 deficiency promotes liver inflammation and tumorigenesis through Toll-like 4 receptor signaling. FASEB J. 2020, 34, 8385–8401. [Google Scholar] [CrossRef]
- Lin, Y.; Yu, L.X.; Yan, H.X.; Yang, W.; Tang, L.; Zhang, H.L.; Liu, Q.; Zou, S.S.; He, Y.Q.; Wang, C.; et al. Gut-derived lipopolysaccharide promotes T-cell-mediated hepatitis in mice through toll-like receptor 4. Cancer Prev. Res. 2012, 5, 1090–1102. [Google Scholar] [CrossRef]
- Chi, G.; Feng, X.X.; Ru, Y.X.; Xiong, T.; Gao, Y.; Wang, H.; Luo, Z.L.; Mo, R.; Guo, F.; He, Y.P.; et al. TLR2/4 ligand-amplified liver inflammation promotes initiation of autoimmune hepatitis due to sustained IL-6/IL-12/IL-4/IL-25 expression. Mol. Immunol. 2018, 99, 171–181. [Google Scholar] [CrossRef]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Bukong, T.N.; Tornai, D.; Babuta, M.; Vlachos, I.S.; Kanata, E.; Catalano, D.; Szabo, G. Neutrophil extracellular traps contribute to liver damage and increase defective low-density neutrophils in alcohol-associated hepatitis. J. Hepatol. 2023, 78, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Chen, S.; Wang, J.; Yu, S.; Li, Y.; Xu, M.; Aboubacar, H.; Li, J.; Shan, T.; et al. Gut-derived lipopolysaccharide promotes alcoholic hepatosteatosis and subsequent hepatocellular carcinoma by stimulating neutrophil extracellular traps through toll-like receptor 4. Clin. Mol. Hepatol. 2022, 28, 522–539. [Google Scholar] [CrossRef]
- Sin, S.Q.; Mohan, C.D.; Goh, R.M.W.J.; You, M.; Nayak, S.C.; Chen, L.; Sethi, G.; Rangappa, K.S.; Wang, L. Hypoxia signaling in hepatocellular carcinoma: Challenges and therapeutic opportunities. Cancer Metastasis Rev. 2022. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial–mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chang, Y.; Liang, X.; Cardinal, J.S.; Huang, H.; Thorne, S.H.; Monga, S.P.S.; Geller, D.A.; Lotze, M.T.; Tsung, A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 2012, 55, 1863–1875. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, N.; Tan, H.Y.; Guo, W.; Chen, F.; Zhong, Z.; Man, K.; Tsao, S.W.; Lao, L.; Feng, Y. Direct inhibition of the TLR4/MyD88 pathway by geniposide suppresses HIF-1α-independent VEGF expression and angiogenesis in hepatocellular carcinoma. Br. J. Pharmacol. 2020, 177, 3240–3257. [Google Scholar] [CrossRef]
- Gao, S.; Chen, T.; Li, L.; Liu, X.; Liu, Y.; Zhao, J.; Lu, Q.; Zeng, Z.; Xu, Q.; Huang, D.; et al. Hypoxia-Inducible Ubiquitin Specific Peptidase 13 Contributes to Tumor Growth and Metastasis via Enhancing the Toll-Like Receptor 4/Myeloid Differentiation Primary Response Gene 88/Nuclear Factor-κB Pathway in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 587389. [Google Scholar] [CrossRef]
- Song, X.; Ma, J. SRRM1 promotes the proliferation, migration, and invasion of hepatocellular carcinoma cells by regulating the JAK/STAT signaling pathway. Tissue Cell 2022, 79, 101954. [Google Scholar] [CrossRef]
- Yu, P.; Cheng, X.; Du, Y.; Huang, L.; Dong, R. TAK-242 can be the potential agents for preventing invasion and metastasis of hepatocellular carcinoma. Med. Hypotheses 2013, 81, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.Q.; Lu, C.W.; Zhang, L.; Yang, J.; Hameed, W.; Chen, W. Toll-like receptor 4 signaling promotes invasion of hepatocellular carcinoma cells through MKK4/JNK pathway. Mol. Immunol. 2015, 68, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ding, F.; Shen, X. Total flavonoids of Radix Tetrastigma suppress inflammation-related hepatocellular carcinoma cell metastasis. Mol. Genet. Genom. 2021, 296, 571–579. [Google Scholar] [CrossRef]
- Yang, L.Y.; Luo, Q.; Lu, L.; Zhu, W.W.; Sun, H.T.; Wei, R.; Lin, Z.F.; Wang, X.Y.; Wang, C.Q.; Lu, M.; et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J. Hematol. Oncol. 2020, 13, 3. [Google Scholar] [CrossRef]
- Zhan, X.; Wu, R.; Kong, X.; You, Y.; He, K.; Sun, X.; Huang, Y.; Chen, W.; Duan, L. Elevated neutrophil extracellular traps by HBV-mediated S100A9-TLR4/RAGE-ROS cascade facilitate the growth and metastasis of hepatocellular carcinoma. Cancer Commun. 2022, 43, 225–245. [Google Scholar] [CrossRef]
- Khanam, A.; Kottilil, S. New Therapeutics for HCC: Does Tumor Immune Microenvironment Matter? Int. J. Mol. Sci. 2023, 24, 437. [Google Scholar] [CrossRef] [PubMed]
- Arelaki, S.; Koletsa, T.; Sinakos, E.; Papadopoulos, V.; Arvanitakis, K.; Skendros, P.; Akriviadis, E.; Ritis, K.; Germanidis, G.; Hytiroglou, P. Neutrophil extracellular traps enriched with IL-1β and IL-17A participate in the hepatic inflammatory process of patients with non-alcoholic steatohepatitis. Virchows Arch. 2022, 481, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-associated neutrophils in hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers 2021, 13, 2899. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, X.D.; Wang, W.Q.; Shen, Y.; Qin, Y.; Ren, Z.G.; Sun, H.C.; Tang, Z.Y. Activation of β-catenin by hypoxia in hepatocellular carcinoma contributes to enhanced metastatic potential and poor prognosis. Clin. Cancer Res. 2010, 16, 2740–2750. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, F.; Li, S.; Cai, J.; Shi, J.; Jiang, Y. TLR4 Influences Hepatitis B Virus Related Hepatocellular Carcinoma by Regulating the Wnt/β-Catenin Pathway. Cell. Physiol. Biochem. 2017, 42, 469–479. [Google Scholar] [CrossRef]
- Son, J.; Kim, M.J.; Lee, J.S.; Kim, J.Y.; Chun, E.; Lee, K.Y. Hepatitis B virus X Protein Promotes Liver Cancer Progression through Autophagy Induction in Response to TLR4 Stimulation. Immune Netw. 2021, 21, e37. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Yang, D.; Yin, C.; Zhang, J. Androgen receptor (AR)-TLR4 crosstalk mediates gender disparities in hepatocellular carcinoma incidence and progression. J. Cancer 2020, 11, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Pope, E.D.; Kimbrough, E.O.; Vemireddy, L.P.; Surapaneni, P.K.; Copland, J.A.; Mody, K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert Opin. Ther. Targets 2019, 23, 473. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt–β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.T.; Jing, Y.Y.; Yu, G.F.; Han, Z.P.; Yu, D.D.; Fan, Q.M.; Ye, F.; Li, R.; Gao, L.; Zhao, Q.D.; et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015, 358, 136–143. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Liu, D.; Liu, J. LPS promotes epithelial–mesenchymal transition and activation of TLR4/JNK signaling. Tumor Biol. 2014, 35, 10429–10435. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.-Y.; Han, Z.-P.; Sun, K.; Zhang, S.-S.; Hou, J.; Liu, Y.; Li, R.; Gao, L.; Zhao, X.; Zhao, Q.-D.; et al. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 2012, 10, 98. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Liu, Y.; Dai, B.; Zhang, Y.H.; Zhang, P.F.; Shi, X.L. Sorafenib inhibits caspase-1 expression through suppressing TLR4/stat3/SUMO1 pathway in hepatocellular carcinoma. Cancer Biol. Ther. 2018, 19, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Uthaya Kumar, D.B.; Chen, C.L.; Liu, J.C.; Feldman, D.E.; Sher, L.S.; French, S.; Dinorcia, J.; French, S.W.; Naini, B.V.; Junrungsee, S.; et al. TLR4 Signaling via NANOG Cooperates with STAT3 to Activate Twist1 and Promote Formation of Tumor-Initiating Stem-Like Cells in Livers of Mice. Gastroenterology 2016, 150, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.R.; Li, J.H.; Zhang, R.; Chen, R.X.; Wang, Y.H. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J. Surg. Oncol. 2018, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular bases of drug resistance in hepatocellular carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef]
- Sasaki, R.; Kanda, T.; Fujisawa, M.; Matsumoto, N.; Masuzaki, R.; Ogawa, M.; Matsuoka, S.; Kuroda, K.; Moriyama, M. Different mechanisms of action of regorafenib and lenvatinib on toll-like receptor-signaling pathways in human hepatoma cell lines. Int. J. Mol. Sci. 2020, 21, 3349. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.P.; Ma, D.K.; Li, Z.; Zhang, H.F. MiR-145 regulates the chemoresistance of hepatic carcinoma cells against 5-fluorouracil by targeting toll-like receptor 4. Cancer Manag. Res. 2020, 12, 6165–6175. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.J.; Li, T.W.; Zhou, L.; Wong, T.L.; Liu, X.; Ma, V.W.S.; Lo, C.M.; Man, K.; Lee, T.K.; Ning, W.; et al. FSTL1 secreted by activated fibroblasts promotes hepatocellular carcinoma metastasis and stemness. Cancer Res. 2021, 81, 5692–5705. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Du, R.; Wang, Z.; Shen, W.; Gao, R.; Jiang, S.; Fang, Y.; Shi, Y.; Chang, A.; Liu, L.; et al. TLR4 increases the stemness and is highly expressed in relapsed human hepatocellular carcinoma. Cancer Med. 2019, 8, 2325–2337. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Dariya, B.; Kasa, P.; Peela, S.; El-Rayes, B.F. Epigenetics in hepatocellular carcinoma. Semin. Cancer Biol. 2022, 86, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xie, Y.; Zhong, X.; Fu, Y.; Huang, Y.; Zhen, Y.; Pan, P.; Wang, H.; Bartlett, D.L.; Billiar, T.R.; et al. Novel chemokine-like activities of histones in tumor metastasis. Oncotarget 2016, 7, 61728–61740. [Google Scholar] [CrossRef]
- Wei, X.; Liu, H.; Li, X.; Liu, X. Over-expression of MiR-122 promotes apoptosis of hepatocellular carcinoma via targeting TLR4. Ann. Hepatol. 2019, 18, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zheng, X.; Fan, Y.; Yang, X.; Li, A.; Qian, J. The contribution of miR-122 to the innate immunity by regulating toll-like receptor 4 in hepatoma cells. BMC Gastroenterol. 2019, 19, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Che, J.; Xu, L.; Yang, W.; Zhou, W.; Zhou, C. Tumor-derived extracellular vesicles containing long noncoding RNA PART1 exert oncogenic effect in hepatocellular carcinoma by polarizing macrophages into M2. Dig. Liver Dis. 2022, 54, 543–553. [Google Scholar] [CrossRef]
- Chen, I.T.; Cheng, A.C.; Liu, Y.T.; Yan, C.; Cheng, Y.C.; Chang, C.F.; Tseng, P.H. Persistent TLR4 Activation Promotes Hepatocellular Carcinoma Growth through Positive Feedback Regulation by LIN28A/Let-7g miRNA. Int. J. Mol. Sci. 2022, 23, 8419. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakou, P.; Tzardi, M.; Valatas, V.; Kanavaros, P.; Karydi, E.; Notas, G.; Xidakis, C.; Kouroumalis, E. Apoptosis and apoptosis related proteins in chronic viral liver disease. Apoptosis 2002, 7, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Kwan, S.Y.; Slayden, A.N.; Coronado, A.R.; Marquez, R.C.; Chen, H.; Wei, P.; Savage, M.I.; Vornik, L.A.; Fox, J.T.; Sei, S.; et al. Treatment Strategies and Mechanisms Associated with the Prevention of NASH-Associated HCC by a Toll-like Receptor 4 Inhibitor. Cancer Prev. Res. 2023, 16, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-X.; Yan, H.-X.; Liu, Q.; Yang, W.; Wu, H.-P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.-Q.; Zou, S.-S.; et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Bin He, H.; Wu, X.L.; Yu, B.; Liu, K.L.; Zhou, G.X.; Qian, G.Q.; da Ju, H.; Chen, X.Y. The effect of desacetyluvaricin on the expression of tlr4 and p53 protein in hepg 2.2.15. Hepat. Mon. 2011, 11, 364–367. [Google Scholar]
- Ding, Y.F.; Peng, Z.X.; Ding, L.; Peng, Y.R. Baishouwu extract suppresses the development of hepatocellular carcinoma via TLR4/MyD88/NF-κB pathway. Front. Pharmacol. 2019, 10, 389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, Q.; Yan, W.; Xiao, D.; Zeng, Z.; Ouyang, Y.; Huang, L.; Cai, J.; Zeng, X.; Chen, Y.J.; et al. CXC195 suppresses proliferation and inflammatory response in LPS-induced human hepatocellular carcinoma cells via regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem. Biophys. Res. Commun. 2015, 456, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.M.; Al-Wahaibi, L.H.; Shehatou, G.S.; El-Emam, A.A. Adamantane-linked isothiourea derivatives suppress the growth of experimental hepatocellular carcinoma via inhibition of TLR4-MyD88-NF-κB signaling. Am. J. Cancer Res. 2021, 11, 350–369. [Google Scholar] [PubMed]
- Qu, L.; Ma, X.; Fan, D. Ginsenoside Rk3 Suppresses Hepatocellular Carcinoma Development through Targeting the Gut-Liver Axis. J. Agric. Food Chem. 2021, 69, 10121–10137. [Google Scholar] [CrossRef]
- Kabel, A.M.; Arab, H.H.; Abd Elmaaboud, M.A. Attenuation of diethyl nitrosamine-induced hepatocellular carcinoma by taxifolin and/or alogliptin: The interplay between toll-like receptor 4, transforming growth factor beta-1, and apoptosis. Hum. Exp. Toxicol. 2021, 40, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Song, I.J.; Yang, Y.M.; Inokuchi-Shimizu, S.; Roh, Y.S.; Yang, L.; Seki, E. The contribution of toll-like receptor signaling to the development of liver fibrosis and cancer in hepatocyte-specific TAK1-deleted mice. Int. J. Cancer 2018, 142, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Elshaer, A.M.; El-kharashi, O.A.; Hamam, G.G.; Nabih, E.S.; Magdy, Y.M.; Abd El Samad, A.A. Involvement of TLR4/CXCL9/PREX-2 pathway in the development of hepatocellular carcinoma (HCC) and the promising role of early administration of lactobacillus plantarum in Wistar rats. Tissue Cell 2019, 60, 38–47. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, R.C.; Zheng, H.; Wang, B.; Liu, Y.; Liu, D.L.; Chen, J.; Xu, W.; Li, A.; Zhu, Y.; et al. Synergistic anti-tumor efficacy of sorafenib and fluvastatin in hepatocellular carcinoma. Oncotarget 2017, 8, 23265–23276. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Chang, A.; Wang, J.; Zhou, W.; Gao, R.; Li, J.; Xu, Y.; Luo, X.; Xiang, R.; Luo, N.; et al. TIFA, an inflammatory signaling adaptor, is tumor suppressive for liver cancer. Oncogenesis 2015, 4, e173. [Google Scholar] [CrossRef]
- Hartwell, H.J.; Petrosky, K.Y.; Fox, J.G.; Horseman, N.D.; Rogers, A.B. Prolactin prevents hepatocellular carcinoma by restricting innate immune activation of c-Myc in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 11455–11460. [Google Scholar] [CrossRef] [PubMed]
- Saber, S.; Ghanim, A.M.H.; El-Ahwany, E.; El-Kader, E.M.A. Novel complementary antitumour effects of celastrol and metformin by targeting IκBκB, apoptosis and NLRP3 inflammasome activation in diethylnitrosamine-induced murine hepatocarcinogenesis. Cancer Chemother. Pharmacol. 2020, 85, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, R.; Huang, Z.; Li, H.; Zhu, H. Lipopolysaccharide-induced toll-like receptor 4 signaling in cancer cells promotes cell survival and proliferation in hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 2223–2236. [Google Scholar] [CrossRef] [PubMed]
- Badawy, M.M.M.; Abdel-Hamid, G.R.; Mohamed, H.E. Antitumor Activity of Chitosan-Coated Iron Oxide Nanocomposite Against Hepatocellular Carcinoma in Animal Models. Biol. Trace Elem. Res. 2022, 201, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Zhe, Y.; Li, Y.; Liu, D.; Su, D.M.; Liu, J.G.; Li, H.Y. Extracellular HSP70-peptide complexes promote the proliferation of hepatocellular carcinoma cells via TLR2/4/JNK1/2MAPK pathway. Tumor Biol. 2016, 37, 13951–13959. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wang, G.; Zhao, H.; Zhang, Y.; Han, Q.; Zhang, C.; Tian, Z.; Zhang, J. TLR4 signaling promotes a COX-2/PGE2/STAT3 positive feedback loop in hepatocellular carcinoma (HCC) cells. Oncoimmunology 2016, 5, e1074376. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, J.; Chen, S.; Zhou, Z.; Lin, N. Lipopolysaccharide promotes angiogenesis in mice model of HCC by stimulating hepatic stellate cell activation via TLR4 pathway. Acta Biochim. Biophys. Sin. 2017, 49, 1029–1034. [Google Scholar] [CrossRef]
- Quiroz Reyes, A.G.; Lozano Sepulveda, S.A.; Martinez-Acuña, N.; Islas, J.F.; Gonzalez, P.D.; Heredia Torres, T.G.; Perez, J.R.; Garza Treviño, E.N. Cancer Stem Cell and Hepatic Stellate Cells in Hepatocellular Carcinoma. Technol. Cancer Res. Treat. 2023, 22, 15330338231163677. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Martinelli, E.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Machairas, N.; Tsilimigras, D.I.; Pawlik, T.M. Current Landscape of Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Cancers 2022, 14, 2018. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; De Vries, I.J.M.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Cheng, C.; Lin, Z.; Jiang, R.; Zhao, W.; Yan, X.; Tang, J.; Yao, K.; Sun, B.; Chen, Y. The attenuated hepatocellular carcinoma-specific Listeria vaccine Lmdd-MPFG prevents tumor occurrence through immune regulation of dendritic cells. Oncotarget 2015, 6, 8822–8838. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.; Egea, J.; Villanueva, L.; Ruiz, M.; Llopiz, D.; Repáraz, D.; Aparicio, B.; Lasarte-cia, A.; Lasarte, J.J.; de Galarreta, M.R.; et al. Cold-inducible rna binding protein as a vaccination platform to enhance immunotherapeutic responses against hepatocellular carcinoma. Cancers 2020, 12, 3397. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yada, N.; Watanabe, T.; Arizumi, T.; Hagiwara, S.; Ueshima, K.; Nishida, N.; Fujita, J.; Kudo, M. Cold-inducible RNA-binding protein promotes the development of liver cancer. Cancer Sci. 2015, 106, 352–358. [Google Scholar] [CrossRef]
- Zheng, Q.C.; Yang, J.; Li, M. Emerging role of Toll-like receptor 4 in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2015, 2, 11. [Google Scholar] [CrossRef]
- Sepehri, Z.; Kiani, Z.; Kohan, F.; Alavian, S.M.; Ghavami, S. Toll like receptor 4 and hepatocellular carcinoma; A systematic review. Life Sci. 2017, 179, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Giannou, A.D.; Lücke, J.; Kleinschmidt, D.; Shiri, A.M.; Steglich, B.; Nawrocki, M.; Zhang, T.; Zazara, D.E.; Kempski, J.; Zhao, L.; et al. A Critical Role of the IL-22-IL-22 Binding Protein Axis in Hepatocellular Carcinoma. Cancers 2022, 14, 6019. [Google Scholar] [CrossRef]
- Kuang, D.M.; Xiao, X.; Zhao, Q.; Chen, M.M.; Li, X.F.; Liu, R.X.; Wei, Y.; Ouyang, F.Z.; Chen, D.P.; Wu, Y.; et al. B7-H1-expressing antigen-presenting cells mediate polarization of protumorigenic Th22 subsets. J. Clin. Investig. 2014, 124, 4657–4667. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, J.X.; Wang, H.; Wang, G.L.; Hu, Q.G.; Zheng, Q.C. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression via treg requires TLR4 signaling. World J. Gastroenterol. 2012, 18, 2938–2947. [Google Scholar] [CrossRef]
- Li, C.X.; Ling, C.C.; Shao, Y.; Xu, A.; Li, X.C.; Ng, K.T.P.; Liu, X.B.; Ma, Y.Y.; Qi, X.; Liu, H.; et al. CXCL10/CXCR3 signaling mobilized-regulatory T cells promote liver tumor recurrence after transplantation. J. Hepatol. 2016, 65, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Lao, X.-M.; Chen, M.-M.; Liu, R.-X.; Wei, Y.; Ouyang, F.-Z.; Chen, D.-P.; Zhao, X.-Y.; Zhao, Q.; Li, X.-F.; et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016, 6, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Tatsumi, T.; Miyagi, T.; Tsunematsu, H.; Aketa, H.; Hosui, A.; Kanto, T.; Hiramatsu, N.; Hayashi, N.; Takehara, T. α-Fetoprotein impairs activation of natural killer cells by inhibiting the function of dendritic cells. Clin. Exp. Immunol. 2011, 165, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ramjiawan, R.R.; Reiberger, T.; Ng, M.R.; Hato, T.; Huang, Y.; Ochiai, H.; Kitahara, S.; Unan, E.C.; Reddy, T.P.; et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 2015, 61, 1591–1602. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ling, C.C.; Yeung, W.H.O.; Pang, L.; Liu, J.; Zhou, J.; Zhang, W.Y.; Liu, X.B.; Ng, T.P.K.; Yang, X.X.; et al. Monocytic MDSC mobilization promotes tumor recurrence after liver transplantation via CXCL10/TLR4/MMP14 signaling. Cell Death Dis. 2021, 12, 489. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.E.; Gan, J.; Zhang, R.D.; Cheng, Y.R.; Huang, G.J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand. J. Gastroenterol. 2011, 46, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef]
- Miura, K.; Ishioka, M.; Minami, S.; Horie, Y.; Ohshima, S.; Goto, T.; Ohnishi, H. Toll-like receptor 4 on macrophage promotes the development of steatohepatitis-related hepatocellular carcinoma in mice. J. Biol. Chem. 2016, 291, 11504–11517. [Google Scholar] [CrossRef]
- Li, W.; Xiao, J.; Zhou, X.; Xu, M.; Hu, C.; Xu, X.; Lu, Y.; Liu, C.; Xue, S.; Nie, L.; et al. STK4 regulates TLR pathways and protects against chronic inflammation-related hepatocellular carcinoma. J. Clin. Investig. 2015, 125, 4239–4254. [Google Scholar] [CrossRef]
- Min, L.; Wang, H.; Qi, H. Astragaloside IV inhibits the progression of liver cancer by modulating macrophage polarization through the TLR4/NF-κB/STAT3 signaling pathway. Am. J. Transl. Res. 2022, 14, 1551–1566. [Google Scholar]
- Pan, C.; Wu, Q.; Wang, S.; Mei, Z.; Zhang, L.; Gao, X.; Qian, J.; Xu, Z.; Zhang, K.; Su, R.; et al. Combination with Toll-like receptor 4 (TLR4) agonist reverses GITR agonism mediated M2 polarization of macrophage in Hepatocellular carcinoma. Oncoimmunology 2022, 11, 2073010. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.T.; Li, C.Y.; Huang, Y.H.; Chang, T.S.; Lin, C.Y.; Chuang, C.H.; Wang, C.Y.; Anuraga, G.; Chang, T.H.; Shih, T.C.; et al. Galectin-1 orchestrates an inflammatory tumor-stroma crosstalk in hepatoma by enhancing TNFR1 protein stability and signaling in carcinoma-associated fibroblasts. Oncogene 2022, 41, 3011–3023. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-Q.; Tang, Z.; Huang, R.; Qu, W.-F.; Fang, Y.; Yang, R.; Tao, C.-Y.; Gao, J.; Wu, X.-L.; Sun, H.-X.; et al. CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023, 9, 25. [Google Scholar] [CrossRef]
- Liu, W.T.; Jing, Y.Y.; Gao, L.; Li, R.; Yang, X.; Pan, X.R.; Yang, Y.; Meng, Y.; Hou, X.J.; Zhao, Q.D.; et al. Lipopolysaccharide induces the differentiation of hepatic progenitor cells into myofibroblasts constitutes the hepatocarcinogenesis-associated microenvironment. Cell Death Differ. 2020, 27, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.M.; Lu, J.F.; Edoo, M.I.A.; Zhou, L.; Xie, H.Y.; Zheng, S.S.; Li, Q.Y. MRC-5 cancer-associated fibroblasts influence production of cancer stem cell markers and inflammation-associated cell surface molecules, in liver cancer cell lines. Int. J. Med. Sci. 2019, 16, 1157–1170. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, J.; Wang, Q.; Chen, P.; Hong, Y.; He, X.; Chen, D.; Liu, H.; Wang, Y.; Cai, X. The Invasive Potential of Hepatoma Cells Induced by Radiotherapy is Related to the Activation of Hepatic Stellate Cells and Could be Inhibited by EGCG Through the TLR4 Signaling Pathway. Radiat. Res. 2022, 197, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, C.A.M.; Scheiner, B.; Korolewicz, J.; Stikas, C.-V.; Gennari, A.; Vincenzi, B.; Openshaw, M.R.; Silletta, M.; Pinter, M.; Cortellini, A.; et al. Efficacy and safety of frontline systemic therapy for advanced HCC: A network meta-analysis of landmark phase III trials. JHEP Rep. 2023, 5, 100702. [Google Scholar] [CrossRef] [PubMed]
- Minaei, N.; Ramezankhani, R.; Tamimi, A.; Piryaei, A.; Zarrabi, A.; Aref, A.R.; Mostafavi, E.; Vosough, M. Immunotherapeutic approaches in Hepatocellular carcinoma: Building blocks of hope in near future. Eur. J. Cell Biol. 2023, 102, 151284. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, 7250–7257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Lv, T.; Xiong, X.; Yan, W.; Liu, M.; Xu, H.; He, Q. Targeting of GSDMD sensitizes HCC to anti-PD-1 by activating cGAS pathway and downregulating PD-L1 expression. J. Immunother. Cancer 2022, 10, e004763. [Google Scholar] [CrossRef]
- Xu, G.; Feng, D.; Yao, Y.; Li, P.; Sun, H.; Yang, H.; Li, C.; Jiang, R.; Sun, B.; Chen, Y. Listeria-based hepatocellular carcinoma vaccine facilitates anti-PD-1 therapy by regulating macrophage polarization. Oncogene 2019, 39, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Uriell-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Himmelsbach, V.; Pinter, M.; Scheiner, B.; Venerito, M.; Sinner, F.; Zimpel, C.; Marquardt, J.U.; Trojan, J.; Waidmann, O.; Finkelmeier, F. Efficacy and Safety of Atezolizumab and Bevacizumab in the Real-World Treatment of Advanced Hepatocellular Carcinoma: Experience from Four Tertiary Centers. Cancers 2022, 14, 1722. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Cortellini, A. Antibiotic Therapy: The Cornerstone of Iatrogenic Resistance to Immune Checkpoint Inhibitors. J. Clin. Oncol. 2023. [Google Scholar] [CrossRef]
- Eng, L.; Sutradhar, R.; Niu, Y.; Liu, N.; Liu, Y.; Kaliwal, Y.; Powis, M.L.; Liu, G.; Peppercorn, J.M.; Bedard, P.L.; et al. Impact of Antibiotic Exposure Before Immune Checkpoint Inhibitor Treatment on Overall Survival in Older Adults with Cancer: A Population-Based Study. J. Clin. Oncol. 2023. [Google Scholar] [CrossRef]
- Donne, R.; Lujambio, A. The liver cancer immune microenvironment: Therapeutic implications for hepatocellular carcinoma. Hepatology 2022, 77, 1773–1796. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, K.; Mitroulis, I.; Chatzigeorgiou, A.; Elefsiniotis, I.; Germanidis, G. The Liver Cancer Immune Microenvironment: Emerging Concepts for Myeloid Cell Profiling with Diagnostic and Therapeutic Implications. Cancers 2023, 15, 1522. [Google Scholar] [CrossRef] [PubMed]
- Montironi, C.; Castet, F.; Haber, P.K.; Pinyol, R.; Torres-Martin, M.; Torrens Fontanals, L.; Mesropian, A.; Wang, H.; Puigvehi, M.; Maeda, M.; et al. Inflamed and non-inflamed classes of HCC: A revised immunogenomic classification. Gut 2022, 72, 129–140. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.K.; Castet, F.; Torres-Martin, M.; Andreu-Oller, C.; Puigvehí, M.; Miho, M.; Radu, P.; Dufour, J.F.; Verslype, C.; Czauderna, C.; et al. Molecular Markers of Response to Anti-PD1 Therapy in Advanced Hepatocellular Carcinoma. Gastroenterology 2023, 164, 72–88.e18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.H.; He, A.R.; Ryoo, B.Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef]
- Gupta, A.; Kurzrock, R.; Adashek, J.J. Evolution of the Targeted Therapy Landscape for Cholangiocarcinoma: Is Cholangiocarcinoma the ‘NSCLC’ of GI Oncology? Cancers 2023, 15, 1578. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, X.; Du, X.; Zhang, H.; Wu, Z.; Ren, K.; Han, X. The interaction between lncRNA SNHG1 and miR-140 in regulating growth and tumorigenesis via the TLR4/NF-kB pathway in cholangiocarcinoma. Oncol. Res. 2019, 27, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Hu, Y.; Hu, M.; Xu, Y.; Chen, M.; Du, C.; Cui, J.; Zheng, P.; Lai, J.; Zhang, Y.; et al. S100A8 facilitates cholangiocarcinoma metastasis via upregulation of VEGF through TLR4/NF-κB pathway activation. Int. J. Oncol. 2019, 56, 101–112. [Google Scholar] [CrossRef]
- Xiao, F.; Xu, F.; Zhang, H.; Shuai, X. Circ_0000591 served as endogenous RNA for miR-326 to promote progression of cholangiocarcinoma via the TLR4/MyD88/IL6 axis. Biochem. Biophys. Res. Commun. 2022, 600, 101–108. [Google Scholar] [CrossRef]
- Foerster, F.; Gairing, S.J.; Ilyas, S.I.; Galle, P.R. Emerging immunotherapy for HCC: A guide for hepatologists. Hepatology 2022, 75, 1604–1626. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Mechanism of Chemoresistance (MOC) | Cellular Function |
|---|---|
| MOC-1a | Uptake carriers |
| MOC-1b | Export pumps |
| MOC-2 | Drug metabolism |
| MOC-3 | Therapeutic/Drug targets |
| MOC-4 | DNA repair |
| MOC-5a | Apoptosis potentiation |
| MOC-5b | Pro-survival signaling |
| MOC-6 | Tumor microenvironment modification |
| MOC-7 | EMT regulation |
| Molecule | Chemotherapeutic | Cell Lines | Signaling Pathway | Outcomes | References |
|---|---|---|---|---|---|
| miR-145 | 5-fluorouracil (5-FU) | SNU449 cells, SNU449/5-FU and Huh7 cells, Huh7/5-FU cells | TLR4/MyD88 | Drug sensitivity | [84] |
| Upregulation of caspase3, caspase9, Bax | |||||
| Downregulation of Bcl-2 | |||||
| Follistatin-like 1 (FSTL1)-neutralizing antibodies | Sorafenib | MHCC97L cells | TLR4/AKT /mTOR/4EBP1 | Drug sensitivity | [85] |
| COX6A2 and FAO inhibition | Sorafenib | CD133+/CD49f+ TICs from human HCCs | TLR4/E2F1/NANOG | Drug sensitivity | [81] |
| Increased cytochrome release | |||||
| LPS | Doxorubicin | SMMC-7721, Hep-3B cells | TLR4/AKT/SOX2 | Drug resistance | [86] |
| Enhancement of apoptosis |
| Molecule | Cell Lines | Signaling Pathways | Outcome | References |
|---|---|---|---|---|
| miR-145 | SNU449 cells, SNU449/5-FU and Huh7 cells, Huh7 /5-FU cells | TLR4/MyD88 | Increase 5-FU sensitivity | [84] |
| Upregulation of Caspase3, Caspase9, Bax | ||||
| Downregulation of Bcl-2 | ||||
| histone | Hepa1-6 cells, HuH7 cells and TLR4−/− mice or TLR4 knockdown cells” | TLR4/ERK/NF-κB/CCL9-10 | Enhancement of migration/invasion and metastasis | [91] |
| miR-122 mimic | Hep3B and MHCC97H cells | PI3K/Akt/NF-κB | Immune escape | [92] |
| Downregulation of VEGF, IL-6, COX-2, PGE2, MMP-9 | ||||
| miR-122 mimic | HepG2 and Huh7 cells | Linkage to 3′ UTR of TLR4 | Downregulation of proliferation | [93] |
| Downregulation of TNFα and IL-6 | ||||
| lncRNA PART1 or miR-372-3p blocker | HB611, Huh7, HCCLM3, Bel 7405, THLE-2 cells and human monocytes THP 1 cells | miR-372-3p/TLR4 axis | M2 TAM polarization | [94] |
| Enhancement of proliferation, migration, EMT | ||||
| let-7g miRNA | PLC5 cells | TLR4/LIN28A/let-7g | Positive feedback loop enhancing stemness and proliferation | [95] |
| Downregulation of TLR4 mRNA |
| Molecule | Cell Lines/Animals | Signaling Pathway | Outcome | Refs |
|---|---|---|---|---|
| LPS | antibiotics + DEN treated rats, TLR4−/− mice, TLR4-mutant mice | LPS/TLR4/NF-κB in HCC cells and Kuppfer cells | TLR4/NF-κB signaling in Kuppfer cells: IL-6, TNFα | [98] |
| TLR4/NF-κB signaling in Kuppfer cells: upregulation of antioxidant enzyme superoxide dismutase (SOD) and anti-apoptotic molecules (A20, Bcl-xl) | ||||
| Desacetyluvaricin (DES) | HepG2.2.15 cells as controls, DES, cisplatin | LPS/P53 | Increased TLR4 expression in DES vs. control (71.94% vs. 37.16%, p < 0.05) | [99] |
| Increased P53 expression (32.6% vs. 3.3%, p < 0.05) | ||||
| Baishouwu extracts | Baishouwu-treated /non-treated rats | TLR4/MyD88/NF-κB in myofibroblasts and HCCs | Downregulation of DEN-related liver inflammation, fibrosis, HCC | [100] |
| CXC195 | HepG2 cells | TLR4/NF-κB TLR4/MAPK TLR4/MyD88/TAK1 | Downregulation of TLR4 expression | [101] |
| Partial downregulation of cell cycle (increase cells in G0/G1 phase) | ||||
| Downregulation of inflammatory cascade: IL-6, CCL-2, CCL-22, EGFR | ||||
| Adamantane byproducts | Thioacetamide (TAA)-induced HCC, Hep-G2 cells, HCC-DOXO rats | TLR4/MyD88/NF-κB | Downregulation of TLR4, MyD88, TRAF-6, p-NF-κB, p65 | [102] |
| Downregulation of liver fibrosis | ||||
| ginsenoside Rk3 | dimethyl nitrosamine-, CCl4-mediated HCC mouse model | LPS/TLR4 signaling | Inhibition of liver damage, fibrosis, and tumor burden | [103] |
| Induction of apoptosis: increased Bax, decreased Bcl-2 expression | ||||
| taxifolin/alogliptin | diethyl nitrosamine-related HCC in rats | Increased survival rate | [104] | |
| Marked upregulation of NRF2 expression | ||||
| Downregulation of IL-1a, TLR4 expression | ||||
| Upregulation of beclin-1, caspase-3, caspase-9, JNK | ||||
| USP13 | SK-HEP-1, HepG2, Huh7, Hep3B | TLR4/MyD88/NF-κB | USP13 silencing: downregulation of TLR4 signaling | [59] |
| BMS345541 (IKK inhibitor), WP1066 (STAT3 inhibitor) | Huh7, PLC5 cells | positive feedback loop: TLR4/LIN28A/let-7g TLR4/STAT3/NF-κB TLR4/PI3K/Akt | Upregulation of TLR4, IL-6 and CCL2 expression | [95] |
| Enhanced survival and proliferation | ||||
| double knockout mice lacking Tak1 in liver | TLR4/MyD88/TNFR | Liver damage, macrophage infiltration, collagen degradation and tumor growth | [105] | |
| L. Plantarum | Wistar rats and/or L. Plantarum-, TAA-treated | TLR4/CXCL9/PREX-2 | Increased mean hep par-1 density | [106] |
| Sorafenib plus Fluvastatin | HepG2, SK-Hep-1 cells, LX-2 cells, DEN-treated rats | HCC cells: TLR4/MAPK Hepatic stellate cells: TLR4/NF-κB/SDF1α/MAPK | HCC cells: Decreased proliferation, Increased apoptosis | [107] |
| Hepatic stellate cells: Decreased activation, fibrosis | ||||
| T2BP | SK-Hep1, HepG2 cells/male NOD/SCID mice | TLR4/NF-κB | p53-independent activation of caspase 3, 8 | [108] |
| Induction of p53-mediated cell cycle arrest | ||||
| miR-122 mimic | Hep3B and MHCC97H cells | PI3K/Akt/NF-κB | Immune escape | [92] |
| Downregulation of VEGF, IL-6, COX-2, PGE2, and MMP-9 | ||||
| PRL | male and female C3H/HeN mice, Hep3B, HuH7, PH5CH8, HepG2, and HuH6 cells | PRL/PRLR signaling | PRL/PRLR-mediated downregulation of IL-1β, TNFα and TLR4 signaling (ubiquitination of “trafosome”) leading to inactivation of c-myc | [109] |
| Celastrol plus metformin | diethylnitrosamine (DEN)-treated BALB/c mice | Drug-mediated downregulation of TNFR, TLR4 mRNA expression leading to NF-κB inactivation | Enhancement of apoptosis | [110] |
| Downregulation of pyroptosis | ||||
| LPS (TLR4 activator) | HL-7702, PLC/PRF/5, HepG2 | LPS/TLR4/MAPK LPS/TLR4/ERK/JNK LPS/TLR4/p38 | Positive correlation of TLR4 and Ki-67 | [111] |
| Increased Bax translocation to mitochondria | ||||
| p38 downregulation induces HCC proliferation | ||||
| Chitosan-coated iron oxide nanocomposite (Fe3O4/Cs) | Fe3O4/Cs- and/or DEN-treated male albino rats | PI3K/Akt/mTOR, MAPK (ERK, JNK, P38) | Fe3O4/Cs-mediated downregulation of TLR4 expression | [112] |
| Downregulation of both PI3K/Akt/mTOR, MAPK signaling | ||||
| Upregulation of caspase-3 | ||||
| Extracellular HSP70-peptide complexes | HepG2 cells | TLR2-TLR4/JNK1/2/MAPK | Upregulation of TLR2- and TLR4-overexpressing cells mediated by cyclin D1 | [113] |
| TLR4 | Male nude BALB/C mice/HepG2.2.15, MHCC97-H, Hep3B cells | TLR4/b-catenin | Reduction of b-catenin-mediated HCC apoptosis | [70] |
| Enhancement of HCC proliferation | ||||
| TLR4 | HepG2, H7402 cells /TLR4−/− mice | TLR4/COX-2/PGE2/STAT3 signaling | Upregulation of cell cycle (more cells in S phase, increased cyclin D1) | [114] |
| anti-IL-17A | methionine-choline deficient (MCD) or high-fat MCD (HFMCD) FGF21 knockout (KO) mice/Hepa1-6-FGF21KD, FL83B-FGF21KD, 3T3-L1 cells | free fatty acids/TLR4/NF-κB /IL-17a | Decreased tumor growth and nodule burden in FGF21 knockout (KO) mice | [115] |
| Dihydrotestosterone (DHT) | DEN- or CCl4-induced HCC/HepG2, H7402, Hepa1-6, and HepG2.2.15” | Androgen receptor (AR)-TLR4 cross-signaling | Enhanced Ki-67 in male wild-type mice compared to female | [72] |
| Ki-67 equalization in TLR4−/− mice |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Lekakis, V.; Davakis, S.; Christodoulou, M.-I.; Germanidis, G.; Theocharis, S. The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks? Cancers 2023, 15, 2795. https://doi.org/10.3390/cancers15102795
Papadakos SP, Arvanitakis K, Stergiou IE, Lekakis V, Davakis S, Christodoulou M-I, Germanidis G, Theocharis S. The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks? Cancers. 2023; 15(10):2795. https://doi.org/10.3390/cancers15102795
Chicago/Turabian StylePapadakos, Stavros P., Konstantinos Arvanitakis, Ioanna E. Stergiou, Vasileios Lekakis, Spyridon Davakis, Maria-Ioanna Christodoulou, Georgios Germanidis, and Stamatios Theocharis. 2023. "The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks?" Cancers 15, no. 10: 2795. https://doi.org/10.3390/cancers15102795