


Cholecystokinin Receptor Antagonist Induces Pancreatic Stellate Cell Plasticity Rendering the Tumor Microenvironment Less Oncogenic

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Peptides and Receptor Antagonists
2.3. Relative Gene Expression by Quantitative RT-PCR
2.4. Migration Assay
2.5. Western Immunoblotting
2.6. Proline and 4-Hydroxyproline Analysis by Mass Spectroscopy
2.7. Stellate Cell Proliferation Assay
2.8. Statistics
3. Results
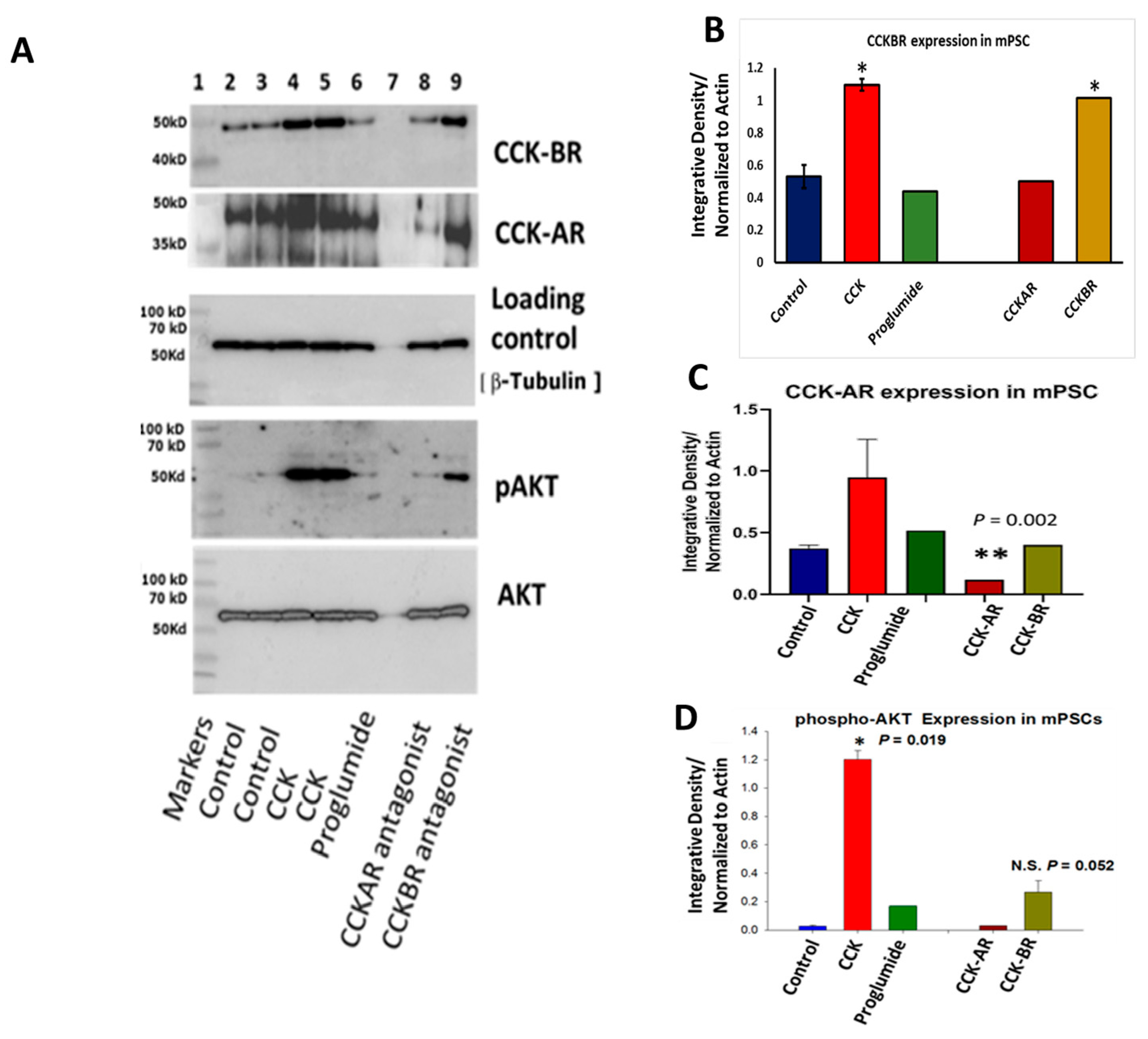
3.1. PSCs Express CCK-A and CCK-B Receptor Protein by Western Blot
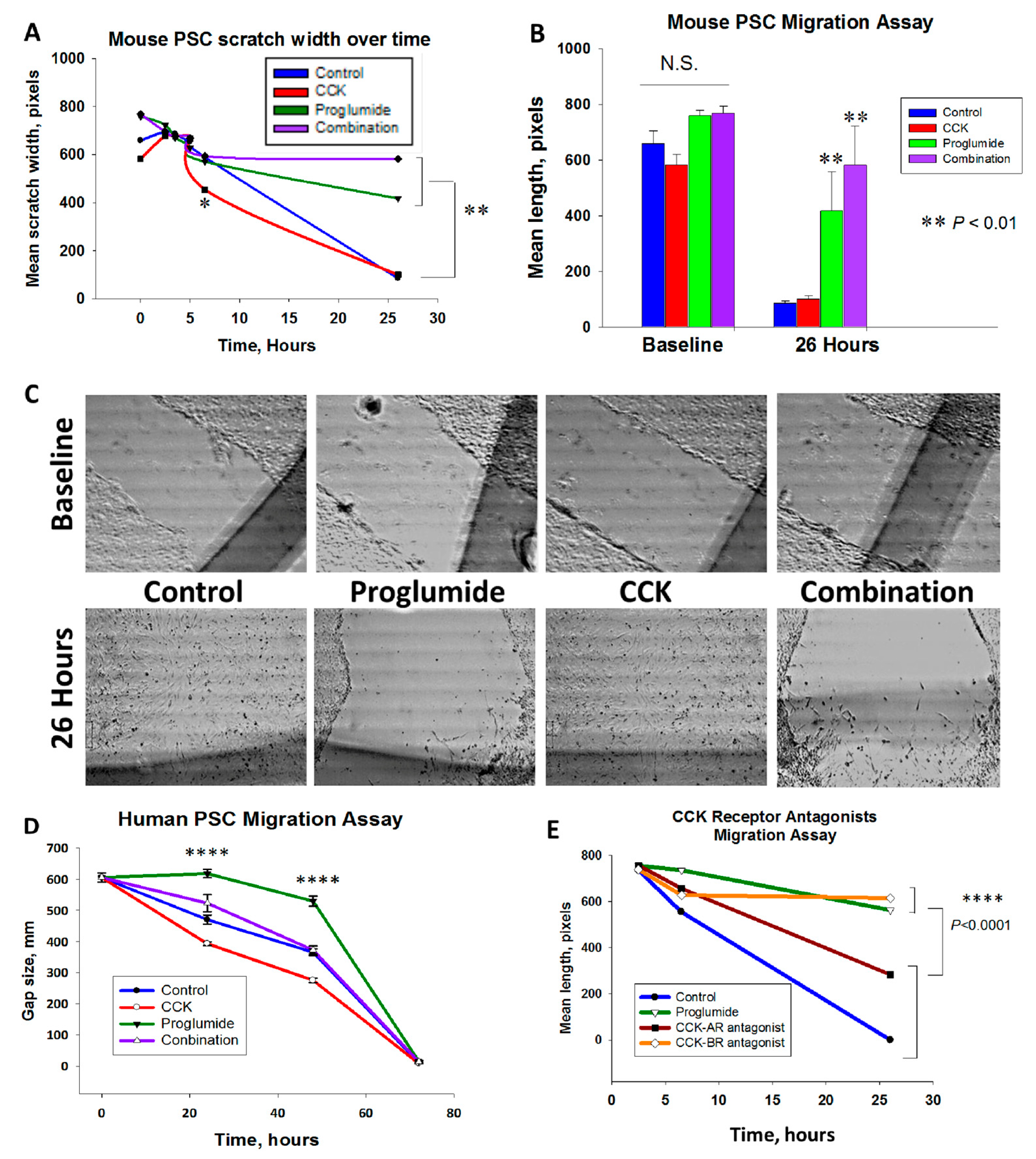
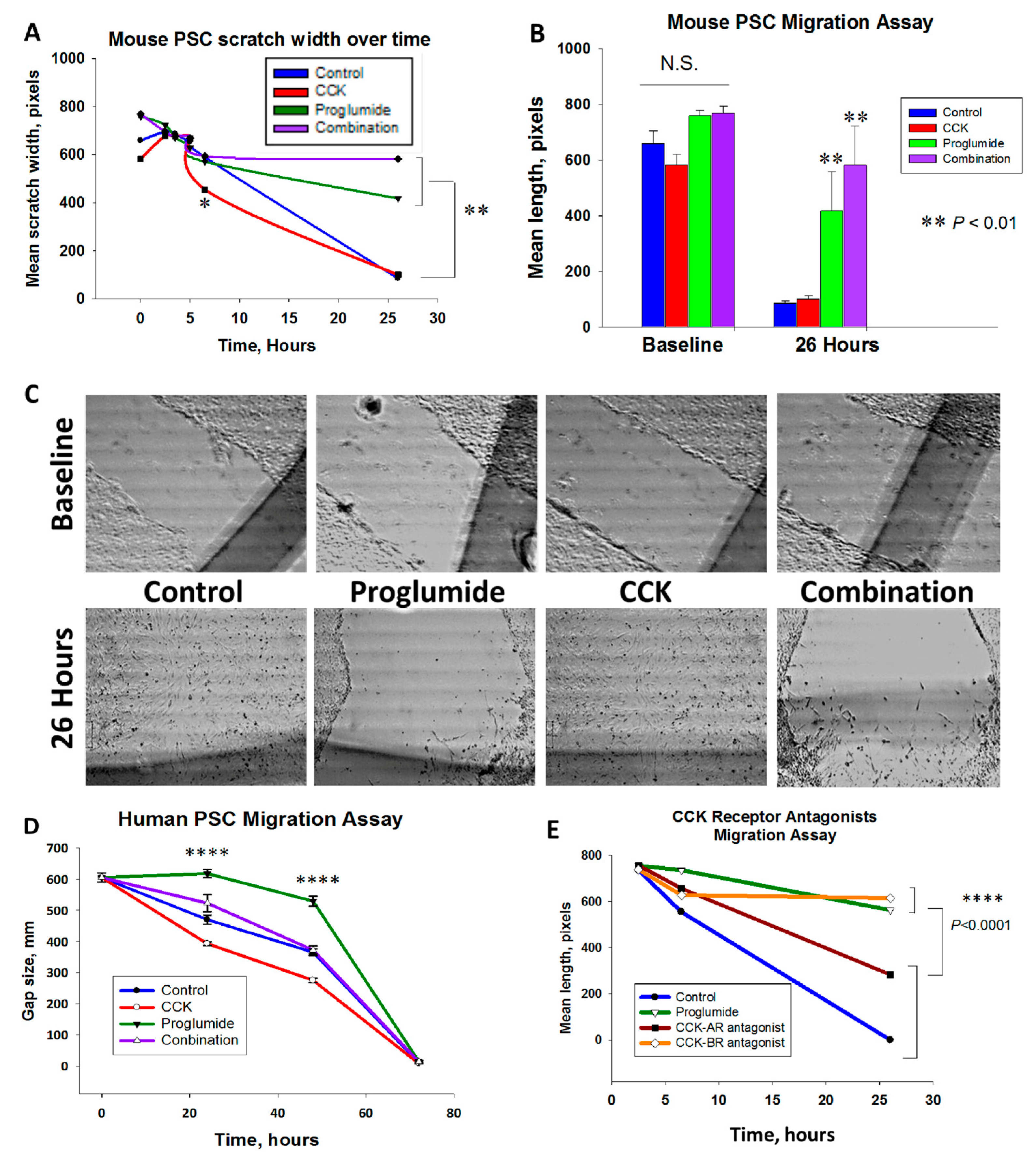
3.2. Proglumide Therapy Significantly Reduced PSC Migration
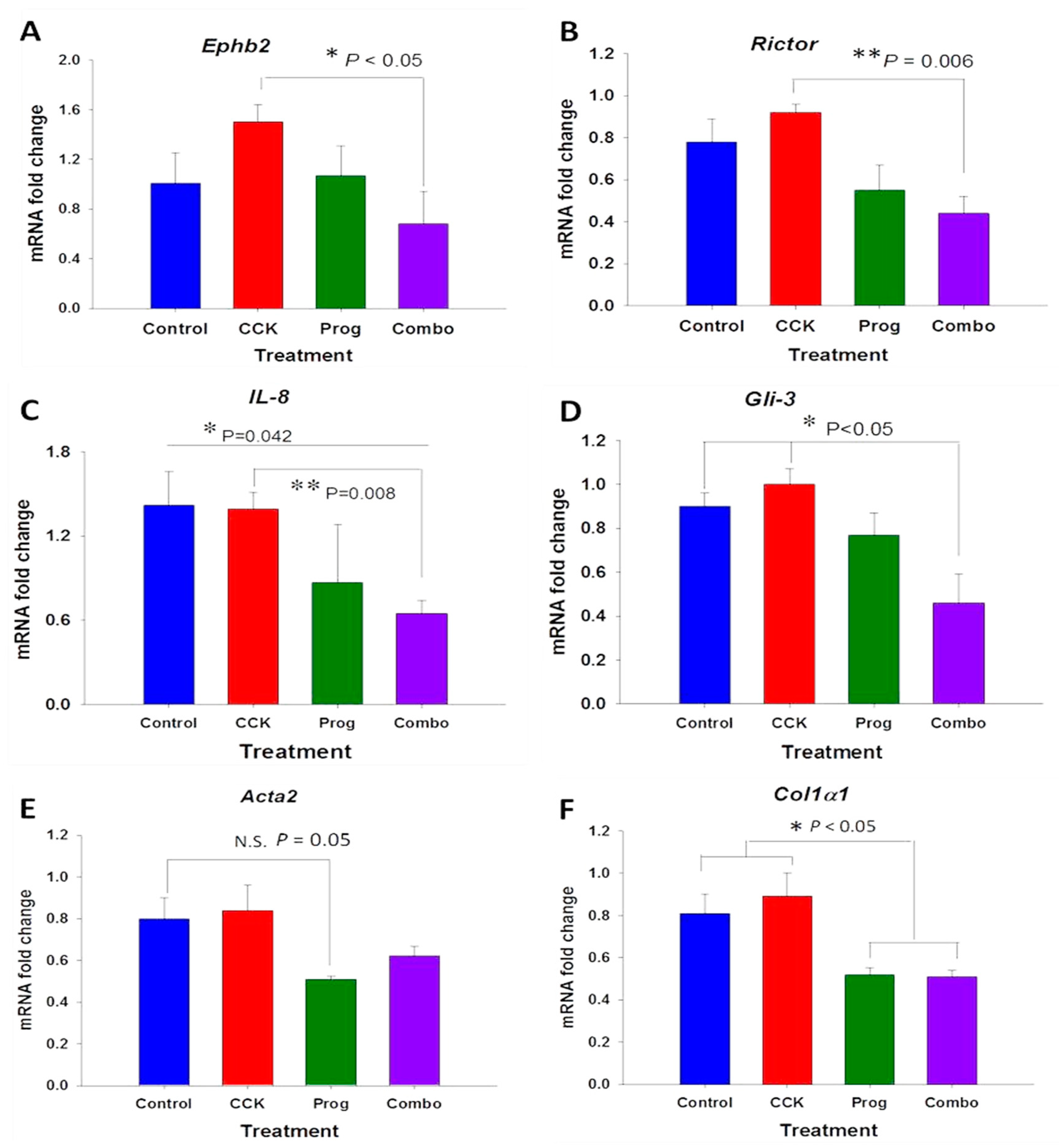
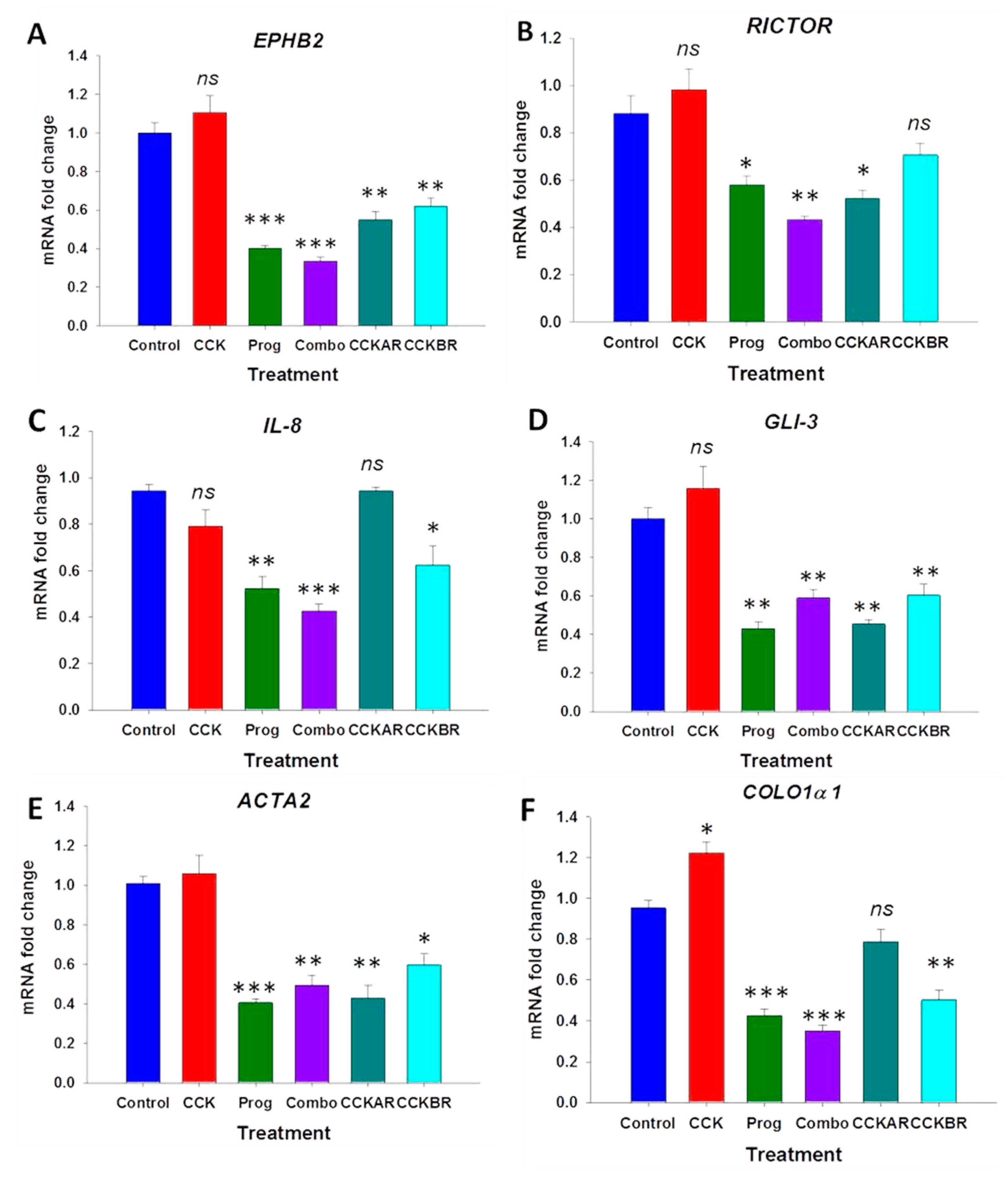
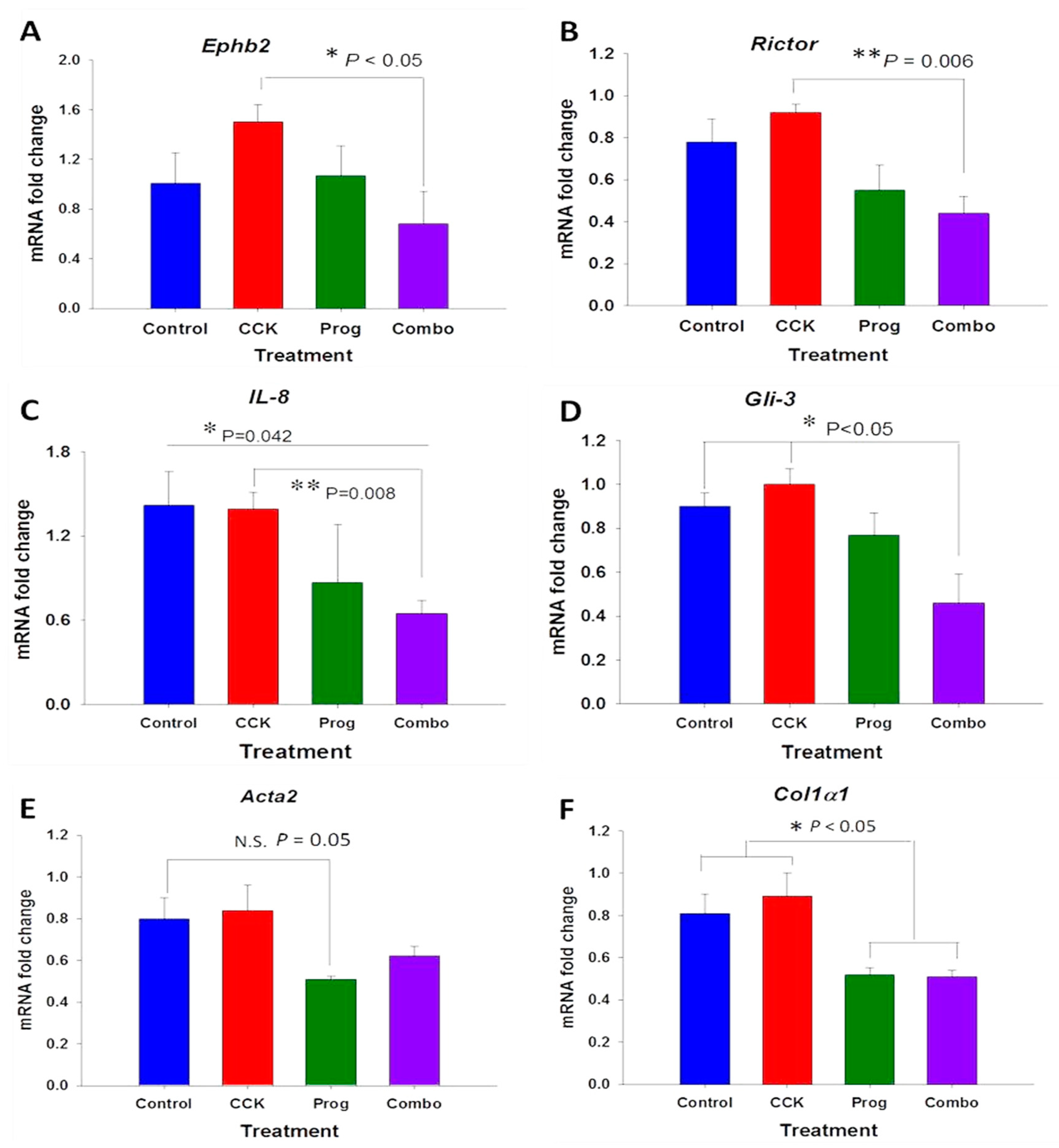
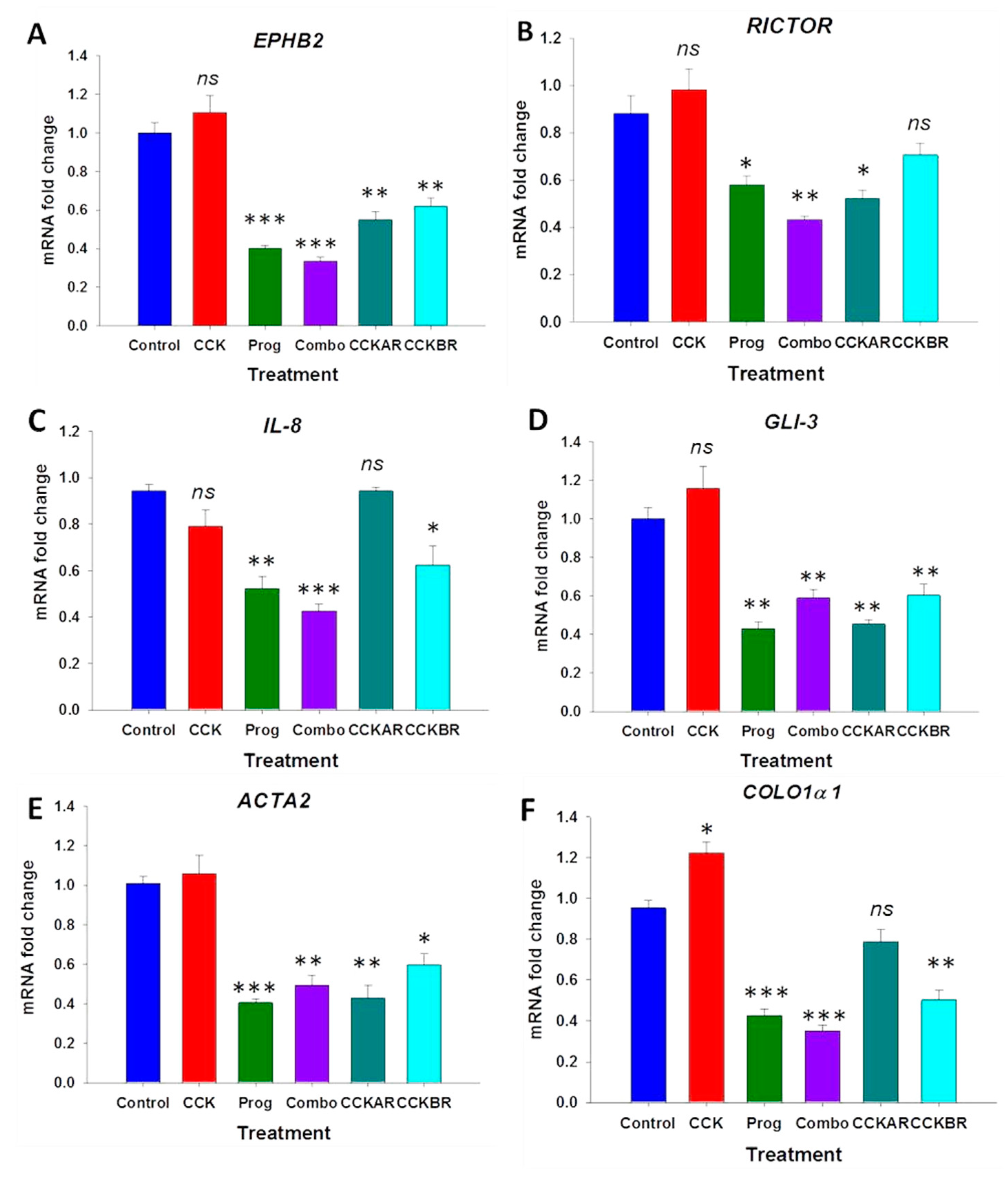
3.3. Effects of CCK and Proglumide Treatment on mPSCs and hPSCs Alters Differently Expressed Genes Associated with the Pancreatic Extracellular Matrix (ECM)
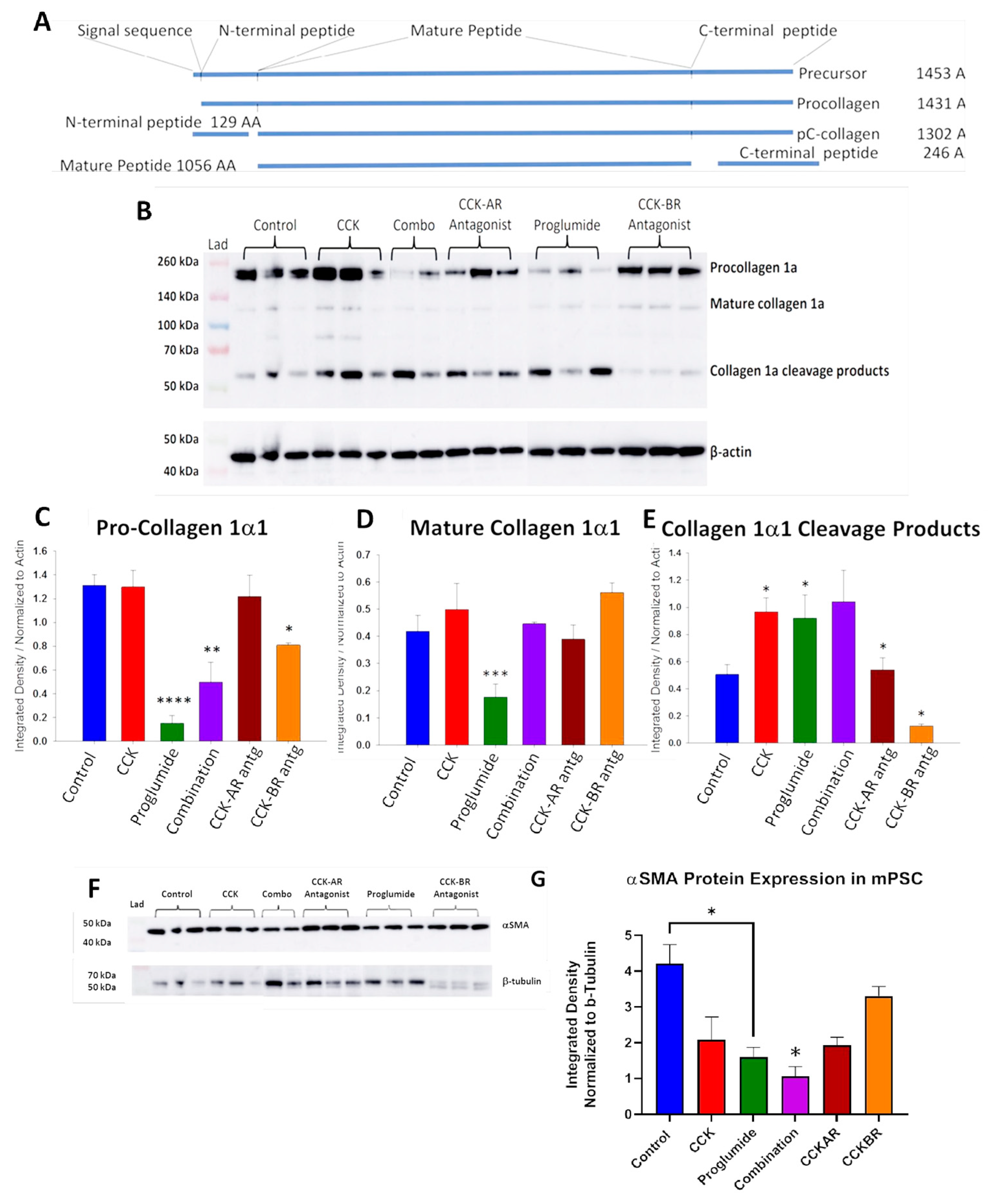
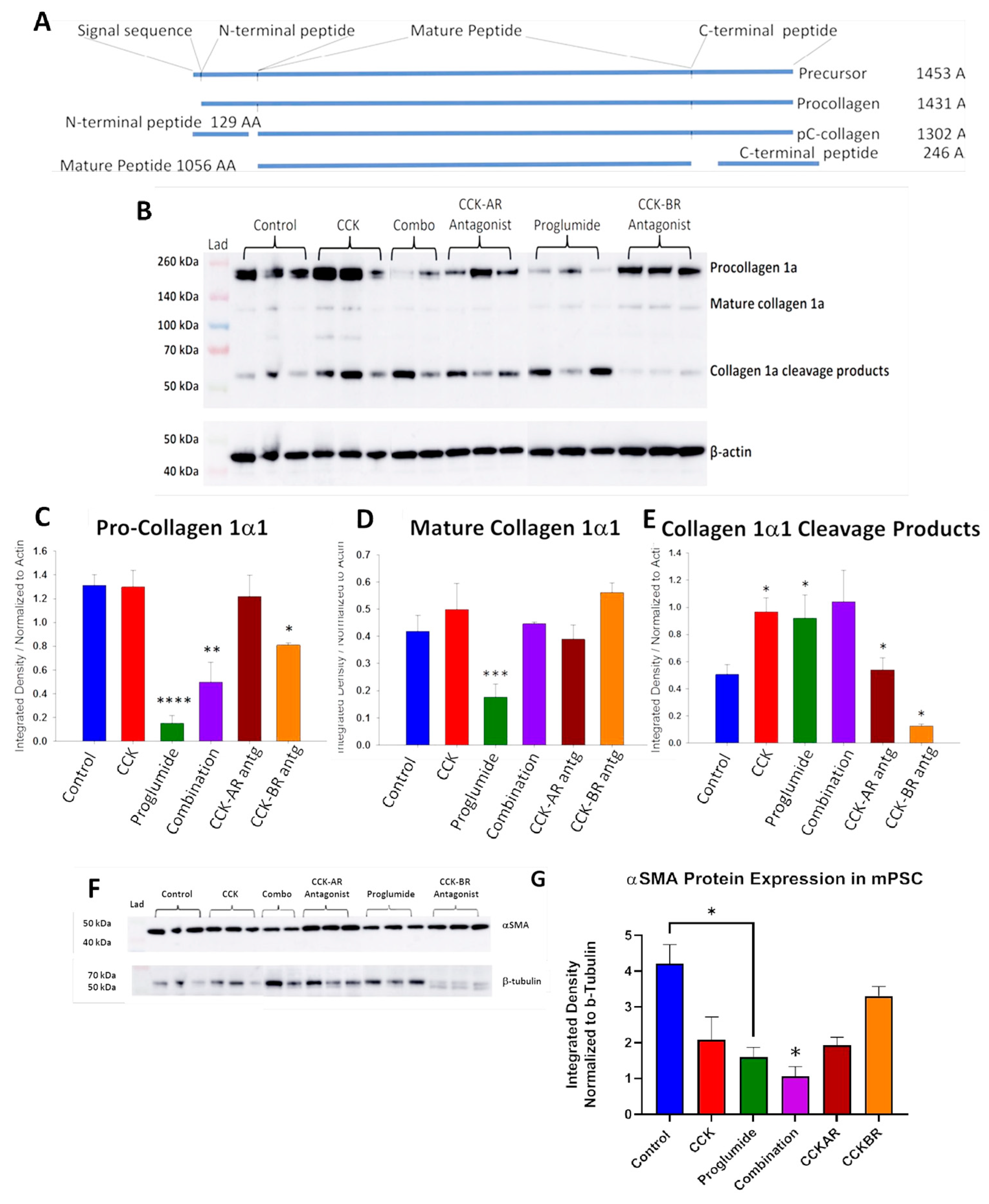
3.4. Evaluation of mPSCs Protein Activation by Western Blotting
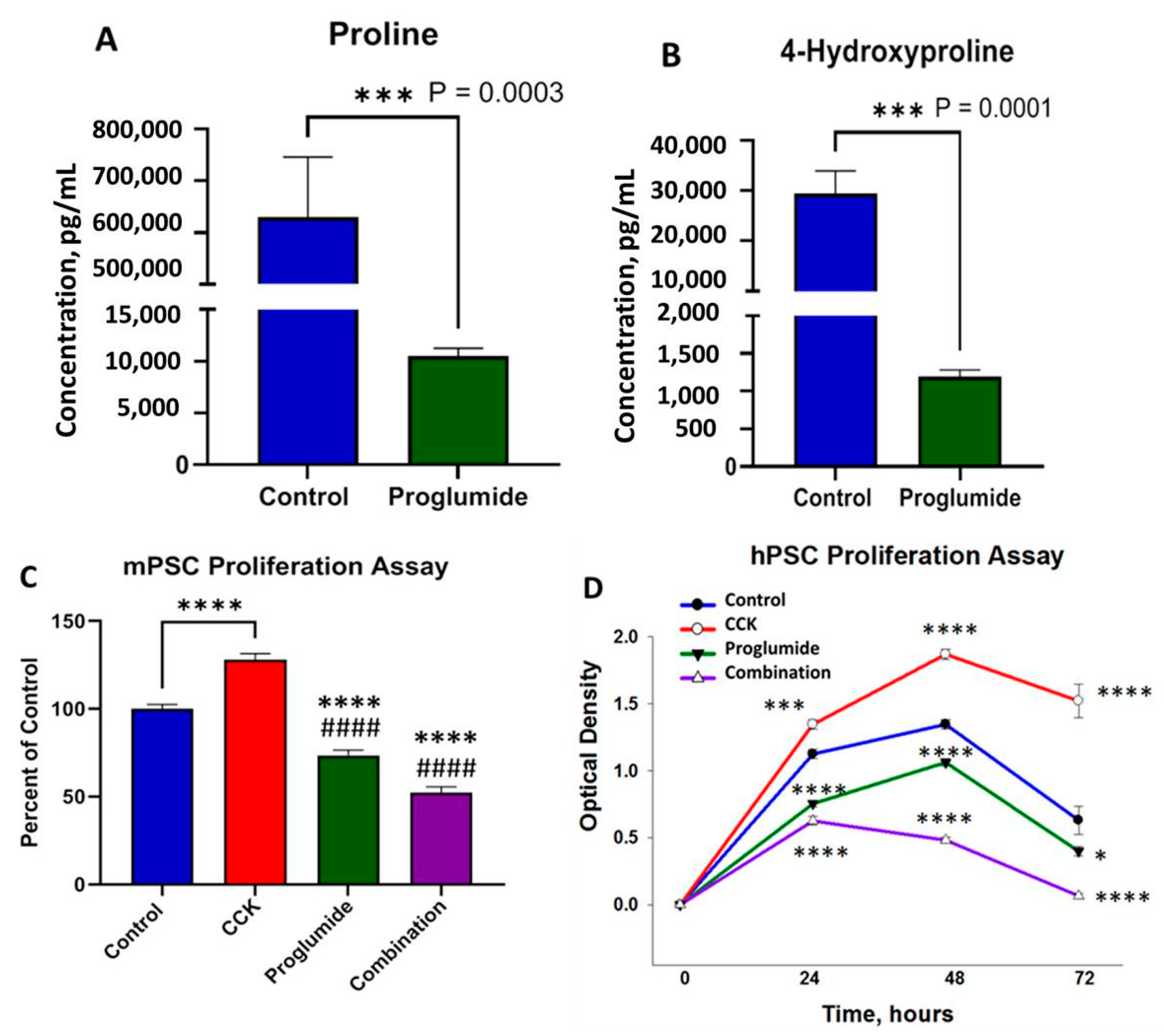
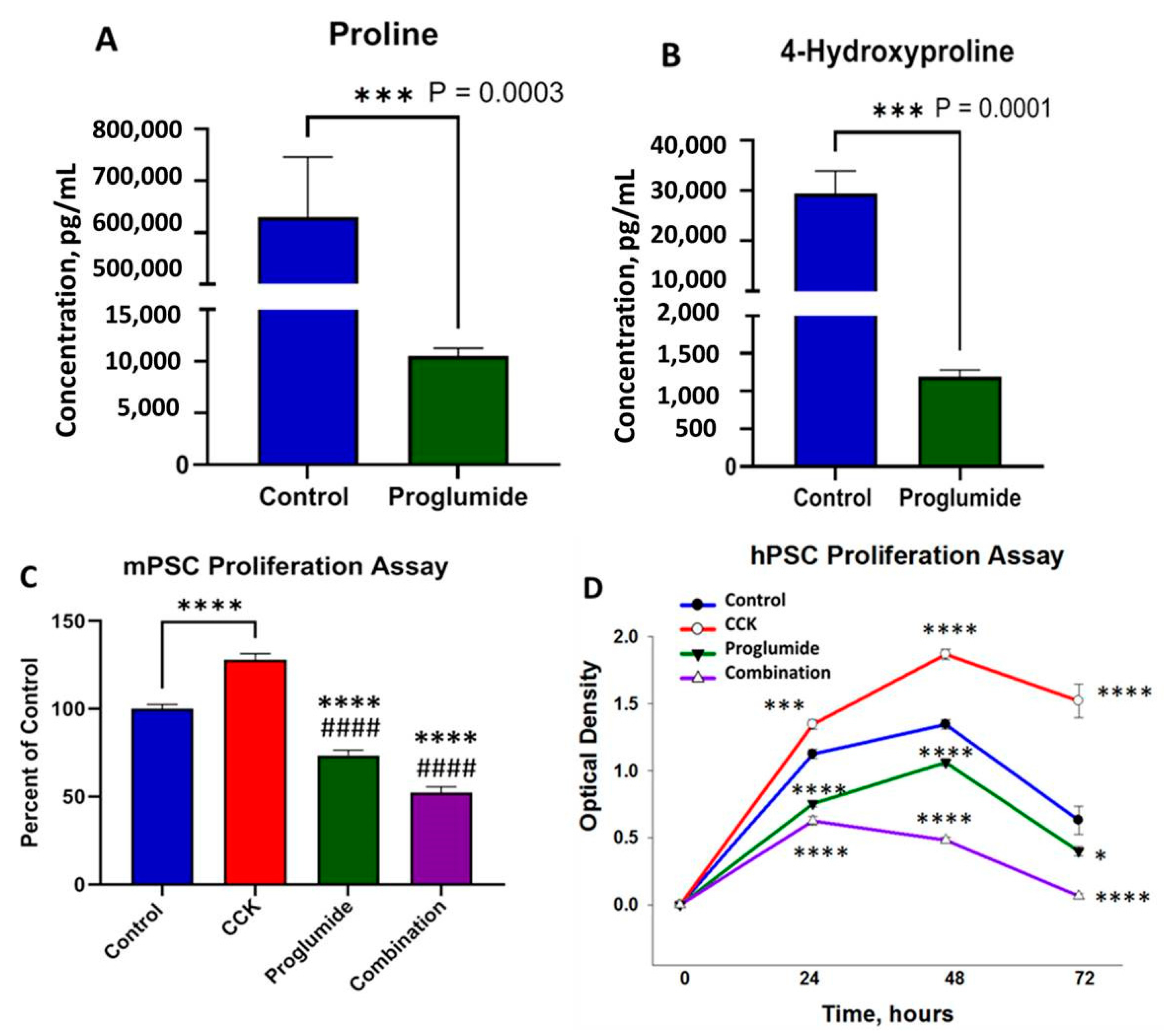
3.5. Proglumide Treated Cells Show Decreased Concentrations of Proline and 4-Hydroxyproline
3.6. Proglumide Decreases PSC Proliferation
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, C.; Xie, K.P. Therapeutic resistance of pancreatic cancer: Roadmap to its reversal. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188461. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; di Magliano, M.P.; Simeone, D.M. Deciphering the role of stroma in pancreatic cancer. Curr. Opin. Gastroenterol. 2013, 29, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat Rev Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Amedei, A.; Niccolai, E.; Prisco, D. Pancreatic cancer: Role of the immune system in cancer progression and vaccine-based immunotherapy. Hum. Vaccin. Immunother. 2014, 10, 3354–3368. [Google Scholar] [CrossRef]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef] [PubMed]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; van Eijck, C.H.; Peppelenbosch, M.P.; Sleijfer, S.; Bruno, M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat. Rev. 2014, 40, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Alewine, C.; Figg, W.D.; Duffy, A. Targeting the microenvironment of pancreatic cancer: Overcoming treatment barriers and improving local immune responses. Clin. Transl. Oncol. 2016, 18, 653–659. [Google Scholar] [CrossRef]
- Van, C.E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar]
- Hecht, J.R.; Papadopoulos, K.P.; Falchook, G.S.; Patel, M.R.; Infante, J.R.; Aljumaily, R.; Wong, D.J.; Autio, K.A.; Wainberg, Z.A.; Bauer, T.M.; et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Investig. New Drugs 2020, 39, 182–192. [Google Scholar] [CrossRef]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Nazemi, M.; Rainero, E. Cross-Talk Between the Tumor Microenvironment, Extracellular Matrix, and Cell Metabolism in Cancer. Front. Oncol. 2020, 10, 239. [Google Scholar] [CrossRef]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells: Aiding and abetting pancreatic cancer progression. Pancreatology 2020, 20, 409–418. [Google Scholar] [CrossRef]
- Apte, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cell: Physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015, 31, 416–423. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Shimosegawa, T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin. Gastroenterol. Hepatol. 2009, 7, S48–S54. [Google Scholar] [CrossRef]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef]
- Ellenrieder, V.; Alber, B.; Lacher, U.; Hendler, S.F.; Menke, A.; Boeck, W.; Wagner, M.; Wilda, M.; Friess, H.; Buchler, M.; et al. Role of MT-MMPs and MMP-2 in pancreatic cancer progression. Int. J. Cancer 2000, 85, 14–20. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef]
- Smith, J.P.; Solomon, T.E. Cholecystokinin and pancreatic cancer: The chicken or the egg? Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G91–G101. [Google Scholar] [CrossRef]
- Smith, J.P.; Cooper, T.K.; McGovern, C.O.; Gilius, E.L.; Zhong, Q.; Liao, J.; Molinolo, A.A.; Gutkind, J.S.; Matters, G.L. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas 2014, 43, 1050–1059. [Google Scholar] [CrossRef]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Blaker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef]
- Phillips, P.A.; Yang, L.; Shulkes, A.; Vonlaufen, A.; Poljak, A.; Bustamante, S.; Warren, A.; Xu, Z.; Guilhaus, M.; Pirola, R.; et al. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17397–17402. [Google Scholar] [CrossRef]
- Wank, S.A.; Pisegna, J.R.; de Weerth, A. Cholecystokinin receptor family. Molecular cloning, structure, and functional expression in rat, guinea pig, and human. Ann. NY Acad. Sci. 1994, 713, 49–66. [Google Scholar] [CrossRef]
- Wank, S.A. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: An exemplary family. Am. J. Physiol. 1998, 274, G607–G613. [Google Scholar]
- Berna, M.J.; Jensen, R.T. Role of CCK/gastrin receptors in gastrointestinal/metabolic diseases and results of human studies using gastrin/CCK receptor agonists/antagonists in these diseases. Curr. Top. Med. Chem. 2007, 7, 1211–1231. [Google Scholar] [CrossRef]
- Hahne, W.F.; Jensen, R.T.; Lemp, G.F.; Gardner, J.D. Proglumide and benzotript: Members of a different class of cholecystokinin receptor antagonists. Proc. Natl. Acad. Sci. USA 1981, 78, 6304–6308. [Google Scholar] [CrossRef]
- Nadella, S.; Burks, J.; Al-Sabban, A.; Inyang, G.; Wang, J.; Tucker, R.D.; Zamanis, M.E.; Bukowski, W.; Shivapurkar, N.; Smith, J.P. Dietary fat stimulates pancreatic cancer growth and promotes fibrosis of the tumor microenvironment through the cholecystokinin receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G699–G712. [Google Scholar] [CrossRef]
- Malchiodi, Z.X.; Cao, H.; Gay, M.D.; Safronenka, A.; Bansal, S.; Tucker, R.D.; Weinberg, B.A.; Cheema, A.; Shivapurkar, N.; Smith, J.P. Cholecystokinin Receptor Antagonist Improves Efficacy of Chemotherapy in Murine Models of Pancreatic Cancer by Altering the Tumor Microenvironment. Cancers 2021, 13, 4949. [Google Scholar] [CrossRef]
- Smith, J.P.; Wang, S.; Nadella, S.; Jablonski, S.A.; Weiner, L.M. Cholecystokinin receptor antagonist alters pancreatic cancer microenvironment and increases efficacy of immune checkpoint antibody therapy in mice. Cancer Immunol. Immunother. 2018, 67, 195–207. [Google Scholar] [CrossRef]
- Mathison, A.; Liebl, A.; Bharucha, J.; Mukhopadhyay, D.; Lomberk, G.; Shah, V.; Urrutia, R. Pancreatic stellate cell models for transcriptional studies of desmoplasia-associated genes. Pancreatology 2010, 10, 505–516. [Google Scholar] [CrossRef]
- Rabiee, A.; Gay, M.D.; Shivapurkar, N.; Cao, H.; Nadella, S.; Smith, C.I.; Lewis, J.H.; Bansal, S.; Cheema, A.; Kwagyan, J.; et al. Safety and Dosing Study of a Cholecystokinin Receptor Antagonist in Non-alcoholic Steatohepatitis. Clin. Pharmacol. Ther. 2022, 112, 1271–1279. [Google Scholar] [CrossRef]
- Gay, M.D.; Safronenka, A.; Cao, H.; Liu, F.H.; Malchiodi, Z.X.; Tucker, R.D.; Kroemer, A.; Shivapurkar, N.; Smith, J.P. Targeting the Cholecystokinin Receptor: A Novel Approach for Treatment and Prevention of Hepatocellular Cancer. Cancer Prev. Res. 2021, 14, 17–29. [Google Scholar] [CrossRef]
- Mimche, P.N.; Lee, C.M.; Mimche, S.M.; Thapa, M.; Grakoui, A.; Henkemeyer, M.; Lamb, T.J. EphB2 receptor tyrosine kinase promotes hepatic fibrogenesis in mice via activation of hepatic stellate cells. Sci. Rep. 2018, 8, 2532. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, S.; Han, M.; Ma, Y.; Feng, S.; Zhao, J.; Lu, H.; Yuan, X.; Cheng, J. miR-185 Inhibits Fibrogenic Activation of Hepatic Stellate Cells and Prevents Liver Fibrosis. Mol. Ther. Nucleic Acids 2018, 10, 91–102. [Google Scholar] [CrossRef]
- Wang, H.C.; Lin, Y.L.; Hsu, C.C.; Chao, Y.J.; Hou, Y.C.; Chiu, T.J.; Huang, P.H.; Tang, M.J.; Chen, L.T.; Shan, Y.S. Pancreatic stellate cells activated by mutant KRAS-mediated PAI-1 upregulation foster pancreatic cancer progression via IL-8. Theranostics 2019, 9, 7168–7183. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, Z.; Wang, P. GLI3 Promotes Invasion and Predicts Poor Prognosis in Colorectal Cancer. BioMed Res. Int. 2021, 2021, 8889986. [Google Scholar] [CrossRef]
- Hyun, J.; Wang, S.; Kim, J.; Rao, K.M.; Park, S.Y.; Chung, I.; Ha, C.S.; Kim, S.W.; Yun, Y.H.; Jung, Y. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 2016, 7, 10993. [Google Scholar] [CrossRef]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef]
- Gao, L.; Lei, X.F.; Miyauchi, A.; Noguchi, M.; Omoto, T.; Haraguchi, S.; Miyazaki, T.; Miyazaki, A.; Kim-Kaneyama, J.R. Hic-5 is required for activation of pancreatic stellate cells and development of pancreatic fibrosis in chronic pancreatitis. Sci. Rep. 2020, 10, 19105. [Google Scholar] [CrossRef]
- Pure, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Lundin, C.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 2009, 101, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Iannarone, V.J.; Cruz, G.E.; Hilliard, B.A.; Barbe, M.F. The answer depends on the question: Optimal conditions for western blot characterization of muscle collagen type 1 depends on desired isoform. J. Biol. Methods 2019, 6, e117. [Google Scholar] [CrossRef]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Murray, E.R.; Menezes, S.; Henry, J.C.; Williams, J.L.; Alba-Castellon, L.; Baskaran, P.; Quetier, I.; Desai, A.; Marshall, J.J.T.; Rosewell, I.; et al. Disruption of pancreatic stellate cell myofibroblast phenotype promotes pancreatic tumor invasion. Cell Rep. 2022, 38, 110227. [Google Scholar] [CrossRef]
- Wang, J.; Zohar, R.; McCulloch, C.A. Multiple roles of alpha-smooth muscle actin in mechanotransduction. Exp. Cell Res. 2006, 312, 205–214. [Google Scholar] [CrossRef]
- Sun, K.H.; Chang, Y.; Reed, N.I.; Sheppard, D. alpha-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFbeta activation or collagen production across multiple models of organ fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Identification of markers for quiescent pancreatic stellate cells in the normal human pancreas. Histochem. Cell Biol. 2017, 148, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef]
- Pauletzki, J.G.; Xu, Q.W.; Shaffer, E.A. Inhibition of gallbladder emptying decreases cholesterol saturation in bile in the Richardson ground squirrel. Hepatology 1995, 22, 325–331. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mGene | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| mEphb2 | CAACGGTGTGATCCTGGACTAC | CACCTGGAAGACATAGATGGCG |
| mHic-5 | GGTCTGGAGAATCTTCAGGAACC | CACCACTGGAAGAGGAGAATGG |
| mGli3 | CTGCGGTATCTCCTCTCATAGG | CAGCACTGTGAAGTCTACACCTG |
| mRheb | GGCAAGTTGTTGGATATGGTGGG | CCAAGATTCTGCCAAAGCCTTTC |
| mRictor | CAGTGTGAGGTCCTTTCCATCC | GCCATAGATGCTTGCGACTGTG |
| mFap | CACCTGATCGGCAATTTGTG | CCCATTCTGAAGGTCGTAGATGT |
| mIL-1β | GGACCTTCCAGGATGAGGACA | GTTCATCTCGGAGCCTGTAGTG |
| mIL-8 | GGTGATATTCGAGACCATTTACTG | GCCAACAGTAGCCTTCACCCAT |
| mCol1a1 | CGCCATCAAGGTCTACTG | ACGGGAATCCATCGGTC |
| mCol4a | GATGGGCTATCCTGGAACCACT | TCTCTCCTCGTTCGCCTTTGG |
| mActa2 | TGCCGAGCGTGAGATTGT | CCCGTCAGGCAGTTCGTAG |
| Forward (5′-3′) | Reverse (5′-3′) | |
|---|---|---|
| hRICTOR | GCCAAACAGCTCACGGTTGTAG | CCAGATGAAGCATTGAGCCACTG |
| hEPHB2 | CGCCATCTATGTCTTCCAGGTG | GATGAGTGGCAACTTCTCCTGG |
| hIL-8 | GAGAGTGATTGAGAGTGGACCAC | CACAACCCTCTGCACCCAGTTT |
| hGLI3 | TCAGCAAGTGGCTCCTATGGTC | GCTCTGTTGTCGGCTTAGGATC |
| hACTA2 | CTATGCCTCTGGACGCACAACT | CAGATCCAGACGCATGATGGCA |
| hCOL1A1 | GATTCCCTGGACCTAAAGGTGC | AGCCTCTCCATCTTTGCCAGCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jolly, G.; Duka, T.; Shivapurkar, N.; Chen, W.; Bansal, S.; Cheema, A.; Smith, J.P. Cholecystokinin Receptor Antagonist Induces Pancreatic Stellate Cell Plasticity Rendering the Tumor Microenvironment Less Oncogenic. Cancers 2023, 15, 2811. https://doi.org/10.3390/cancers15102811
Jolly G, Duka T, Shivapurkar N, Chen W, Bansal S, Cheema A, Smith JP. Cholecystokinin Receptor Antagonist Induces Pancreatic Stellate Cell Plasticity Rendering the Tumor Microenvironment Less Oncogenic. Cancers. 2023; 15(10):2811. https://doi.org/10.3390/cancers15102811
Chicago/Turabian StyleJolly, Gurbani, Tetyana Duka, Narayan Shivapurkar, Wenqiang Chen, Sunil Bansal, Amrita Cheema, and Jill P. Smith. 2023. "Cholecystokinin Receptor Antagonist Induces Pancreatic Stellate Cell Plasticity Rendering the Tumor Microenvironment Less Oncogenic" Cancers 15, no. 10: 2811. https://doi.org/10.3390/cancers15102811
APA StyleJolly, G., Duka, T., Shivapurkar, N., Chen, W., Bansal, S., Cheema, A., & Smith, J. P. (2023). Cholecystokinin Receptor Antagonist Induces Pancreatic Stellate Cell Plasticity Rendering the Tumor Microenvironment Less Oncogenic. Cancers, 15(10), 2811. https://doi.org/10.3390/cancers15102811