
Crosstalk between ILC3s and Microbiota: Implications for Colon Cancer Development and Treatment with Immune Check Point Inhibitors
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
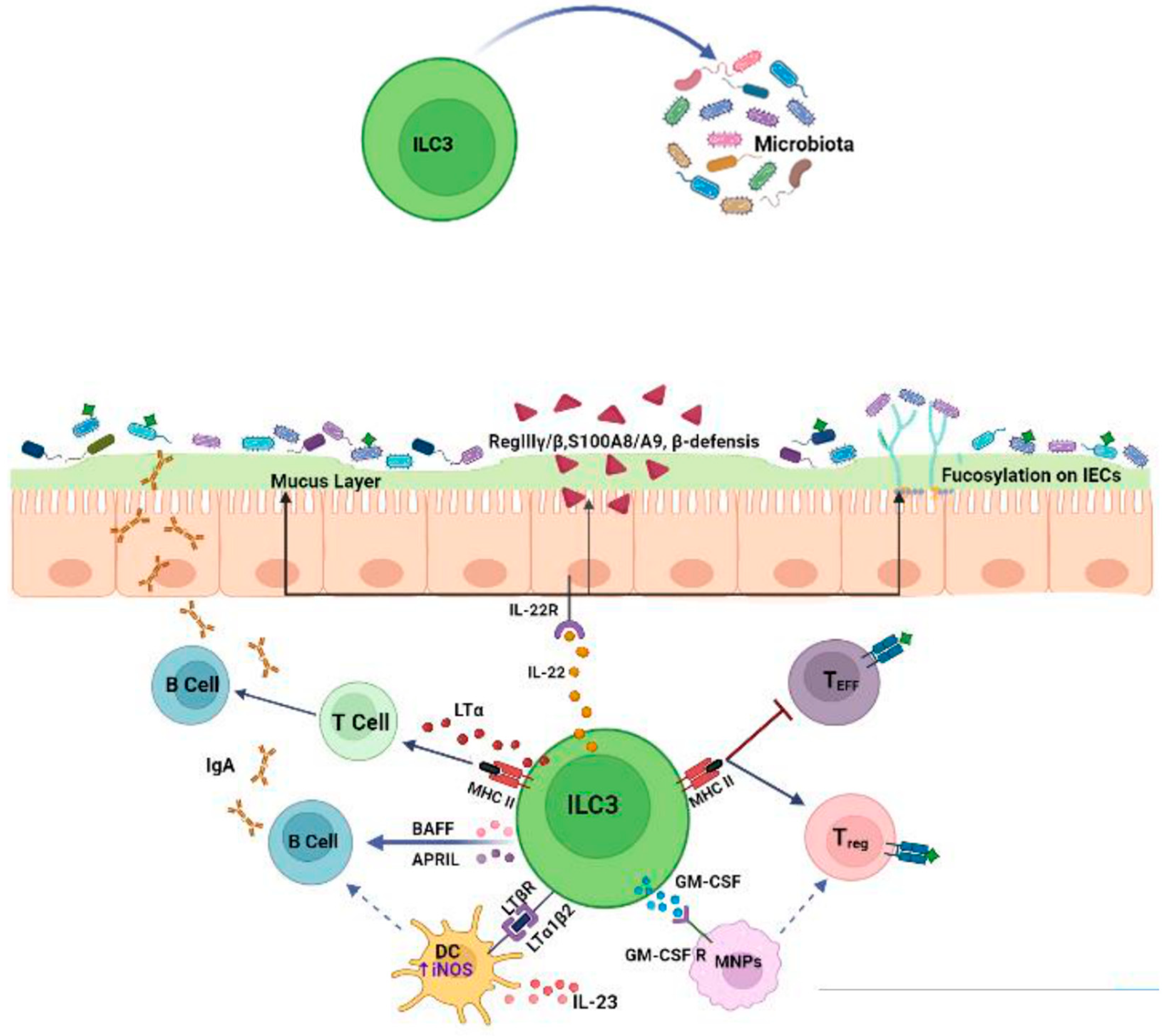
2. Crosstalk between ILC3s and Microbiota
2.1. Effects of the Microbiota on ILC3s
2.2. Effects of ILC3s on the Microbiota
3. ILC3s, Microbiota and Colon Cancer
4. ILC3s, Microbiota and CRC Immunotherapy
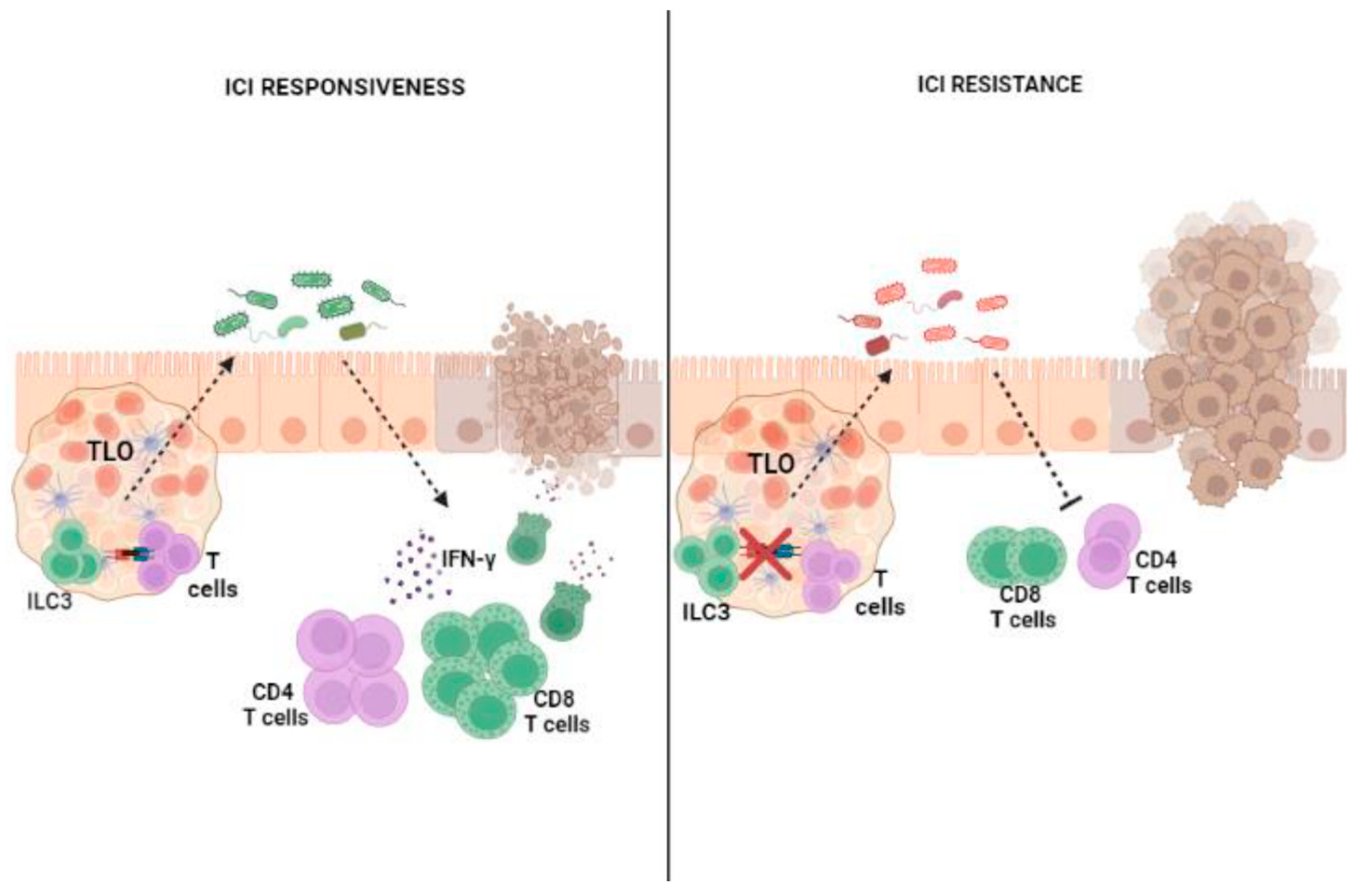
4.1. Colon Cancer and Response to ICI Therapy
4.2. Influence of ILC3–Microbiota Interactions on the Outcomes of ICI Therapy
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The anti-bacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Umesaki, Y.; Setoyama, H.; Matsumoto, S.; Okada, Y. Expansion of alpha beta T-cell receptor-bearing intestinal intraepithe-lial lymphocytes after microbial colonization in germ-free mice and its independence from thymus. Immunology 1993, 79, 32–37. [Google Scholar]
- Hapfelmeier, S.; Lawson, M.A.; Slack, E.; Kirundi, J.K.; Stoel, M.; Heikenwalder, M.; Cahenzli, J.; Velykoredko, Y.; Balmer, M.L.; Endt, K.; et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010, 328, 1705–1709. [Google Scholar] [CrossRef]
- Sawa, S.; Lochner, M.; Satoh-Takayama, N.; Dulauroy, S.; Bérard, M.; Kleinschek, M.; Cua, D.; Di Santo, J.P.; Eberl, G. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011, 12, 320–326. [Google Scholar] [CrossRef]
- Zhong, C.; Zheng, M.; Zhu, J. Lymphoid tissue inducer—A divergent member of the ILC family. Cytokine Growth Factor Rev. 2018, 42, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; d’Hargues, Y.; Göppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Hoorweg, K.; Peters, C.P.; Cornelissen, F.; Aparicio-Domingo, P.; Papazian, N.; Kazemier, G.; Mjösberg, J.M.; Spits, H.; Cupedo, T. Functional Differences between Human NKp44(-) and NKp44(+) RORC(+) Innate Lymphoid Cells. Front. Immunol. 2012, 3, 72. [Google Scholar] [CrossRef] [PubMed]
- Marchalot, A.; Mjösberg, J. Innate lymphoid cells in colorectal cancer. Scand. J. Immunol. 2022, 95, 13156. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Itzkowitz, S.H. Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Crellin, N.K.; Trifari, S.; Kaplan, C.D.; Satoh-Takayama, N.; Di Santo, J.P.; Spits, H. Regulation of cytokine secretion in human CD127(+)LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 2010, 33, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Fuchs, A.W.; Vermi, F.; Facchetti, K.; Otero, J.K.; Lennerz, J.M.; Doherty, J.C.; Mills, M.; Colonna, A. human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Vosshenrich, C.A.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Glatzer, T.; Killig, M.; Meisig, J.; Ommert, I.; Luetke-Eversloh, M.; Babic, M.; Paclik, D.; Blüthgen, N.; Seidl, R.; Seifarth, C.; et al. RORγt+ innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp. Immunity 2013, 38, 1223–1235. [Google Scholar] [CrossRef]
- Carrega, P.; Loiacono, F.; Di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulases gut im-munity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef]
- Qiu, J.; Zhou, L. Aryl hydrocarbon receptor promotes RORγt+ group 3 ILCs and controls intestinal immunity and inflammation. Semin. Immunopathol. 2013, 35, 657–670. [Google Scholar] [CrossRef]
- Qiu, J.; Guo, X.; Chen, Z.M.; He, L.; Sonnenberg, G.F.; Artis, D.; Fu, Y.X.; Zhou, L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013, 39, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bostick, J.W.; Zhou, L. Regulation of Innate Lymphoid Cells by Aryl Hydrocarbon Receptor. Front. Immunol. 2018, 8, 1909. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T. Metabolic control of innate lymphoid cell migration. Front. Immunol. 2019, 10, 2010. [Google Scholar] [CrossRef]
- Chun, E.; Lavoie, S.; Fonseca-Pereira, D.; Bae, S.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Gallini Comeau, C.A.; Glickman, J.N.; Fuller, M.H.; et al. Metabolite-Sensing Receptor Ffar2 Regulates Colonic Group 3 Innate Lymphoid Cells and Gut Immunity. Immunity 2019, 51, 871–884.e6. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef]
- Bhatt, B.; Zeng, P.; Zhu, H.; Sivaprakasam, S.; Li, S.; Xiao, H.; Dong, L.; Shiao, P.; Kolhe, R.; Patel, N.; et al. Gpr109a limits microbiota-induced IL-23 production to constrain IL-C3-mediaded colonic inflammation. J. Immunol. 2018, 200, 2905–2914. [Google Scholar] [CrossRef]
- Kim, S.H.; Cho, B.H.; Kiyono, H.; Jang, Y.S. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci. Rep. 2017, 7, 3980. [Google Scholar] [CrossRef]
- Bostick, J.W.; Wang, Y.; Shen, Z.; Ge, Y.; Brown, J.; Chen, Z.E.; Mohamadzadeh, M.; Fox, J.G.; Zhou, L. Dichotomous regulation of group 3 innate lymphoid cells by nongastric Helicobacter species. Proc. Natl. Acad. Sci. USA 2019, 116, 24760–24769. [Google Scholar] [CrossRef]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef]
- Shaw, M.H.; Kamada, N.; Kim, Y.G.; Núῆez, G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258. [Google Scholar] [CrossRef]
- Ahn, Y.O.; Weeres, M.A.; Neulen, M.L.; Choi, J.; Kang, S.H.; Heo, D.S.; Bergerson, R.; Blazar, B.R.; Miller, J.S.; Ver-neris, M.R. Human group 3 innate lymphoid cells express DR3 and respond to TL1A with enhanced IL-22 production and IL-2-dependent proliferation. Eur. J. Immunol. 2015, 45, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, J.G.; Woo, V.; Viladomiu, M.; Putzel, G.; Lima, S.; Diehl, G.E.; Marderstein, A.R.; Gandara, J.; Perez, A.R.; Withers, D.R.; et al. Microbiota-Induced TNF-like Ligand 1A Drives Group 3 Innate Lymphoid Cell-Mediated Barrier Protection and Intestinal T Cell Activation during Colitis. Immunity 2018, 49, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, F.M.; von Burg, N.; Ivanek, R.; Teufel, C.; Horvath, E.; Peter, A.; Turchinovich, G.; Staehli, D.; Eichlisberger, T.; Gomez de Agüero, M.; et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat. Commun. 2020, 11, 1794. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Artis, D. Innate lymphoid cell interactions with microbiota: Implications for intestinal health and disease. Immunity 2012, 37, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yin, X.; Wang, G.; Elinav, E.; Hao, L.; Zhao, L.; Flavell, R.A. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J. Immunol. 2013, 190, 5306–5312. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef]
- Goto, Y.; Obata, T.; Kunisawa, J.; Sato, S.; Ivanov, I.; Lamichhane, A.; Takeyama, N.; Kamioka, M.; Sakamoto, M.; Matsu-ki, T.; et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014, 345, 1254009. [Google Scholar] [CrossRef]
- Tumanov, A.V.; Koroleva, E.P.; Guo, X.; Wang, Y.; Kruglov, A.; Nedospasov, S.; Fu, Y.X. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 2011, 10, 44–53. [Google Scholar] [CrossRef]
- Ganal-Vonarburg, S.C.; Duerr, C.U. The interaction of intestinal microbiota and innate lymphoid cells in health and disease throughout life. Immunology 2020, 159, 39–51. [Google Scholar] [CrossRef]
- Kruglov, A.A.; Grivennikov, S.I.; Kuprash, D.V.; Winsauer, C.; Prepens, S.; Seleznik, G.M.; Eberl, G.; Littman, D.R.; Heikenwalder, M.; Tumanov, A.V.; et al. Non redundant function of soluble LTα3 produced by innate lymphoid cells in intestinal homeostasis. Science 2013, 342, 1243–1246. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Monticelli, L.A.; Fung, T.C.; Ziegler, C.G.; Grunberg, S.; Sinha, R.; Mantegazza, A.R.; Ma, H.L.; Craw-ford, A.; Angelosanto, J.M.; et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 2013, 498, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Von Burg, N.; Chappaz, S.; Baerenwaldt, A.; Horvath, E.; Bose Dasgupta, S.; Ashok, D.; Pieters, J.; Tacchini-Cottier, F.; Rolink, A.; Acha-Orbea, H.; et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl. Acad. Sci. USA 2014, 111, 12835–12840. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Lyu, M.; Suzuki, H.; Kang, L.; Gaspal, F.; Zhou, W.; Goc, J.; Zhou, L.; Zhou, J.; Zhang, W.; JRI Live Cell Bank; et al. ILC3s select microbiota-specific regulatory T cells to establish tolerance in the gut. Nature 2022, 610, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef]
- Zhou, L.; Chu, C.; Teng, F.; Bessman, N.J.; Goc, J.; Santosa, E.K.; Putzel, G.G.; Kabata, H.; Kelsen, J.R.; Baldassano, R.N.; et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 2019, 568, 405–409. [Google Scholar] [CrossRef]
- Kedmi, R.; Najar, T.A.; Mesa, K.R.; Grayson, A.; Kroehling, L.; Hao, Y.; Hao, S.; Pokrovskii, M.; Xu, M.; Talbot, J.; et al. A RORγt+ cell instructs gut microbiota-specific Treg cell differentiation. Nature 2022, 610, 737–743. [Google Scholar] [CrossRef]
- Melo-Gonzalez, F.; Kammoun, H.; Evren, E.; Dutton, E.E.; Papadopoulou, M.; Bradford, B.M.; Tanes, C.; Fardus-Reid, F.; Swann, J.R.; Bittinger, K.; et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J. Exp. Med. 2019, 216, 728–742. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Köller, Y.; McCoy, K.D. The bilateral responsiveness between intestinal microbes and IgA. Trends. Immunol. 2015, 36, 460–470. [Google Scholar] [CrossRef]
- Magri, G.; Miyajima, M.; Bascones, S.; Mortha, A.; Puga, I.; Cassis, L.; Barra, C.M.; Comerma, L.; Chudnovskiy, A.; Gentile, M.; et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat. Immunol. 2014, 15, 354–364. [Google Scholar] [CrossRef]
- Goto, Y.; Panea, C.; Nakato, G.; Cebula, A.; Lee, C.; Diez, M.G.; Laufer, T.M.; Ignatowicz, L.; Ivanov, I.I. Segmented fila-mentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014, 40, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Goc, J.; Lv, M.; Bessman, N.J.; Flamar, A.L.; Sahota, S.; Suzuki, H.; Teng, F.; Putzel, G.G.; Eberl, G.; JRI Live Cell Bank; et al. Dysregulation of ILC3s unleashes progression and immunotherapy re-sistance in colon cancer. Cell 2021, 184, 5015–5030. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Ogino, T.; Kayama, H.; Okuzaki, D.; Nishimura, J.; Fujino, S.; Miyoshi, N.; Takahashi, H.; Uemura, M.; Matsuda, C.; et al. Human NKp44+ Group 3 Innate Lymphoid Cells Associate with Tumor-Associated Tertiary Lymphoid Structures in Colorectal Cancer. Cancer Immunol. Res. 2020, 8, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Overacre-Delgoffe, E.; Bumgarner, H.J.; Cillo, A.R.; Burr, A.H.P.; Tometich, J.T.; Bhattacharjee, A.; Bruno, T.C.; Vignali, D.A.A.; Hand, T.W. Microbiota-specific T follicular helper cells drive tertiary lymphoid structures and anti-tumor immunity against colorectal cancer. Immunity 2021, 54, 2812–2824. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pokrovskii, M.; Ding, Y.; Yi, R.; Au, C.; Harrison, O.J.; Galan, C.; Belkaid, Y.; Bonneau, R.; Littman, D.R. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 2018, 554, 373–377. [Google Scholar] [CrossRef]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 6, 917–931. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, T.; Lu, X.; Xu, Z.; Qu, J.; Zhang, Z.; Shi, G.; Shen, S.; Hou, Y.; Chen, Y.; et al. Fungal-induced glycolysis in macrophages promotes colon cancer by enhancing innate lymphoid cell secretion of IL-22. EMBO J. 2021, 40, e105320. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W., Jr.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef]
- Gelsomino, F.; Barbolini, M.; Spallanzani, A.; Pugliese, G.; Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat. Rev. 2016, 51, 19–26. [Google Scholar] [CrossRef]
- Yuza, K.; Nagahashi, M.; Watanabe, S.; Takabe, K.; Wakai, T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget 2017, 8, 112103–112115. [Google Scholar] [CrossRef]
- Hutchins, G.; Southward, K.; Handley, K.; Magill, L.; Beaumont, C.; Stahlschmidt, J.; Richman, S.; Chambers, P.; Seymour, M.; Kerr, D.; et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J. Clin. Oncol. 2011, 29, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, A.; Kircher, S.; Shah, H.; Mulcahy, M.; Benson, A. Updates on immunotherapy for colorectal cancer. J. Gastr. Oncol. 2018, 9, 160–169. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Le, D.T.; Diaz, L.A., Jr.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.H.; Kavan, P.; et al. Pembrolizumab for previously treated, microsatellite instability-high/mismatch repair-deficient advanced colorectal cancer: Final analysis of KEYNOTE-164. Eur. J. Cancer 2023, 186, 185–195. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. KEYNOTE-177 Investigators. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch re-pair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Lenz, H.J.; Van Cutsem, E.; Luisa Limon, M.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Ro-berti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 5, 91–97. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Ale-gre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Suda, W.; Luo, C.; Kawaguchi, T.; Motoo, I.; Narushima, S.; Kiguchi, Y.; Yasuma, K.; Watanabe, E.; Tanoue, T.; et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Cottrell, T.R.; Thompson, E.D.; Forde, P.M.; Stein, J.E.; Duffield, A.S.; Anagnostou, V.; Rekhtman, N.; Anders, R.A.; Cuda, J.D.; Illei, P.B. Pathologic features of response to neoadjuvant anti-PD-1 in resected non-small-cell lung carcinoma: A proposal for quantitative immune-related pathologic response criteria (irPRC). Ann. Oncol. 2018, 29, 1853–1860. [Google Scholar] [CrossRef]
- Remark, R.; Lupo, A.; Lifano, M.; Biton, J.; Ouakrim, H.; Stefani, A.; Cremer, I.; Goc, J.; Régnard, J.F.; Dieu-Nosjean, M.C.; et al. Immune contexture and histological response after neoadjuvant chemotherapy predict clinical outcome of lung cancer patients. Oncoimmunology 2016, 5, 1255394. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef]
- Eisenring, M.; vom Berg, J.; Kristiansen, G.; Saller, E.; Becher, B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 2010, 11, 1030–1038. [Google Scholar] [CrossRef]
- Nussbaum, K.; Burkhard, S.H.; Ohs, I.; Mair, F.; Klose, C.S.N.; Arnold, S.J.; Diefenbach, A.; Tugues, S.; Becher, B. Tissue mi-croenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J. Exp. Med. 2017, 214, 2331–2347. [Google Scholar] [CrossRef]
- Cupedo, T.; Jansen, W.; Kraal, G.; Mebius, R.E. Induction of secondary and tertiary lymphoid structures in the skin. Immunity 2004, 21, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Geindreau, M.; Perrichet, A.; Truntzer, C.; Ballot, E.; Boidot, R.; Racoeur, C.; Barsac, E.; Chalmin, F.; Hibos, C.; et al. Recruitment and activation of type 3 innate lymphoid cells promote antitumor immune responses. Nat. Immunol. 2022, 23, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is al-tered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, 000145. [Google Scholar] [CrossRef]
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal. Immunol. 2021, 14, 317–330. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- He, Y.; Fu, L.; Li, Y.; Wang, W.; Gong, M.; Zhang, J.; Dong, X.; Huang, J.; Wang, Q.; Mackay, C.R.; et al. Gut microbial me-tabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8+ T cell immunity. Cell Metab. 2021, 33, 988–1000. [Google Scholar] [CrossRef]
- Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021, 374, 1632–1640. [Google Scholar] [CrossRef]
- Nomura, M.; Nagatomo, R.; Doi, K.; Shimizu, J.; Baba, K.; Saito, T.; Matsumoto, S.; Inoue, K.; Muto, M. Association of Short-Chain Fatty Acids in the Gut Microbiome with Clinical Response to Treatment With Nivolumab or Pembrolizumab in Patients With Solid Cancer Tumors. JAMA Netw Open. 2020, 3, 202895. [Google Scholar] [CrossRef]
- Fachi, J.L.; Sécca, C.; Rodrigues, P.B.; Mato, F.C.P.; Di Luccia, B.; Felipe, J.S.; Pral, L.P.; Rungue, M.; Rocha, V.M.; Sato, F.T.; et al. Acetate coordinates neutrophil and ILC3 responses against C. difficile through FFAR2. J. Exp. Med. 2020, 217, 20190489. [Google Scholar] [CrossRef]
- Hu, C.; Xu, B.; Wang, X.; Wan, W.H.; Lu, J.; Kong, D.; Jin, Y.; You, W.; Sun, H.; Mu, X.; et al. Gut microbiota-derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology 2023, 77, 48–64. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 2011, 141, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Otero, K.; Colonna, M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. USA 2010, 107, 10961–10966. [Google Scholar] [CrossRef] [PubMed]
- Campana, S.; Di Carlo, E.; De Pasquale, C.; Barberi, C.; Oliveri, D.; Migliore, G.S.; Cannavò, S.P.; Galletti, B.; Pende, D.; Carrega, P.; et al. Dendritic cell recognition by group 3 innate lymphoid cells through DNAX accessory molecule 1 triggers proinflammatory reciprocal cell activation. J. Allergy Clin Immunol. 2019, 144, 1118–1122.e6. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Strauss, O.; Kokkinou, E.; Bruchard, M.; Tripathi, K.P.; Schlums, H.; Carrasco, A.; Mazzurana, L.; Konya, V.; Villablanca, E.J.; et al. Cytokines regulate the antigen-presenting characteristics of human circulating and tissue-resident intestinal ILCs. Nat. Commun. 2020, 11, 2049. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drommi, F.; Calabrò, A.; Vento, G.; Pezzino, G.; Cavaliere, R.; Omero, F.; Muscolino, P.; Granata, B.; D’Anna, F.; Silvestris, N.; et al. Crosstalk between ILC3s and Microbiota: Implications for Colon Cancer Development and Treatment with Immune Check Point Inhibitors. Cancers 2023, 15, 2893. https://doi.org/10.3390/cancers15112893
Drommi F, Calabrò A, Vento G, Pezzino G, Cavaliere R, Omero F, Muscolino P, Granata B, D’Anna F, Silvestris N, et al. Crosstalk between ILC3s and Microbiota: Implications for Colon Cancer Development and Treatment with Immune Check Point Inhibitors. Cancers. 2023; 15(11):2893. https://doi.org/10.3390/cancers15112893
Chicago/Turabian StyleDrommi, Fabiana, Alessia Calabrò, Grazia Vento, Gaetana Pezzino, Riccardo Cavaliere, Fausto Omero, Paola Muscolino, Barbara Granata, Federica D’Anna, Nicola Silvestris, and et al. 2023. "Crosstalk between ILC3s and Microbiota: Implications for Colon Cancer Development and Treatment with Immune Check Point Inhibitors" Cancers 15, no. 11: 2893. https://doi.org/10.3390/cancers15112893