


Interactions of IDO and the Kynurenine Pathway with Cell Transduction Systems and Metabolism at the Inflammation–Cancer Interface


Abstract
:Simple Summary
Abstract
1. Introduction
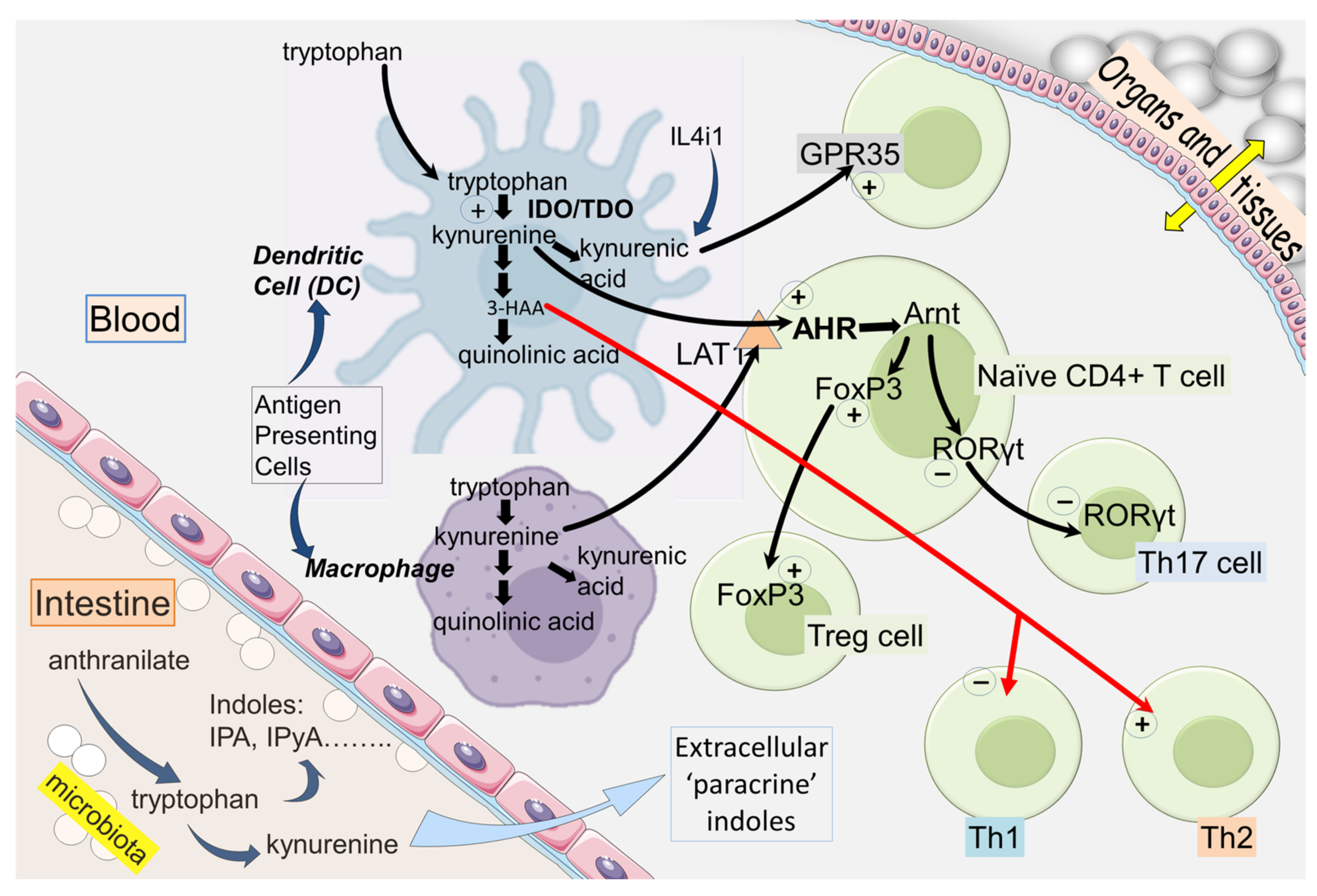
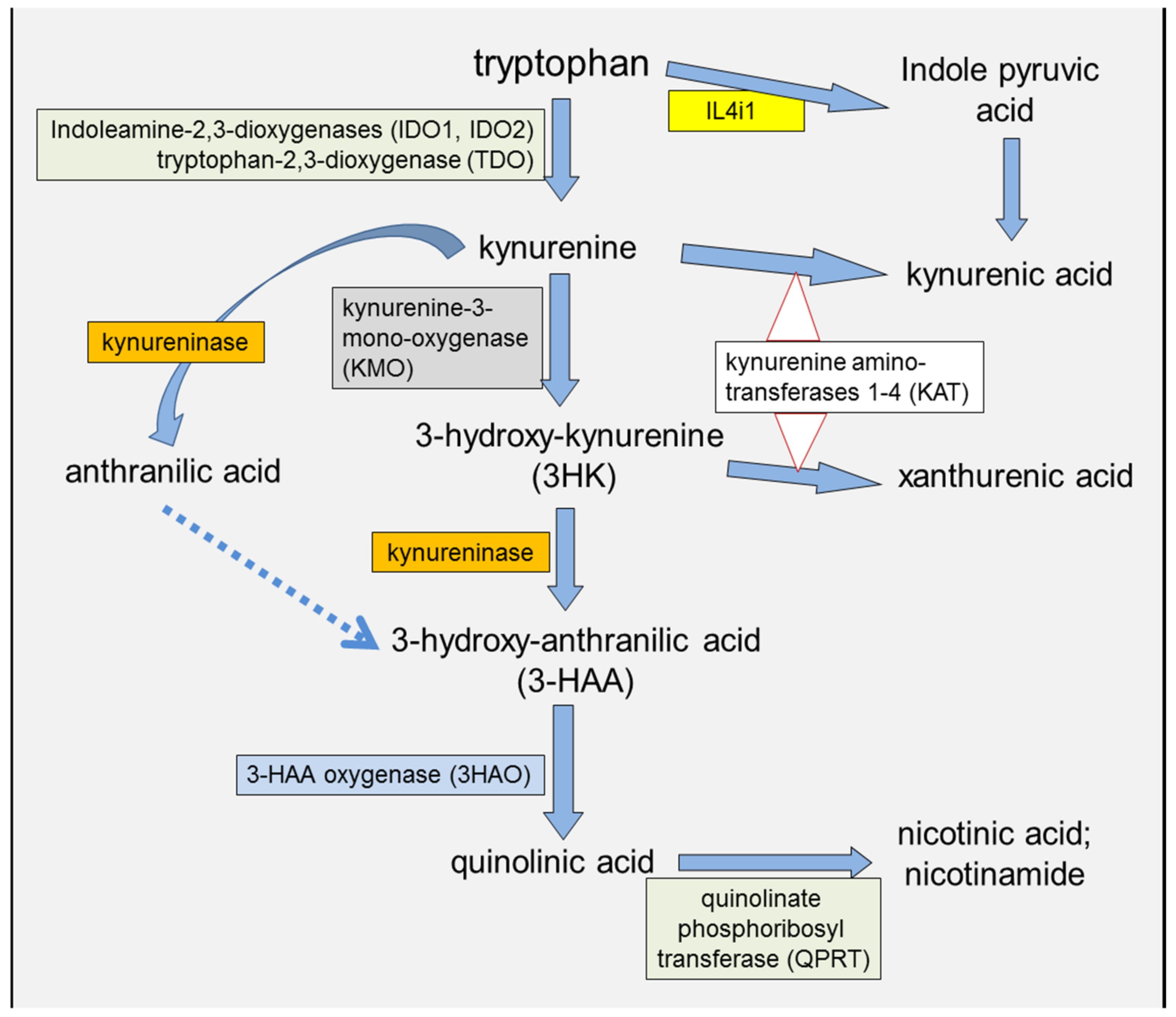
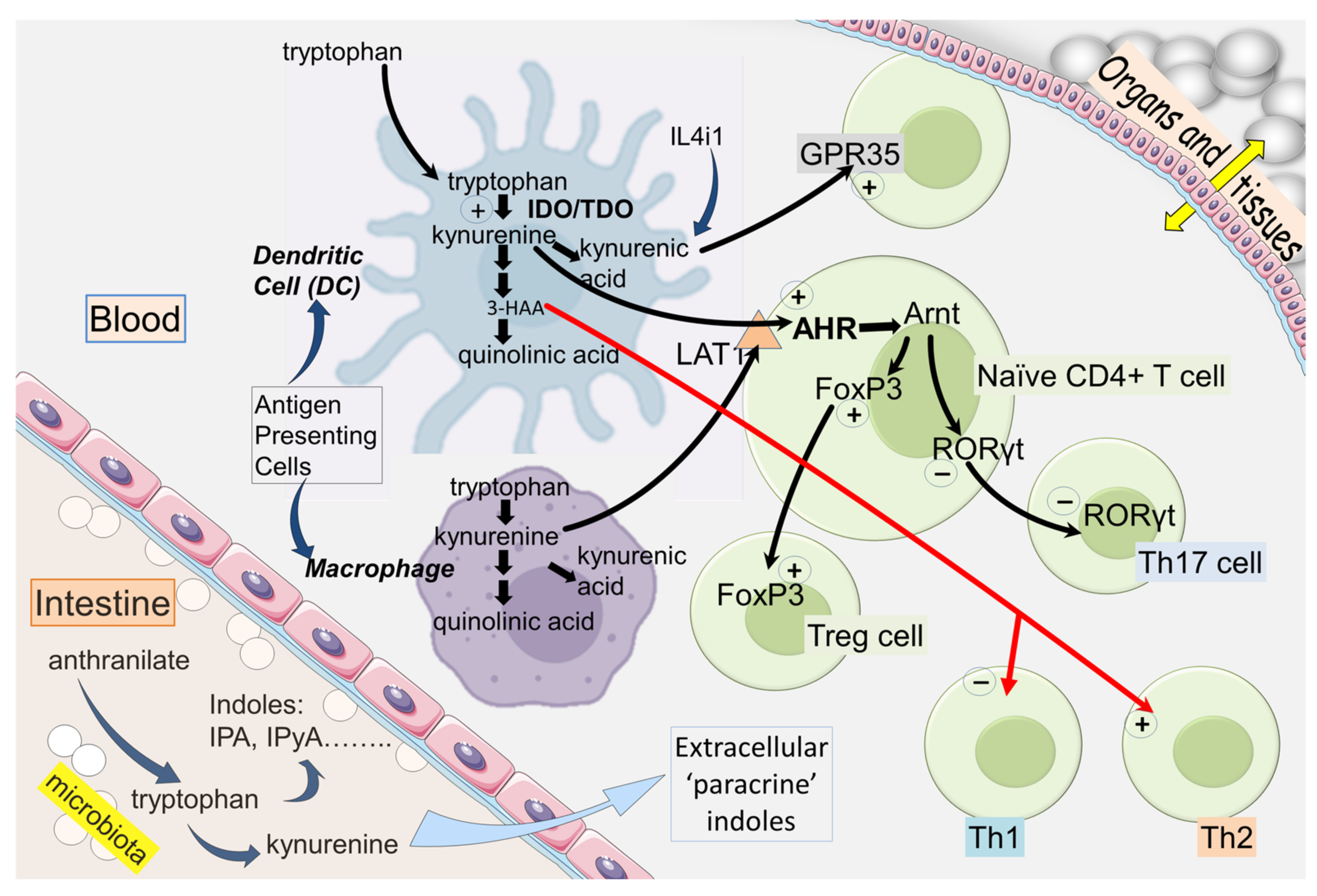
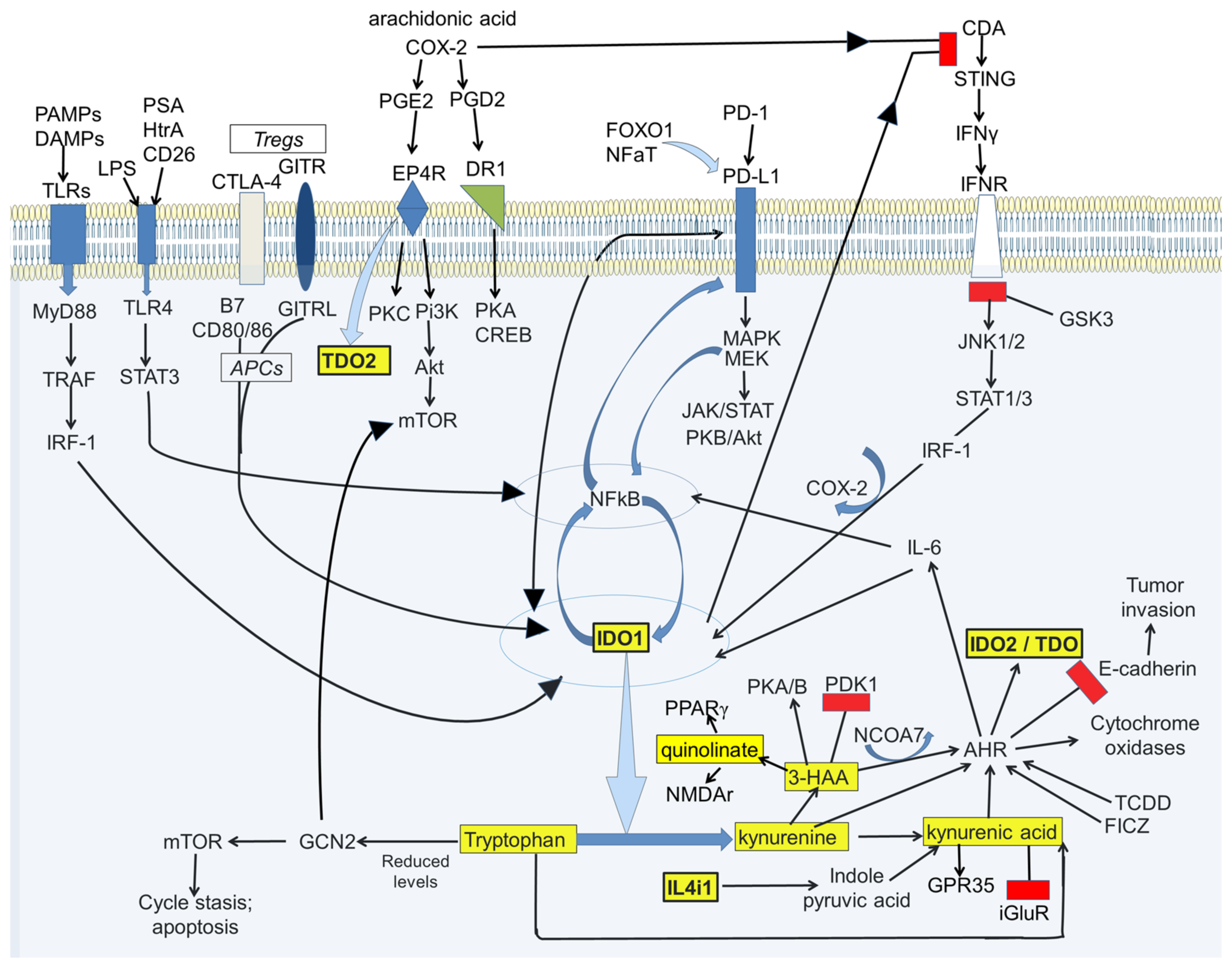
2. IDO and Kynurenine in the Immune System
2.1. IDO and Immunological Tolerance
2.2. T Cell Exhaustion
2.3. Kynurenic and 3-Hydroxyanthranilic Acids
2.4. IDO Modulation and Cancer Susceptibility
2.5. KMO Involvement
3. Kynurenine Pathway Interactions with Transduction Pathways
3.1. AHR
3.2. AHR and Homing
3.3. Programmed Cell Death Protein-1 (PD-1/PD-L1)
3.4. NFκB
3.5. Therapeutic Implications
3.6. Epigenetics and PD-1
3.7. FoxP3
3.8. FOXO1
3.9. miRNA
4. Metabolic Interactions
4.1. Anaerobic Glycolysis
4.2. Glutamine Metabolism
4.3. Miscellaneous Metabolic Factors
4.4. COX and Oxidative Stress
4.5. HIF-1α
4.6. Pyruvate Dehydrogenase Lipoamide Kinase Isozyme 1 (PDK1)
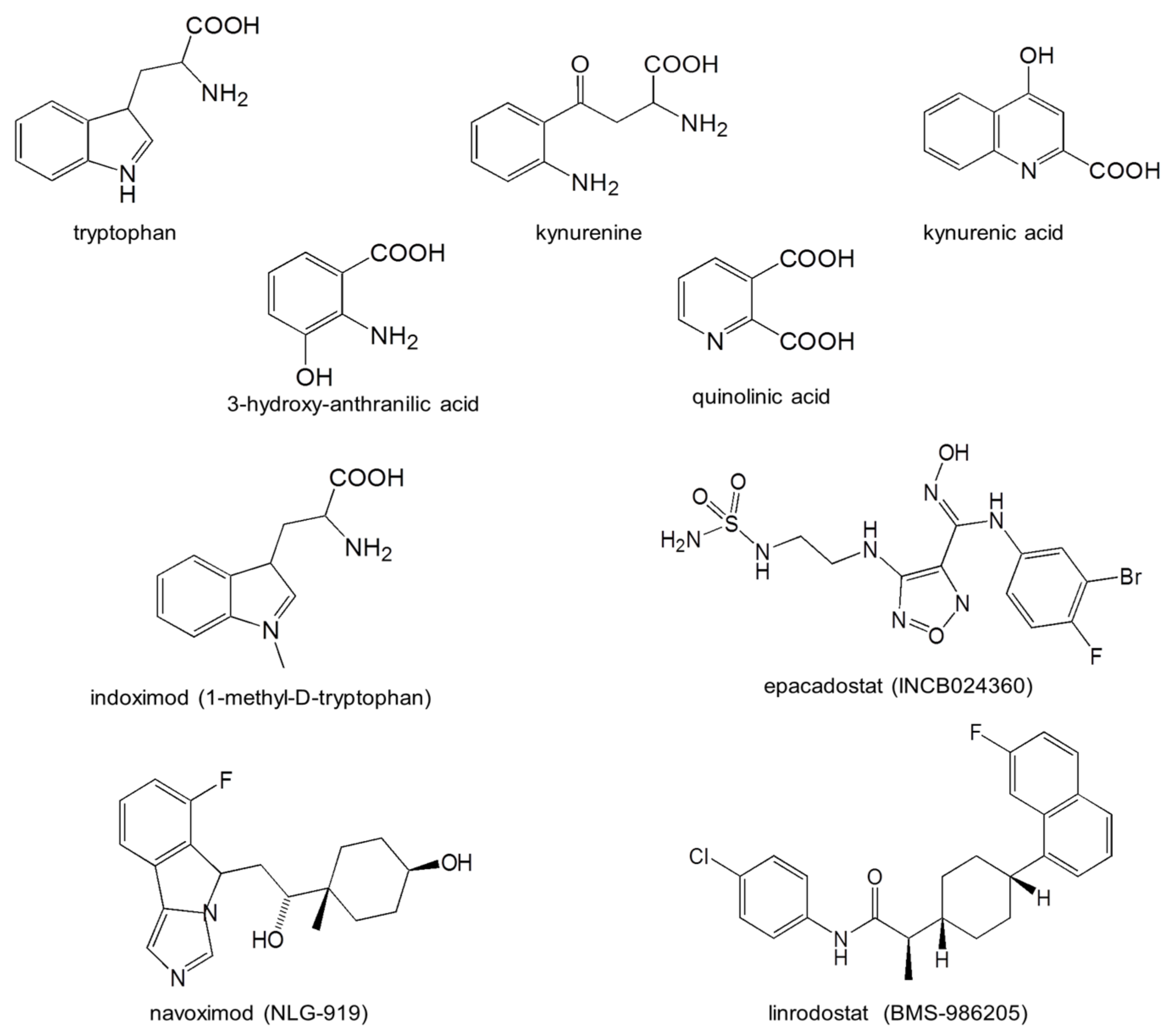
4.7. Status of Drug Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Target | Trial Target and Phase | Trial ID (NCT) | Reference and Comment |
|---|---|---|---|---|
| Indoximod (1-methyl-D-tryptophan; NLG-8919) | IDO1 activity inhibition; stimulates mTORC to modulate downstream transduction; promotes kynurenate formation | Metastatic breast cancer Phase 2 | NCT02913430 | NLG-802 is a prodrug for this, which entered Phase 1 in 2020 |
| Indoximod (1-methyl-D-tryptophan) | As above | Glioblastoma Phase 2 | NCT04049669 | |
| Indoximod (1-methyl-D-tryptophan) | As above | Metastatic prostate cancer; Phase 2 | NCT01560923 | [224] Combination with sipuleucel-T yielded radiological and clinical improvement |
| Indoximod (1-methyl-D-tryptophan) | As above | Metastatic pancreatic cancer; Phase 2 | NCT02077881 | [225] |
| Indoximod (1-methyl-D-tryptophan) | As above | Metastatic breast cancer; Phase 2 | NCT01792050 | [226] No improvement compared with taxane |
| Epacadostat INCB024360 | IDO1 inhibitor. IC50 = 93 nM in direct enzyme assay; IC50 = 12.5 nM in HeLa cell-based assay | Prostate cancer; Phase 1 and 2 | NCT 03493945 | [227,228] |
| Epacadostat INCB024360 | As above | Gliomas; Phase 2 | NCT03532295 | |
| Epacadostat INCB024360 | As above | Endometrial cancer; Phase 2 | NCT04463771 | |
| Epacadostat INCB024360 | As above | Head and neck cancer; Phase 2 | NCT03463161 |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, Y.S.; Ogbechi, J.; Clanchy, F.; Williams, R.O.; Stone, T.W. Kynurenine metabolites in peripheral disorders and neuroinflammation. Front. Immunol. 2020, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Meininger, V.; Brew, B.J. Implications for the kynurenine pathway and quinolinic acid in amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Tran, D.Q.; Pesu, M.; Davidson, T.S.; Ramsey, H.; O’Shea, J.J.; Shevach, E.M. CD4(+)FoxP3(+) regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J. Exp. Med. 2008, 205, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Volpi, C.; Bianchi, R.; Vacca, C.; Orabona, C.; Pallotta, M.T.; Boon, L.; Gizzi, S.; Fioretti, M.C.; Grohmann, U.; et al. Cutting edge: Autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J. Immunol. 2008, 181, 5194–5198. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Orabona, C.; Grohmann, U.; Puccetti, P. TGF-β and kynurenines as the key to infectious tolerance. Trends Mol. Med. 2009, 15, 41–49. [Google Scholar] [CrossRef]
- Yoshida, R.; Imanishi, J.; Oku, T.; Kishida, T.; Hayaishi, O. Induction of pulmonary indoleamine 2,3-dioxygenase by interferon. Proc. Natl. Acad. Sci. USA 1981, 78, 129–132. [Google Scholar] [CrossRef]
- Yasui, H.; Takai, K.; Yoshida, R.; Hayaishi, O. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: Its possible occurrence in cancer patients. Proc. Natl. Acad. Sci. USA 1986, 83, 6622–6626. [Google Scholar] [CrossRef]
- Stone, T.W.; Clanchy, F.I.L.; Huang, Y.-S.; Chiang, N.Y.; Darlington, L.G.; Williams, R.O. An integrated cytokine and kynurenine network as the basis of neuroimmune communication. Front. Neurosci. 2022, 16, 1002004. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, D.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9, 1981. [Google Scholar] [CrossRef]
- Ogbechi, J.; Wright, H.L.; Balin, S.; Topping, L.M.; Kristina, Z.; Huang, Y.S.; Pantazi, E.; Swart, M.; Windell, D.; Marin, E.; et al. LAT1 enables T cell activation under inflammatory conditions: A new therapeutic target for rheumatoid arthritis. J. Autoimmun. 2023, in press. [Google Scholar] [CrossRef]
- Ogbechi, J.; Huang, Y.-S.; Clanchy, F.I.L.; Pantazi, E.; Topping, L.M.; Darlington, L.G.; Williams, R.O.; Stone, T.W. Modulation of immune cell function; IDO expression and kynurenine production by the quorum sensor 2-heptyl-3-hydroxy-4-quinolone (PQS). Front. Immunol. 2022, 13, 1001956. [Google Scholar] [CrossRef]
- Stone, T.W.; Perkins, M.N. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef]
- Stone, T.W. The neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the action of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. Actions of kynurenic acid and quinolinic acid in the rat hippocampus in vivo. Exp. Neurol. 1985, 88, 570–579. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, W.; Liang, J.; Guo, J.; Ji, J.; Yao, Y.; Zheng, M.Z.; Cai, Z.J.; Lu, L.R.; Wang, J.L. IL4I1 Is a Novel Regulator of M2 macrophage polarization that can inhibit T cell activation via L-tryptophan and arginine depletion and IL-10 production. PLoS ONE 2015, 10, e0142979. [Google Scholar] [CrossRef]
- Sadik, A.; Patterson, L.F.S.; Ozturk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270. [Google Scholar] [CrossRef]
- Hezaveh, K.; Shinde, R.S.; Klotgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340. [Google Scholar] [CrossRef]
- Boulland, M.L.; Marquet, J.; Molinier-Frenkel, V.; Moller, P.; Guiter, C.; Lasoudris, F.; Copie-Berman, C.; Baia, M.; Gaulard, P.; Leroy, K.; et al. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood 2007, 110, 220–227. [Google Scholar] [CrossRef]
- Mason, J.M.; Naidu, M.D.; Barcia, M.; Porti, D.; Chavan, S.S.; Chu, C.C. IL-4-induced gene-1 is a leukocyte L-amino acid oxidase with an unusual acidic pH preference and lysosomal localization. J. Immunol. 2004, 173, 4561–4567. [Google Scholar] [CrossRef]
- Marquet, J.; Lasoudris, F.; Cousin, C.; Puiffe, M.L.; Martin-Garcia, N.; Baud, V.; Chereau, F.; Farcet, J.P.; Molinier-Frenkel, V.; Castellano, F. Dichotomy between factors inducing the immunosuppressive enzyme IL-4-induced gene 1 (IL4I1) in B lymphocytes and mononuclear phagocytes. Eur. J. Immunol. 2020, 40, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Carbonnelle-Puscian, A.; Copie-Bergman, C.; Baia, M.; Martin-Garcia, N.; Allory, Y.; Haioun, C.; Cremades, A.; Abd-Alsamad, I.; Farcet, J.P.; Gaulard, P.; et al. The novel immunosuppressive enzyme IL4I1 is expressed by neoplastic cells of several B-cell lymphomas and by tumor-associated macrophages. Leukemia 2009, 23, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Lasoudris, F.; Cousin, C.; Prevost-Blondel, A.; Martin-Garcia, N.; Abd-Alsamad, I.; Ortonne, N.; Farcet, J.P.; Castellano, F.; Molinier-Frenkel, V. IL4i1: An inhibitor of the CD8(+) antitumor T-cell response in vivo. Eur. J. Immunol. 2011, 41, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, I.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccett, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef]
- Liu, H.; Huang, L.; Bradley, J.; Liu, K.; Bardhan, K.; Ron, D.; Mellor, A.L.; Munn, D.H.; McGaha, T.L. GCN2-dependent metabolic stress is essential for endotoxemic cytokine induction and pathology. Mol. Cell. Biol. 2014, 34, 428–438. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Mark, M.W.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORct function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Read, J.E.; Amjadi, P.; Green, P.; Syed, K.; Manka, S.W.; Brennan, F.M.; Gregory, B.; et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by CTLA-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis Rheumatol. 2014, 66, 2344–2354. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Metz, R.; Muller, A.J.; Merlo, L.M.F.; Mandik-Nayak, L. IDO2 in immunomodulation and autoimmune disease. Front. Immunol. 2014, 5, 585. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; DuHadaway, J.B.; Montgomery, J.D.; Peng, W.D.; Murray, P.J.; Prendergast, G.C.; Caton, A.J.; Muller, A.J.; Mandik-Nayak, L. Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses. Front. Immunol. 2020, 11, 1861. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Peng, W.; DuHadaway, J.B.; Montgomery, J.D.; Prendergast, G.C.; Muller, A.J.; Mandik-Nayak, L. The Immunomodulatory Enzyme IDO2 Mediates Autoimmune Arthritis through a Nonenzymatic Mechanism. J. Immunol. 2022, 208, 571–581. [Google Scholar] [CrossRef]
- Pan, S.; Zhou, Y.; Wang, Q.S.; Wang, Y.L.; Tian, C.Y.; Wang, T.Q.; Huang, L.Y.; Nan, J.S.; Li, L.L.; Yang, S.Y. Discovery and structure-activity relationship studies of 1-aryl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione derivatives as potent dual inhibitors of indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO). Eur. J. Med. Chem. 2020, 207, 112703. [Google Scholar] [CrossRef]
- Pan, L.; Zheng, Q.; Chen, Y.; Yang, R.; Yang, Y.; Li, Z.J.; Meng, X.B. Design, synthesis and biological evaluation of novel naphthoquinone derivatives as IDO1 inhibitors. Eur. J. Med. Chem. 2018, 157, 423–436. [Google Scholar] [CrossRef]
- Cui, G.N.; Lai, F.F.; Wang, X.Y.; Chen, X.G.; Xu, B.L. Design, synthesis and biological evaluation of indole-2-carboxylic acid derivatives as IDO1/TDO dual inhibitors. Eur. J. Med. Chem. 2020, 188, 111985. [Google Scholar] [CrossRef]
- Serafini, M.; Torre, E.; Aprile, S.; Del Grosso, E.; Gesu, A.; Griglio, A.; Colombo, G.; Travelli, C.; Paiella, S.; Adamo, A.; et al. Discovery of highly potent benzimidazole derivatives as indoleamine 2,3-dioxygenase-1 (ido1) inhibitors: From structure-based virtual screening to in Vivo Pharmacodynamic Activity. J. Med. Chem. 2020, 63, 3047–3065. [Google Scholar] [CrossRef]
- He, X.; He, G.; Chu, Z.X.; Wu, H.; Wang, J.; Ge, Y.R.; Shen, H.; Zhang, S.; Shan, J.X.; Peng, K.W.; et al. Discovery of the First Potent IDO1/IDO2 Dual Inhibitors: A Promising Strategy for Cancer Immunotherapy. J. Med. Chem. 2021, 64, 17950–17968. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Q.; Yang, N.F.; Shi, Q.; Su, H.F.; Lin, T.S.; He, Z.L.; Wang, W.X.; Guo, H.Q.; Shen, P.P. Crosstalk between IL-15R alpha(+) tumor-associated macrophages and breast cancer cells reduces CD8(+) T cell recruitment. Cancer Commun. 2022, 42, 536–557. [Google Scholar] [CrossRef]
- Delpoux, A.; Michelini, R.H.; Verma, S.; Lai, C.Y.; Omilusik, K.D.; Utzschneider, D.T.; Redwood, A.J.; Goldrath, A.W.; Benedict, C.A.; Hedricj, S.M. Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8(+) T cells. J. Exp. Med. 2018, 215, 575–594. [Google Scholar] [CrossRef]
- Jancewicz, I.; Szarkowska, J.; Konopinski, R.; Stachowiak, M.; Swiatek, M.; Blachnio, K.; Kubala, S.; Oksinska, P.; Konopinski, R.; Stachowiak, M.; et al. PD-L1 Overexpression, SWI/SNF Complex Deregulation, and Profound Transcriptomic Changes Characterize Cancer-Dependent Exhaustion of Persistently Activated CD4(+) T Cells. Cancers 2021, 13, 4148. [Google Scholar] [CrossRef]
- Campos, J.S.; Henrickson, S.E. Defining and targeting patterns of T cell dysfunction in inborn errors of immunity. Front. Immunol. 2022, 13, 932715. [Google Scholar] [CrossRef]
- Ford, B.R.; Vignali, P.D.A.; Rittenhouse, N.L.; Scharping, N.E.; Peralta, R.; Lontos, K.; Frisch, A.T.; Delgoffe, G.M.; Poholek, A.C. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci. Immunol. 2022, 7, eabj9123. [Google Scholar] [CrossRef] [PubMed]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Guenther, J. Kynurenic Acid: The Janus-Faced Role of an immunomodulatory tryptophan metabolite and its link to pathological conditions. Front. Immunol. 2018, 8, 1957. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Langner, E.; Makuch-Kocka, A.; Szelest, M.; Szalast, K.; Marciniak, S.; Plech, T. Effect of tryptophan-derived AHR ligands; kynurenine; kynurenic acid and FICZ; on proliferation; cell cycle regulation and cell death of melanoma cells-in vitro studies. Int. J. Mol. Sci. 2020, 21, 7946. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic acid and cancer: Facts and controversies. Cell. Mol. Life Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef] [PubMed]
- Tiszlavicz, Z.; Németh, B.; Fülöp, F.; Vécsei, L.; Tápai, K.; Ocsovszky, I.; Mandi, Y. Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-α (TNF-α) production by mononuclear cells; HMGB1 production by monocytes and HNP1-3 secretion by neutrophils. Arch. Pharmacol. 2011, 383, 447–455. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef]
- Resta, F.; Masi, A.; Sili, M.; Laurino, A.; Moroni, F.; Mannaioni, G. Kynurenic acid and zaprinast induce analgesia by modulating HCN channels through GPR35 activation. Neuropharmacology 2016, 108, 136–143. [Google Scholar] [CrossRef]
- Wyant, G.A.; Yu, W.; Doulamis, I.P.; Nomoto, R.S.; Saeed, M.Y.; Duignan, T.; McCully, J.D.; Kaelin, W.G. Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science 2022, 377, 621–629. [Google Scholar] [CrossRef]
- Kvaratskhelia, E.; Maisuradze, E.; Dabrundashvili, N.G.; Natsvlishvili, N.; Zhuravliova, E.; Mikeladze, D.G. N-Methyl-D-Aspartate and sigma-ligands change the production of interleukins 8 and 10 in lymphocytes through modulation of the NMDA glutamate receptor. Neuro-Immunomodulation 2009, 16, 201–207. [Google Scholar] [CrossRef]
- Lombardi, G.; Dianzani, C.; Miglio, G.; Canonico, P.L.; Fantozzi, R. Characterization of ionotropic glutamate receptors in human lymphocytes. Br. J. Pharmacol. 2001, 133, 936–944. [Google Scholar] [CrossRef]
- Bhandage, A.K.; Jin, Z.; Hellgren, C.; Korol, S.V.; Nowak, K.; Williamsson, L.; Sundstrom-Poromaa, I.; Birnir, B. AMPA, NMDA and kainate glutamate receptor subunits are expressed in human peripheral blood mononuclear cells (PBMCs) where the expression of GluK4 is altered by pregnancy and GluN2D by depression in pregnant women. J. Neuroimmunol. 2017, 305, 51–58. [Google Scholar] [CrossRef]
- Mashkina, A.P.; Cizkova, D.; Vanicky, I.; Boldyrev, A.A. NMDA receptors are expressed in lymphocytes activated both in vitro and in vivo. Cell. Mol. Neurobiol. 2010, 30, 901–907. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Kazey, V.I.; Leinsoo, T.A.; Mashkina, A.P.; Tyulina, O.V.; Johnson, P.; Tuneva, J.O.; Chittur, S. Rodent lymphocytes express functionally active glutamate receptors. Biochem. Biophys. Res. Commun. 2004, 324, 133–139. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Bryushkova, E.A.; Vladychenskaya, E.A. NMDA Receptors in Immune Competent Cells. Biochemistry 2012, 77, 128–134. [Google Scholar] [CrossRef]
- Nohara, L.L.; Stanwood, S.R.; Omilusik, K.D.; Jefferies, W.A. Tweeters; woofers and horns: The complex orchestration of calcium currents in T lymphocytes. Front. Immunol. 2015, 6, 234. [Google Scholar] [CrossRef]
- Turski, W.A.; Gramsbergen, J.B.; Traitler, H.; Schwarcz, R. Rat brain slices produce and liberate kynurenic acid upon exposure to L-kynurenine. J. Neurochem. 1989, 52, 1629–1636. [Google Scholar] [CrossRef]
- Heredi, J.; Cseh, E.K.; Berko, A.M.; Veres, G.; Zadori, D.; Toldi, J.; Kis, Z.; Vecsei, L.; Ono, E.; Gellert, L. Investigating KYNA production and kynurenergic manipulation on acute mouse brain slice preparations. Brain Res. Bull. 2019, 146, 185–191. [Google Scholar] [CrossRef]
- Correale, J. Immunosuppressive Amino-Acid Catabolizing Enzymes in Multiple Sclerosis. Front. Immunol. 2021, 11, 600428. [Google Scholar] [CrossRef]
- Gargaro, M.; Vacca, C.; Massari, S.; Scalisi, G.; Manni, G.; Mondanelli, G.; Mazza, E.M.C.; Bicciato, S.; Pallotta, M.T.; Orabona, C. Engagement of nuclear coactivator 7 by 3-hydroxyanthranilic acid enhances activation of aryl hydrocarbon receptor in immunoregulatory dendritic cells. Front. Immunol. 2019, 10, 1973. [Google Scholar] [CrossRef]
- Oh, G.S.; Pae, H.O.; Choi, B.M.; Chae, S.C.; Lee, H.S.; Ryu, D.G.; Chung, H.T. 3-Hydroxyanthranilic acid, one of metabolites of tryptophan via indoleamine 2,3-dioxygenase pathway, suppresses inducible nitric oxide synthase expression by enhancing heme oxygenase-1 expression. Biochem. Biophys. Res. Commun. 2004, 320, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.F.; Shi, Z.P.; Liu, D.; Zhang, S.Y.; Zhu, H.; Wang, Y.G.; Mi, J. 3-hydroxyanthranilic acid increases the sensitivity of hepatocellular carcinoma to sorafenib by decreasing tumor cell stemness. Cell Death Discov. 2021, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.; Shi, Z.; Shangguan, C.; Zhang, J.; Yuan, Y.; Chen, L.; Liu, W.; Meng, S.; Xiong, W.; Mi, J. The kynurenine derivative 3-HAA sensitizes hepatocellular carcinoma to sorafenib by upregulating phosphatases. Theranostics 2021, 11, 6006–6018. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Mo, J.H.; Gong, X.; Rossetto, C.; Jang, A.; Beck, L.; Elliott, G.I.; Kufareva, I.; Abagyan, R.; Broide, D.H.; et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 18619–18624. [Google Scholar] [CrossRef]
- Hosooka, T.; Hosokawa, Y.; Matsugi, K.; Shinohara, M.; Senga, Y.; Tamori, Y.; Aoki, C.; Matsui, S.; Sasaki, T.; Kitamura, T.; et al. The PDK1-FoxO1 signaling in adipocytes controls systemic insulin sensitivity through the 5-lipoxygenase- leukotriene B-4 axis. Proc. Natl. Acad. Sci. USA 2020, 117, 11674–11684. [Google Scholar] [CrossRef]
- Zuo, H.; Tell, G.S.; Vollset, S.E.; Ueland, P.M.; Nygard, O.; Midttun, O.; Meyer, K.; Ulvik, A.; Eussen, S.J.P.M. Interferon-gamma-Induced Inflammatory Markers and the Risk of Cancer: The Hordaland Health Study. Cancer 2014, 120, 3370–3377. [Google Scholar] [CrossRef]
- Dugue, P.A.; Hodge, A.M.; Ulvik, A.; Ueland, P.M.; Midttun, O.; Rinaldi, S.; Macinnis, R.J.; Li, S.X.; Meyer, K.; Navionis, A.S.; et al. Association of Markers of Inflammation, the Kynurenine Pathway and B Vitamins with Age and Mortality, and a Signature of Inflammaging. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 826–836. [Google Scholar] [CrossRef]
- Clanchy, F.I.L.; Huang, I.-S.; Ogbechi, J.; Darlington, L.G.; Williams, R.O.; Stone, T.W. Induction of IDO1 and Kynurenine by Serine Proteases Subtilisin, Prostate Specific Antigen, CD26 and HtrA: A New Form of Immunosuppression? Front. Immunol. 2022, 13, 832989. [Google Scholar] [CrossRef]
- Forrest, C.M.; McNair, K.; Vincenten, M.C.; Darlington, L.G.; Stone, T.W. Selective depletion of tumour suppressors Deleted in Colorectal Cancer (DCC) and neogenin by environmental and endogenous serine proteases: Linking diet and cancer. BMC Cancer 2016, 16, 772. [Google Scholar] [CrossRef]
- McNair, K.; Forrest, C.M.; Vincenten, M.; Darlington, L.G.; Stone, T.W. Serine protease modulation of dependence receptors and EMT protein expression. Cancer Biol. Ther. 2019, 20, 349–367. [Google Scholar] [CrossRef]
- Stone, T.W. Dependence and Guidance Receptors—DCC and neogenin—In partial EMT and the actions of serine proteases. Front. Oncol. 2020, 10, 94. [Google Scholar] [CrossRef]
- Feng, X.; Shen, P.; Wang, Y.; Li, Z.Y.; Bian, J. Synthesis and in vivo antitumor evaluation of an orally active potent phosphonamidate derivative targeting IDO1/IDO2/TDO. Biochem. Pharmacol. 2019, 168, 214–223. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Hunt, N.H.; Ball, H.J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: Characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. [Google Scholar] [CrossRef]
- Huang, Y.S.; Tseng, W.Y.; Clanchy, F.; Topping, L.M.; Ogbechi, J.; McNamee, K.; Perocheau, D.; Chiang, N.Y.; Ericsson, P.; Sundstedt, A.; et al. Pharmacological modulation of T cell immunity results in long-term remission of autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2021, 118, e2100939118. [Google Scholar] [CrossRef]
- Mondanelli, G.; Coletti, A.; Greco, F.A.; Pallotta, M.T.; Orabona, C.; Iacono, A.; Belladonna, M.L.; Albini, E.; Panfili, E.; Fallarino, F.; et al. Positive allosteric modulation of indoleamine 2,3-dioxygenase 1 restrains neuroinflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 3848–3857. [Google Scholar] [CrossRef]
- Peng, Y.P.; Zhang, J.J.; Liang, W.B.; Tu, M.; Lu, Z.P.; Wei, J.S.; Jiang, K.R.; Gao, W.T.; Wu, J.L.; Xu, Z.K.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar] [CrossRef]
- Mitra, D.; Horick, N.K.; Brackett, D.G.; Mouw, K.W.; Hornick, J.L.; Ferrone, S.; Hong, T.S.; Mamon, H.; Clark, J.W.; Parikh, A.R.; et al. High IDO1 expression is associated with poor outcome in patients with anal cancer treated with definitive chemoradiotherapy. Oncologist 2019, 24, e275–e283. [Google Scholar] [CrossRef]
- Jin, H.J.; Zhang, Y.R.; You, H.Y.; Tao, X.M.; Wang, C.; Jin, G.Z.; Wang, N.; Ruan, H.Y.; Gu, D.S.; Huo, X.S.; et al. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration, and invasion of human hepatocellular carcinoma. Sci. Rep. 2015, 5, 10466. [Google Scholar] [CrossRef]
- Forrest, C.M.; Khalil, O.S.; Pisar, M.; Darlington, L.G.; Stone, T.W. Prenatal inhibition of the tryptophan-kynurenine pathway alters synaptic plasticity and protein expression in the rat hippocampus. Brain Res. 2013, 1504, 1–15. [Google Scholar] [CrossRef]
- Zhou, L.L.; Mu, L.; Jiang, W.Y.; Yang, Q. QPRT Acts as an Independent Prognostic Factor in Invasive Breast Cancer. J. Oncol. 2022, 2022, 6548644. [Google Scholar] [CrossRef]
- Ala, M. The footprint of kynurenine pathway in every cancer: A new target for chemotherapy. Eur. J. Pharmacol. 2021, 896, 173921. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.C.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Bankoti, J.; Rase, B.; Simones, T.; Shepherd, D.M. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol. Appl. Pharmacol. 2010, 246, 18–28. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.D.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6; STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K. Tolerogenic phenotype of IFN-gamma-induced IDO+ dendritic cells is maintained via an autocrine IDO-kynurenine/AhR-IDO loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef]
- Wang, Z.; Monti, S.; Sherr, D.H. The diverse and important contributions of the AHR to cancer and cancer immunity. Curr. Opin. Toxicol. 2017, 2, 93–102. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2(-) Human Breast Cancer Cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef]
- Gargaro, M.; Manni, G.; Scalisi, G.; Puccetti, P.; Fallarino, F. Tryptophan metabolites at the crossroad of immune-cell interaction via the aryl hydrocarbon receptor: Implications for tumor immunotherapy. Int. J. Mol. Sci. 2021, 22, 4644. [Google Scholar] [CrossRef]
- Pallotta, M.T.; Fallarino, F.; Matino, D.; Macchiarula, A.; Orabona, C. AhR-mediated, non-genomic modulation of IDO1 function. Front. Immunol. 2014, 5, 497. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L.J. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Patterson, L.F.S.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.K.; Pennington, J.M.; Bisson, W.H.; Kolluri, S.K.; Kerkvliet, N.I. TCDD, FICZ, and Other High Affinity AhR Ligands Dose-Dependently Determine the Fate of CD41 T Cell Differentiation. Toxicol. Sci. 2018, 161, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.J.; Croze, E.; Yamaguchi, K.D.; Tran, T.; Reder, A.T.; Litvak, V.; Volkert, M.R. Induction of a Unique Isoform of the NCOA7 Oxidation Resistance Gene by Interferon beta-1b. J. Interferon Cytokine Res. 2015, 35, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Dean, J.W.; Fu, Z.; Oliff, K.N.; Bostick, J.W.; Ye, J.; Chen, Z.; Muhlbauer, M.; Zhou, L. Ahr-Foxp3-ROR gamma-t axis controls gut homing of CD4(+) T cells by regulating GPR15. Sci. Immunol. 2020, 5, eaaz7277. [Google Scholar] [CrossRef]
- Ye, J.; Qiu, J.; Bostick, J.W.; Ueda, A.; Schjerven, H.; Li, S.; Jobin, C.; Chen, Z.E.; Zhou, L. The Aryl Hydrocarbon Receptor Preferentially Marks and Promotes Gut Regulatory T Cells. Cell Rep. 2017, 21, 2277–2290. [Google Scholar] [CrossRef]
- Ohnmacht, C.; Park, J.H.; Cording, S.; Wing, J.B.; Atarashi, K.; Obata, Y.; Gaboriau-Routhiau, V.; Marques, R.; Dulauroy, S.; Fedoseeva, M.; et al. The microbiota regulates type 2 immunity through RORgamma(+) T cells. Science 2015, 349, 989–993. [Google Scholar] [CrossRef]
- Swaminathan, G.; Nguyen, L.P.; Namkoong, H.; Pan, J.; Haileselassie, Y.; Patel, A.; Ji, A.R.; Mikhail, D.M.; Dinh, T.; Singh, H.; et al. The aryl hydrocarbon receptor regulates expression of mucosal trafficking receptor GPR15. Mucosal Immunol. 2021, 14, 852–861. [Google Scholar] [CrossRef]
- Kim, S.V.; Xiang, W.V.; Kwak, C.; Yang, Y.; Lin, X.W.; Ota, M.; Sarpel, U.; Rifkin, D.B.; Xu, R.; Littman, D.R. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 2013, 340, 1456–1459. [Google Scholar] [CrossRef]
- Nguyen, L.P.; Pan, J.; Dinh, T.T.; Hadeiba, H.; O’Hara, E., 3rd; Ebtikar, A.; Hertweck, A.; Gokmen, M.R.; Lord, G.M.; Jenner, R.G.; et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat. Immunol. 2015, 16, 207–213. [Google Scholar] [CrossRef]
- Rudra, D.; de Roos, P.; Chaudhry, A.; Niec, R.E.; Arvey, A.; Samstein, R.M.; Leslie, C.; Shaffer, S.A.; Goodlett, D.R.; Rudensky, A.Y. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 2012, 13, 1010–1019. [Google Scholar] [CrossRef]
- Lu, L.; Barbi, J.; Pan, F. The regulation of immune tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef]
- Nagai, Y.; Lam, L.; Greene, M.I.; Zhang, H.T. FOXP3 and Its Cofactors as Targets of Immunotherapies. Engineering 2019, 5, 115–121. [Google Scholar] [CrossRef]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 exploits a pre- existent enhancer landscape for regulatory T cell lineage specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef]
- Pavlick, K.P.; Ostanin, D.V.; Furr, K.L.; Laroux, F.S.; Brown, C.M.; Gray, L.; Kevil, C.G.; Grisham, M.B. Role of T-cell-associated lymphocyte function-associated antigen-1 in the pathogenesis of experimental colitis. Intern. Immunol. 2006, 18, 389–398. [Google Scholar] [CrossRef]
- Wolchok, J.D. PD-1 blockers. Cell 2015, 162, 937. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, E.W.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Cao, W.; Lu, J.; Li, L.; Qiu, C.; Qin, X.; Wang, T.; Li, S.; Zhang, J.; Xu, J. Activation of the Aryl Hydrocarbon Receptor Ameliorates Acute Rejection of Rat Liver Transplantation by Regulating Treg Proliferation and PD-1 Expression. Transplantation 2022, 106, 2172–2181. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Ann. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, C.; Xian, J.; Zhang, M.; Cao, Y.; Cao, Y. Expression of programmed cell death protein 1 (PD-1) and indoleamine 2,3-dioxygenase (IDO) in the tumor microenvironment and in tumor draining lymph nodes of breast cancer. Hum. Pathol. 2018, 75, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; He, X.L.; Qi, L.; Shi, W.; Yuan, L.W.; Huang, M.Y.; Xu, Y.L.; Chen, X.; Gu, L.; Zhang, L.L.; et al. Myricetin inhibits interferon-gamma-induced PD-L1 and IDO1 expression in lung cancer cells. Biochem. Pharmacol. 2022, 197, 114940. [Google Scholar] [CrossRef] [PubMed]
- Anzai, H.; Yoshimoto, S.; Okamura, K.; Hiraki, A.; Hashimoto, S. IDO1-mediated Trp-kynurenine-AhR signal activation induces stemness and tumor dormancy in oral squamous cell carcinomas. Oral Sci. Int. 2022, 19, 31–43. [Google Scholar] [CrossRef]
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280. [Google Scholar] [CrossRef]
- Donizy, P.; Wu, C.L.; Kopczynski, J.; Pieniazek, M.; Biecek, P.; Rys, J.; Hoang, M.P. Prognostic Role of Tumoral PD-L1 and IDO1 Expression, and Intratumoral CD8+ and FoxP3+Lymphocyte Infiltrates in 132 Primary Cutaneous Merkel Cell Carcinomas. Int. J. Mol. Sci. 2021, 11, 5489. [Google Scholar] [CrossRef]
- Xiang, Z.; Zhou, Z.J.; Song, S.Z.; Li, J.; Ji, J.; Yan, R.L.; Wang, J.X.; Cai, W.; Hu, W.J.; Zang, L.; et al. Dexamethasone suppresses immune evasion by inducing GR/STAT3 mediated downregulation of PD-L1 and IDO1 pathways. Oncogene 2021, 40, 5002–5012. [Google Scholar] [CrossRef]
- Kotecki, N.; Vuagnat, P.; O’Neil, B.H.; Jalal, S.; Rottey, S.; Prenen, H.; Benhadj, K.A.; Xia, M.; Szpurka, A.M.; Saha, A.; et al. A Phase I Study of an IDO-1 Inhibitor (LY3381916) as Monotherapy and in Combination with an Anti-PD-L1 Antibody (LY3300054) in Patients With Advanced Cancer. J. Immunother. 2021, 44, 264–275. [Google Scholar] [CrossRef]
- Jung, M.Y.; Aibaidula, A.; Brown, D.A.; Himes, B.T.; Garcia, L.M.C.; Parney, I.F. Superinduction of immunosuppressive glioblastoma extracellular vesicles by IFN-gamma through PD-L1 and IDO1. Neuro-Oncol. Adv. 2022, 4, vdac017. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat. Med. 2021, 27, 2212–2220. [Google Scholar] [CrossRef]
- Abdulla, M.; Alexsson, A.; Sundstrom, C.; Ladenvall, C.; Mansouri, L.; Lindskog, C.; Berglund, M.; Cavelier, L.; Enblad, G.; Hollander, P.; et al. PD-L1 and IDO1 are potential targets for treatment in patients with primary diffuse large B-cell lymphoma of the CNS. Acta Oncol. 2021, 60, 531–538. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.Q.; Fang, Y.; Lv, J.D.; Zhang, T.Z.; Fiskesund, R.; Xie, J.; Liu, J.Y.; Yin, X.N.; et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef]
- Amobi-McCloud, A.; Muthuswamy, R.; Battaglia, S.; Yu, H.; Liu, T.; Wang, J.; Putluri, V.; Singh, P.K.; Qian, F.; Huang, R.; et al. IDO1 Expression in Ovarian Cancer Induces PD-1 in T cells via Aryl Hydrocarbon Receptor Activation. Front. Immunol. 2021, 12, 678999. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Humid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Iwasaki, T.; Kohashi, K.; Toda, Y.; Ishihara, S.; Yamada, Y.; Oda, Y. Association of PD-L1 and IDO1 expression with JAK-STAT pathway activation in soft-tissue leiomyosarcoma. J. Cancer Res. Clin. Oncol. 2021, 147, 1451–1463. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Rosen, D.B.; Grein, J.; Tedesco, D.; Joyce-Shaikh, B.; Ueda, R.; Semana, M.; Bauer, M.; Bang, K.; Stevenson, C.; et al. GITR Agonism Enhances Cellular Metabolism to Support CD8(+) T-cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol. Res. 2018, 6, 1199–1211. [Google Scholar] [CrossRef]
- Ishihara, S.; Yamada, Y.; Iwasaki, T.; Yoshimoto, M.; Toda, Y.; Kohashi, K.; Yamamoto, H.; Matsumoto, Y.; Nakashima, Y.; Oda, Y. PD-L1 and IDO-1 expression in undifferentiated pleomorphic sarcoma: The associations with tumor infiltrating lymphocytes; dMMR and HLA class I. Oncol. Rep. 2021, 45, 379–389. [Google Scholar] [CrossRef]
- Takada, K.; Toyokawa, G.; Kinoshita, F.; Jogo, T.; Kohashi, K.; Wakasu, S.; Ono, Y.; Tanaka, K.; Oba, T.; Osoegawa, A.; et al. Expression of PD-L1, PD-L2, and IDO1 on tumor cells and density of CD8-positive tumor-infiltrating lymphocytes in early-stage lung adenocarcinoma according to histological subtype. J. Cancer Res. Clin. Oncol. 2020, 146, 2639–2650. [Google Scholar] [CrossRef]
- Toda, Y.; Kohashi, K.; Yamada, Y.; Yoshimoto, M.; Ishihara, S.; Ito, Y.; Iwasaki, T.; Yamamoto, H.; Matsumoto, Y.; Nakashima, Y.; et al. PD-L1 and IDO1 expression and tumor-infiltrating lymphocytes in osteosarcoma patients: Comparative study of primary and metastatic lesions. J. Cancer Res. Clin. Oncol. 2020, 146, 2607–2620. [Google Scholar] [CrossRef]
- Swainson, L.A.; Ahn, H.; Pajanirassa, P.; Khetarpal, V.; Deleage, C.; Estes, J.D.; Hunt, P.W.; Munoz-Sanjuan, I.G.; McCune, J. Kynurenine 3-Monooxygenase Inhibition during Acute Simian Immunodeficiency Virus Infection Lowers PD-1 Expression and Improves Post-Combination Antiretroviral Therapy CD4(+) T cell Counts and Body Weight. J. Immunol. 2019, 203, 899–910. [Google Scholar] [CrossRef]
- Miyazaki, T.; Chung, S.Y.; Sakai, H.; Ohata, H.; Obata, Y.; Shiokawa, D.; Mizoguchi, Y.; Kubo, T.; Ishikawa, H.; Taniguchi, H.; et al. Stemness and immune evasion conferred by the TDO2-AHR pathway are associated with liver metastasis of colon cancer. Cancer Sci. 2022, 113, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Ludovini, V.; Bianconi, F.; Siggillino, A.; Vannucci, J.; Baglivo, S.; Berti, V.; Tofanetti, F.R.; Reda, M.S.; Bellezza, G.; Mandarano, M.; et al. High PD-L1/IDO-2 and PD-L2/IDO-1 co-expression levels are associated with worse overall survival in resected non-small cell lung cancer patients. Genes 2021, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Hacking, S.; Chavarria, H.; Jin, C.; Perry, A.; Nasim, M. Landscape of Immune Checkpoint Inhibition in Carcinosarcoma (MMMT): Analysis of IDO-1; PD-L1 and PD-1. Pathol. Res. Pract. 2020, 216, 152847. [Google Scholar] [CrossRef] [PubMed]
- Tao, B.B.; Shi, J.H.; Shuai, S.; Zhou, H.Y.; Zhang, H.X.; Li, B.; Wang, X.Q.; Li, G.H.; He, H.; Zhong, J. CYB561D2 up-regulation activates STAT3 to induce immunosuppression and aggression in gliomas. J. Transl. Med. 2021, 19, 338. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Bally, A.P.R.; Tang, Y.; Lee, J.T.; Barwick, B.G.; Martinez, R.; Evavold, B.D.; Boss, J.M. Conserved Region C Functions To Regulate PD-1 Expression and Subsequent CD8 T Cell Memory. J. Immunol. 2017, 198, 205–217. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, Y.; Auger-Messier, M.; Molkentin, J.D. Interaction Between NFκB and NFAT Coordinates Cardiac Hypertrophy and Pathological Remodeling. Circ. Res. 2012, 110, 1077–1086. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, J.Z.; Jiang, R.; Liu, S.F.; He, Y.Y.; van der Vorst, E.P.C.; Weber, C.; Doring, Y.; Yan, Y. Regulatory T cell-related gene indicators in pulmonary hypertension. Front. Pharmacol. 2022, 31, 908783. [Google Scholar] [CrossRef]
- Hsu, F.T.; Chen, T.C.; Chuang, H.Y.; Chang, Y.F.; Hwang, J.J. Enhancement of adoptive T cell transfer with single low dose pretreatment of doxorubicin or paclitaxel in mice. Oncotarget 2015, 6, 44134–44150. [Google Scholar] [CrossRef]
- Kaiser, H.; Parker, E.; Hamrick, M.W. Kynurenine Signaling through the Aryl Hydrocarbon Receptor: Implications for Aging and Healthspan. Exp. Gerontol. 2020, 130, 110797. [Google Scholar] [CrossRef]
- Welz, B.; Bikker, R.; Junemann, J.; Christmann, M.; Neumann, K.; Weber, M.; Hoffmeister, L.; Preuss, K.; Pich, A.; Huber, R.; et al. Proteome and Phosphoproteome Analysis in TNF Long Term-Exposed Primary Human Monocytes. Int. J. Mol. Sci. 2019, 20, 1241. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR Signaling Axis Facilitates Anoikis Resistance and Metastasis in Triple-Negative Breast Cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef]
- Lee, C.H.; Bae, J.H.; Choe, E.J.; Park, J.M.; Park, S.S.; Cho, H.J.; Song, B.J.; Baek, M.C. Macitentan improves antitumor immune responses by inhibiting the secretion of tumor-derived extracellular vesicle PD-L1. Theranostics 2022, 12, 1971–1987. [Google Scholar] [CrossRef]
- Moshofsky, K.B.; Cho, H.J.; Wu, G.M.; Romine, K.A.; Newman, M.T.; Kosaka, Y.; McWeeney, S.K.; Lind, E.F. Acute myeloid leukemia-induced T-cell suppression can be reversed by inhibition of the MAPK pathway. Blood Adv. 2019, 3, 3038–3051. [Google Scholar] [CrossRef]
- Guan, L.; Wu, B.; Li, T.; Beer, L.A.; Sharma, G.; Li, M.; Lee, C.N.; Liu, S.J.; Yang, C.S.; Huang, L.L.; et al. HRS phosphorylation drives immunosuppressive exosome secretion and restricts CD8(+) T-cell infiltration into tumors. Nat. Commun. 2022, 13, 4078. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Kroemer, G.; Tang, D.L. Targeting HSP90 sensitizes pancreas carcinoma to PD-1 blockade. Oncoimmunology 2022, 11, 2068488. [Google Scholar] [CrossRef]
- Yang, A.O.; Li, M.Y.; Zhang, Z.H.; Wang, J.Y.; Xing, Y.; Ri, M.; Jin, C.H.; Xu, G.H.; Piao, L.X.; Jin, H.L.; et al. Erianin regulates programmed cell death ligand 1 expression and enhances cytotoxic T lymphocyte activity. J. Ethnopharmacol. 2021, 273, 113598. [Google Scholar] [CrossRef]
- Ando, S.; Araki, K. CD8 T-cell heterogeneity during T-cell exhaustion and PD-1-targeted immunotherapy. Int. Immunol. 2022, 34, 571–577. [Google Scholar] [CrossRef]
- Bae, J.; Hideshima, T.; Zhang, G.L.; Zhou, J.; Keskin, D.B.; Munshi, N.C.; Anderson, K.C. Identification and characterization of HLA-A24-specific XBP1; CD138 (Syndecan-1) and CS1 (SLAMF7) peptides inducing antigens-specific memory cytotoxic T lymphocytes targeting multiple myeloma. Leukemia 2018, 32, 752–764. [Google Scholar] [CrossRef]
- Bae, J.; Hideshima, T.; Tai, Y.-T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018, 32, 1932–1947. [Google Scholar] [CrossRef]
- Zammarchi, F.; Havenith, K.; Bertelli, F.; Vijayakrishnan, B.; Chivers, S.; van Berkel, P.H. CD25-targeted antibody-drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors via antitumor immunity. J. Immunother. Cancer. 2020, 8, e000860. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Molecular targets of FoxP3(+) regulatory T cells. Mini-Rev. Med. Chem. 2007, 7, 1136–1143. [Google Scholar] [CrossRef]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M.G. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N.Y. Acad. Sci. 2018, 1417, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Chen, H.M.; Mathis, D.; Benoist, C. FoxP3 scanning mutagenesis reveals functional variegation and mild mutations with atypical autoimmune phenotypes. Proc. Natl. Acad. Sci. USA 2018, 115, E253–E262. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Chen, H.M.; Mathis, D.; Benoist, C. Different molecular complexes that mediate transcriptional induction and repression by Foxp3. Nat. Immunol. 2017, 18, 1238–1248. [Google Scholar] [CrossRef]
- van Loosdregt, J.; Coffer, P.J. Post-translational modification networks regulating FOXP3 function. Trends Immunol. 2014, 35, 368–378. [Google Scholar] [CrossRef]
- Nakahira, K.; Morita, A.; Kim, N.S.; Yanagihara, I. Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS ONE 2013, 8, e77099. [Google Scholar] [CrossRef]
- Deng, G.; Nagai, Y.; Xiao, Y.; Li, Z.; Dai, S.; Ohtani, T.; Banham, A.; Li, B.; Wu, S.L.; Hancock, W.; et al. Pim-2 kinase influences regulatory T cell function and stability by mediating Foxp3 protein N-terminal phosphorylation. J. Biol. Chem. 2015, 290, 20211–20220. [Google Scholar] [CrossRef]
- Li, Z.; Lin, F.; Zhuo, C.; Deng, G.; Chen, Z.; Yin, S.; Gao, Z.M.; Piccioni, M.; Tsun, A.; Cai, S.J.; et al. Pim1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J. Biol. Chem. 2014, 289, 26872–26881. [Google Scholar] [CrossRef]
- Luszczak, S.; Simpson, B.S.; Stopka-Farooqui, U.; Sathyadevan, V.K.; Echeverria, L.M.C.; Kumar, C.; Costa, H.; Haider, A.; Freeman, A.; Jameson, C.; et al. Co-targeting PIM and PI3K/mTOR using multikinase inhibitor AUM302 and a combination of AZD-1208 and BEZ235 in prostate cancer. Sci. Rep. 2020, 10, 14380. [Google Scholar] [CrossRef]
- Tanaka, S.; Pfleger, C.; Lai, J.F.; Roan, F.; Sun, S.C.; Ziegler, S.F. KAP1 regulates regulatory T cell function and proliferation in both FOXP3-dependent and -independent manners. Cell Rep. 2018, 23, 796–807. [Google Scholar] [CrossRef]
- Lozano, T.; Gorraiz, M.; Lasarte-Cia, A.; Ruiz, M.; Rabal, O.; Oyarzabal, J.; Hervas-Stubbs, S.; Llopiz, D.; Sarobe, P.; Prieto, J.; et al. Blockage of FOXP3 transcription factor dimerization and FOXP3/AML1 interaction inhibits T regulatory cell activity: Sequence optimization of a peptide inhibitor. Oncotarget 2017, 8, 71709–71724. [Google Scholar] [CrossRef]
- Liu, X.; Ji, B.; Sun, M.; Wu, W.; Huang, L.; Sun, A.; Zong, Y.; Xia, S.; Shi, L.; Qian, H.; et al. Cell-penetrable mouse Forkhead box protein 3 alleviates experimental arthritis in mice by up-regulating regulatory T cells. Clin. Exp. Immunol. 2015, 181, 87–99. [Google Scholar] [CrossRef]
- Wang, Z.; Qi, Y.; Feng, Y.; Xu, H.; Wan, J.; Zhang, L.; Zhang, J.; Hou, X.; Feng, G.; Shang, E. The N6-methyl-adenosine writer WTAP contributes to the induction of immune tolerance post kidney transplantation by targeting regulatory T cells. Lab. Investig. 2022, 102, 1268–1279. [Google Scholar] [CrossRef]
- Zeng, R.; Peng, B.; Peng, E.M. Downregulated copper homeostasis-related gene FOXO1 as a novel indicator for the prognosis and immune response of breast cancer. J. Immunol. Res. 2022, 2022, 9140461. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yu, W.; Wu, Q.; Lian, J.; Li, F.; Liu, S.; Li, A.; He, Z.; Liu, J.; et al. Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-kappa B signaling. J. Immunother. Cancer 2019, 7, 215. [Google Scholar] [CrossRef]
- Luu, T.T.; Sondergaard, J.N.; Pena-Perez, L.; Kharazi, S.; Krstic, A.; Meinke, S.; Schmied, L.; Frengen, N.; Heshmati, Y.; Kierczak, M.; et al. FOXO1 and FOXO3 Cooperatively Regulate Innate Lymphoid Cell Development. Front. Immunol. 2022, 13, 854312. [Google Scholar] [CrossRef]
- Kesarwani, P.; Kant, S.; Zhao, Y.; Prabhu, A.; Buelow, K.L.; Miller, C.R.; Chinnaiyan, P. Quinolinate promotes macrophage-induced immune tolerance in glioblastoma through the NMDA/PPARγ signaling axis. Nat. Commun. 2023, 14, 1459. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Li, F.; Ping, Y.; Qin, G.; Zhang, C.; Zhang, Y. miR-143 Regulates Memory T cell Differentiation by Reprogramming T cell Metabolism. J. Immunol. 2018, 201, 2165–2175. [Google Scholar] [CrossRef]
- Huang, Q.; Xia, J.; Wang, L.; Wang, X.; Ma, X.; Deng, Q.; Lu, Y.; Kumar, M.; Zhou, Z.Y.; Li, L.; et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Haematol. Oncol. 2018, 11, 58. [Google Scholar] [CrossRef]
- Lou, Q.; Liu, R.; Yang, X.; Li, W.; Huang, L.; Wei, L.; Tan, H.; Xiang, N.; Chan, K.; Chen, J.; et al. miR-448 targets IDO1 and regulates CD8(+) T cell response in human colon cancer. J. Immunother. Cancer 2019, 7, 210. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.W.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.W.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8(+) T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, Y.; Wei, J.; Jiang, L.; Mao, J.; Chang, C.H.; Wu, D. Targeting T cell metabolism for immunotherapy. J. Leukoc. Biol. 2021, 110, 1081–1090. [Google Scholar] [CrossRef]
- Tan, S.Y.; Kelkar, Y.; Hadjipanayis, A.; Shipstone, A.; Wynn, T.A.; Hall, J.P. Metformin and 2-Deoxyglucose Collaboratively Suppress Human CD4(+) T Cell Effector Functions and Activation-Induced Metabolic Reprogramming. J. Immunol. 2020, 205, 957–967. [Google Scholar] [CrossRef]
- Maciver, N.J.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Coloff, J.L.; Rathmell, J.C. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 2008, 84, 949–957. [Google Scholar] [CrossRef]
- Li, W.H.; Xu, M.; Li, Y.; Huang, Z.W.; Zhou, J.; Zhao, Q.Y.; Le, K.H.; Dong, F.; Wan, C.; Yi, P.F. Comprehensive analysis of the association between tumor glycolysis and immune/inflammation function in breast cancer. J. Transl. Med. 2020, 18, 92. [Google Scholar] [CrossRef]
- Dimeloe, S.; Burgener, A.V.; Grahlert, J.; Hess, C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology 2017, 150, 35–44. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; can der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Post-transcriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M. Therapeutic Targeting of Tumor Cells and Tumor Immune Microenvironment Vulnerabilities. Front. Oncol. 2022, 12, 816504. [Google Scholar] [CrossRef]
- Balmer, M.L.; Hess, C. Starving for survival-how catabolic metabolism fuels immune function. Curr. Opin. Immunol. 2017, 46, 8–13. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Thompson, C.B. Regulation of T lymphocyte metabolism. J. Immunol. 2004, 172, 4661–4665. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Gubser, P.M.; Bantug, G.R.; Razik, L.; Fischer, M.; Dimeloe, S.; Hoenger, G.; Durovic, B.; Jauch, A.; Hess, C. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 2013, 14, 1064–1072. [Google Scholar] [CrossRef]
- Garige, M.; Ghosh, S.; Norris, A.; Li, G.Y.; Poncet, S.; Chou, C.K.; Wu, W.W.; Shen, R.F.; Sourbier, C. PD-L1 Mediates IFN gamma-Regulation of Glucose but Not of Tryptophan Metabolism in Clear Cell Renal Cell Carcinoma. Front. Oncol. 2022, 12, 858379. [Google Scholar] [CrossRef]
- Toriyama, K.; Kuwahara, M.; Kondoh, H.; Mikawa, T.; Takemori, N.; Konishi, A.; Yorozuya, T.; Yamada, T.; Soga, T.; Shiraishi, A.; et al. T cell-specific deletion of Pgam1 reveals a critical role for glycolysis in T cell responses. Commun. Biol. 2020, 3, 394. [Google Scholar] [CrossRef]
- Li, G.; Liu, L.; Yin, Z.; Ye, Z.; Shen, N. Glutamine metabolism is essential for the production of IL-17A in gamma-delta T cells and skin inflammation. Tissue Cell 2021, 71, 101569. [Google Scholar] [CrossRef]
- Yu, Q.; Tu, H.; Yin, X.; Peng, C.; Dou, C.; Yang, W.; Wu, W.; Guan, X.; Li, J.; Yan, H.; et al. Targeting glutamine metabolism ameliorates autoimmune hepatitis via inhibiting T cell activation and differentiation. Front. Immunol. 2022, 13, 880262. [Google Scholar] [CrossRef]
- Wu, J.; Li, G.; Li, L.; Li, D.; Dong, Z.; Jiang, P. Asparagine enhances LCK signalling to potentiate CD8(+) T-cell activation and anti-tumour responses. Nat. Cell Biol. 2021, 23, 75–86. [Google Scholar] [CrossRef]
- Hope, H.; Brownlie, R.J.; Fife, M.; Steele, L.; Lorger, M.; Salmond, R.J. Coordination of asparagine uptake and asparagine synthetase expression modulates CD8(+) T cell activation. JCI Insight 2021, 6, e137761. [Google Scholar] [CrossRef]
- Wesch, D.; Kabelitz, D.; Oberg, H.H. Tumor resistance mechanisms and their consequences on gamma delta T cell activation. Immunol. Rev. 2020, 298, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.N.; Yerneni, S.; Sarkar, S.N.; Orr, B.; Muthuswamy, R.; Voyten, J.; Modugno, F.; Jiang, W.J.; Grimm, M.; Bsse, P.H.; et al. Helicase-Driven Activation of NF kappa B-COX2 Pathway Mediates the Immunosuppressive Component of dsRNA-Driven Inflammation in the Human Tumor Microenvironment. Cancer Res. 2018, 78, 4292–4302. [Google Scholar] [CrossRef] [PubMed]
- Casaril, A.M.; Domingues, M.; Bampi, S.R.; Lourenco, D.D.; Smaniotto, T.A.; Segatto, N.; Vieira, B.; Seixas, F.K.; Collares, T.; Lemardao, E.J.; et al. The antioxidant and immunomodulatory compound 3-[(4-chlorophenyl)selanyl]-1-methyl-1H-indole attenuates depression-like behavior and cognitive impairment developed in a mouse model of breast tumor. Brain Behav. Immun. 2020, 84, 229–241. [Google Scholar] [CrossRef]
- Leon-Letelier, R.A.; Sater, A.A.H.; Chen, Y.H.; Park, S.; Wu, R.R.; Irajizad, E.; Dennison, J.B.; Katayama, H.; Vykoukal, J.V.; Hanash, S.; et al. Kynureninase Upregulation Is a Prominent Feature of NFR2-Activated Cancers and Is Associated with Tumor Immunosuppression and Poor Prognosis. Cancers 2023, 15, 834. [Google Scholar] [CrossRef] [PubMed]
- Iachininoto, M.G.; Nuzzolo, E.R.; Bonanno, G.; Mariotti, A.; Procoli, A.; Locatelli, F.; DeCristofaro, R.; Rutella, S. Cyclooxygenase-2 (COX-2) Inhibition Constrains Indoleamine 2,3-Dioxygenase 1 (IDO1) Activity in Acute Myeloid Leukaemia Cells. Molecules 2013, 18, 10132–10145. [Google Scholar] [CrossRef]
- Erkes, D.A.; Field, C.O.; Capparelli, C.; Tiago, M.; Purwin, T.J.; Chervoneva, I.; Berger, A.C.; Hartsough, E.J.; Villanueva, J.; Aplin, A.E. The next-generation BET inhibitor, PLX51107, delays melanoma growth in a CD8-mediated manner. Pigment Cell Melanoma Res. 2019, 32, 687–696. [Google Scholar] [CrossRef]
- Bassal, N.K.; Hughes, B.P.; Costabile, M. Prostaglandin D-2 is a novel repressor of IFN gamma induced indoleamine-2,3-dioxygenase via the DP1 receptor and cAMP pathway Prostaglandins Leukot. Essent. Fat. Acids 2016, 110, 48–54. [Google Scholar] [CrossRef]
- Bassal, N.K.; Hughes, B.P.; Costabile, M. Arachidonic acid and its COX1/2 metabolites inhibit interferon-gamma mediated induction of indoleamine-2,3 dioxygenase in THP-1 cells and Human monocytes. Prostaglandins Leukot. Essent. Fat. Acids 2012, 87, 119–126. [Google Scholar] [CrossRef]
- Ochs, K.; Ott, M.; Rauschenbach, K.J.; Deumelandt, K.; Sahm, F.; Opitz, C.A.; von Deimling, A.; Wick, W.; Platten, M. Tryptophan-2,3-dioxygenase is regulated by prostaglandin E2 in malignant glioma via a positive signaling loop involving prostaglandin E receptor-4. J. Neurochem. 2016, 136, 1142–1154. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef]
- Costabile, M.; Bassal, N.K.; Gerber, J.P.; Hughes, B.P. Inhibition of indoleamine 2,3-dioxygenase activity by fatty acids and prostaglandins: A structure function analysis. Prostaglandins Leukot. Essent. Fat. Acids 2017, 122, 7–15. [Google Scholar] [CrossRef]
- Muthuswamy, R.; Okada, N.J.; Jenkins, F.J.; McGuire, K.; McAuliffe, P.F.; Zeh, H.J.; Bartlett, D.L.; Wallace, C.; Watkins, S.; Henning, J.D.; et al. Epinephrine promotes COX-2-dependent immune suppression in myeloid cells and cancer tissues. Brain Behav. Immun. 2017, 62, 78–86. [Google Scholar] [CrossRef]
- Basu, G.; Tinder, T.L.; Bradley, J.M.; Tu, T.; Hattrup, C.L.; Pockaj, B.A.; Mukherjee, P. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: Role of IDO. J. Immunol. 2006, 177, 2391–2402. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, H.K.; Lee, K.J.; Jung, J.Y.; Hur, G.Y.; Jung, K.H.; Kim, J.H.; Shin, C.; Shim, J.J.; In, K.H.; et al. The Immune Tolerance of Cancer is Mediated by IDO That is Inhibited by COX-2 Inhibitors Through Regulatory T cells. J. Immunother. 2009, 32, 22–28. [Google Scholar] [CrossRef]
- Cesario, A.; Rocca, B.; Rutella, S. The Interplay between Indoleamine 2,3-Dioxygenase 1 (IDO1) andCyclooxygenase (COX)-2 In Chronic Inflammation and Cancer. Current Med. Chem. 2011, 18, 2263–2271. [Google Scholar] [CrossRef]
- Jeong, Y.I.; Jung, I.D.; Lee, J.S.; Lee, C.M.; Lee, J.D.; Park, Y.M. (-)-Epigallocatechin gallate suppresses indoleamine 2,3-dioxygenase expression in murine dendritic cells: Evidences for the COX-2 and STAT1 as potential targets. Biochem. Biophys. Res. Commun. 2007, 354, 1004–1009. [Google Scholar] [CrossRef]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 Induction by IFN gamma and TNF alpha Self-Limits Type-1 Immunity in the Human Tumor Microenvironment. Cancer Immunol. Res. 2016, 4, 303–311. [Google Scholar] [CrossRef]
- Lemos, H.; Ou, R.; McCardle, C.; Lin, Y.J.; Calver, J.; Minett, J.; Chadli, A.; Huang, L.; Mellor, A.L. Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J. Immunother. Cancer 2020, 8, e001182. [Google Scholar] [CrossRef]
- Ramsay, G.; Cantrell, D. Environmental and metabolic sensors that control T cell biology. Front. Immunol. 2015, 6, 99. [Google Scholar] [CrossRef]
- Xu, Y.X.; Cao, C.; Zhu, Z.; Wang, Y.; Tan, Y.; Xu, X. Novel Hypoxia-Associated Gene Signature Depicts Tumor Immune Microenvironment and Predicts Prognosis of Colon Cancer Patients 2022. Front. Genet. 2022, 13, 901734. [Google Scholar] [CrossRef]
- Huang, B.N.; Phelan, J.D.; Preite, S.; Gomez-Rodriguez, J.; Johansen, K.H.; Shibata, H.; Shaffer, A.L.; Xu, Q.; Jeffrey, B.; Kirby, M.; et al. In vivo CRISPR screens reveal a HIF-1 alpha-mTOR-network regulates T follicular helper versus Th1 cells. Nat. Commun. 2022, 13, 805. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.R.; Sadik, A.; Tykocinski, L.O.; Dietze, J.; Poschet, G.; Heiland, I.; Opitz, C.A. Hypoxia Inducible Factor 1-alpha. Front. Immunol. 2019, 10, 2762. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Wirthgen, E.; Leonard, A.K.; Scharf, C.; Domanska, G. The immunomodulatory 1-methyltryptophan drives tryptophan catabolism toward the kynurenic acid branch. Front. Immunol. 2020, 11, 313. [Google Scholar] [CrossRef]
- Tang, K.; Wu, Y.H.; Song, Y.; Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for caner immunotherapy. J. Haematol. Oncol. 2021, 14, 68. [Google Scholar] [CrossRef]
- Pires, A.S.; Sundaram, G.; Heng, B.; Krishnamurthy, S.; Brew, B.J.; Guillemin, G.J. Recent advances in clinical trials targeting the kynurenine pathway. Pharmacol. Ther. 2022, 236, 108055. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Majjigapu, S.R.; Caldelari, D.; Dilek, N.; Reichenbach, P.; Ascencao, K.; Irving, M.; Coukos, J.; Vogel, P.; Zoete, V.; et al. 1,2,3-Triazoles as inhibitors of indoleamine 2,3-dioxygenase 2 (IDO2). Bioorg. Med. Chem. Lett. 2016, 26, 4330–4333. [Google Scholar] [CrossRef]
- He, G.C.; Wan, S.; Wu, Y.Z.; Chu, Z.X.; Shen, H.; Zhang, S.; Chen, L.Y.; Bao, Z.J.; Gu, S.H.; Huang, J.Z.; et al. Discovery of the First Selective IDO2 Inhibitor as Novel Immunotherapeutic Avenues for Rheumatoid Arthritis. J. Med. Chem. 2022, 65, 14348–14365. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Z.L.; Zhang, J.F.; Ren, C.Y.; Wang, Y.X. Dual-target inhibitors of indoleamine 2, 3 dioxygenase 1 (Ido1): A promising direction in cancer immunotherapy. Eur. J. Med. Chem. 2022, 238, 114524. [Google Scholar] [CrossRef]
- Yamasuge, W.; Yamamoto, Y.; Fujigaki, H.; Hoshi, M.; Nakamoto, K.; Kunisawa, K.; Mouri, A.; Nabeshima, T.; Saito, K. Indoleamine 2,3-dioxygenase 2 depletion suppresses tumor growth in a mouse model of Lewis lung carcinoma. Cancer Sci. 2019, 110, 3061–3067. [Google Scholar] [CrossRef]
- Rover, S.; Cesura, A.M.; Huguenin, P.; Kettler, R.; Szente, A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J. Med. Chem. 1997, 40, 4378–4385. [Google Scholar] [CrossRef]
- Mole, D.J.; Webster, S.P.; Uings, I.; Zheng, X.Z.; Binnie, M.; Wilson, K.; Hutchinson, J.P.; Mirguet, O.; Walker, A.; Beaufils, B.; et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 2016, 22, 202–209. [Google Scholar] [CrossRef]
- Walker, A.L.; Ancellin, N.; Beaufils, B.; Bergeal, M.; Binnie, M.; Bouillot, A.; Clapham, D.; Denis, A.; Haslan, C.P.; Holmes, D.S.; et al. Development of a Series of Kynurenine 3-Monooxygenase Inhibitors Leading to a Clinical Candidate for the Treatment of Acute Pancreatitis. J. Med. Chem. 2017, 60, 3383–3404. [Google Scholar] [CrossRef]
- Jha, G.G.; Gupta, S.; Tagawa, S.T.; Koopmeiners, J.S.; Vivek, S.; Dudek, A.Z.; Miller, J.S. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod, or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35, S3066. [Google Scholar] [CrossRef]
- Bahary, N.; Wang-Gillam, A.; Haraldsdottir, S.; Somer, B.G.; Lee, J.S.; O’Rourke, A.M.; Nayak-Kapoor, A.; Beatty, G.L.; Liu, M.; Delman, D.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of patients with metastatic pancreas cancer. J. Clin. Oncol. 2018, 36, S4015. [Google Scholar] [CrossRef]
- Mariotti, V.; Han, H.; Ismail-Khan, R.; Tang, S.J.; Dillon, P.; Montero, A.J.; Poklepovic, A.; Melin, S.; Ibrahim, N.K.; Kennedy, E.; et al. Effect of Taxane Chemotherapy With or Without Indoximod in Metastatic Breast Cancer A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 61–69. [Google Scholar] [CrossRef]
- Redman, J.M.; Steinbrg, S.M.; Gulley, J.L. Quick efficacy seeking trial (QuEST1): A novel combination immunotherapy study designed for rapid clinical signal assessment metastatic castration-resistant prostate cancer. J. Immunother. Cancer 2018, 6, 91. [Google Scholar] [CrossRef]
- von Amsberg, G.; Alsdorf, W.; Karagiannis, P.; Coym, A.; Kaune, M.; Werner, S.; Graefen, M.; Bokemeyer, C.; Merkens, L.; Dyshlovoy, S.A. Immunotherapy in Advanced Prostate Cancer—Light at the End of the Tunnel? Int. J. Mol. Sci. 2022, 23, 2569. [Google Scholar] [CrossRef]
- Smethurst, D. A pharmacologic perspective on newly emerging T-cell manipulation technologies. Br. J. Clin. Pharmacol. 2013, 76, 173–187. [Google Scholar] [CrossRef]
- Fattori, S.; Roux, H.; Connen, E.; Robert, L.; Gorvel, L.; Le Roy, A.; Houacine, J.; Foussat, A.; Chretien, A.S.; Olive, D. Therapeutic targeting of tumor-infiltrating regulatory T cells in breast cancer. Cancer Res. 2022, 82, 3868–3879. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stone, T.W.; Williams, R.O. Interactions of IDO and the Kynurenine Pathway with Cell Transduction Systems and Metabolism at the Inflammation–Cancer Interface. Cancers 2023, 15, 2895. https://doi.org/10.3390/cancers15112895
Stone TW, Williams RO. Interactions of IDO and the Kynurenine Pathway with Cell Transduction Systems and Metabolism at the Inflammation–Cancer Interface. Cancers. 2023; 15(11):2895. https://doi.org/10.3390/cancers15112895
Chicago/Turabian StyleStone, Trevor W., and Richard O. Williams. 2023. "Interactions of IDO and the Kynurenine Pathway with Cell Transduction Systems and Metabolism at the Inflammation–Cancer Interface" Cancers 15, no. 11: 2895. https://doi.org/10.3390/cancers15112895
APA StyleStone, T. W., & Williams, R. O. (2023). Interactions of IDO and the Kynurenine Pathway with Cell Transduction Systems and Metabolism at the Inflammation–Cancer Interface. Cancers, 15(11), 2895. https://doi.org/10.3390/cancers15112895