
Deciphering the Role of the Anaphylatoxin C3a: A Key Function in Modulating the Tumor Microenvironment
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Transfection
2.3. Cell Culture and Stimulation
2.4. Cytotoxicity Assay
2.5. qRT-PCR (Sybergreen) Analyses
2.6. Immunofluorescence Staining and Microscopy
2.7. Immunohistochemical Staining
2.8. Statistical Analysis
3. Results
3.1. Cell Viability
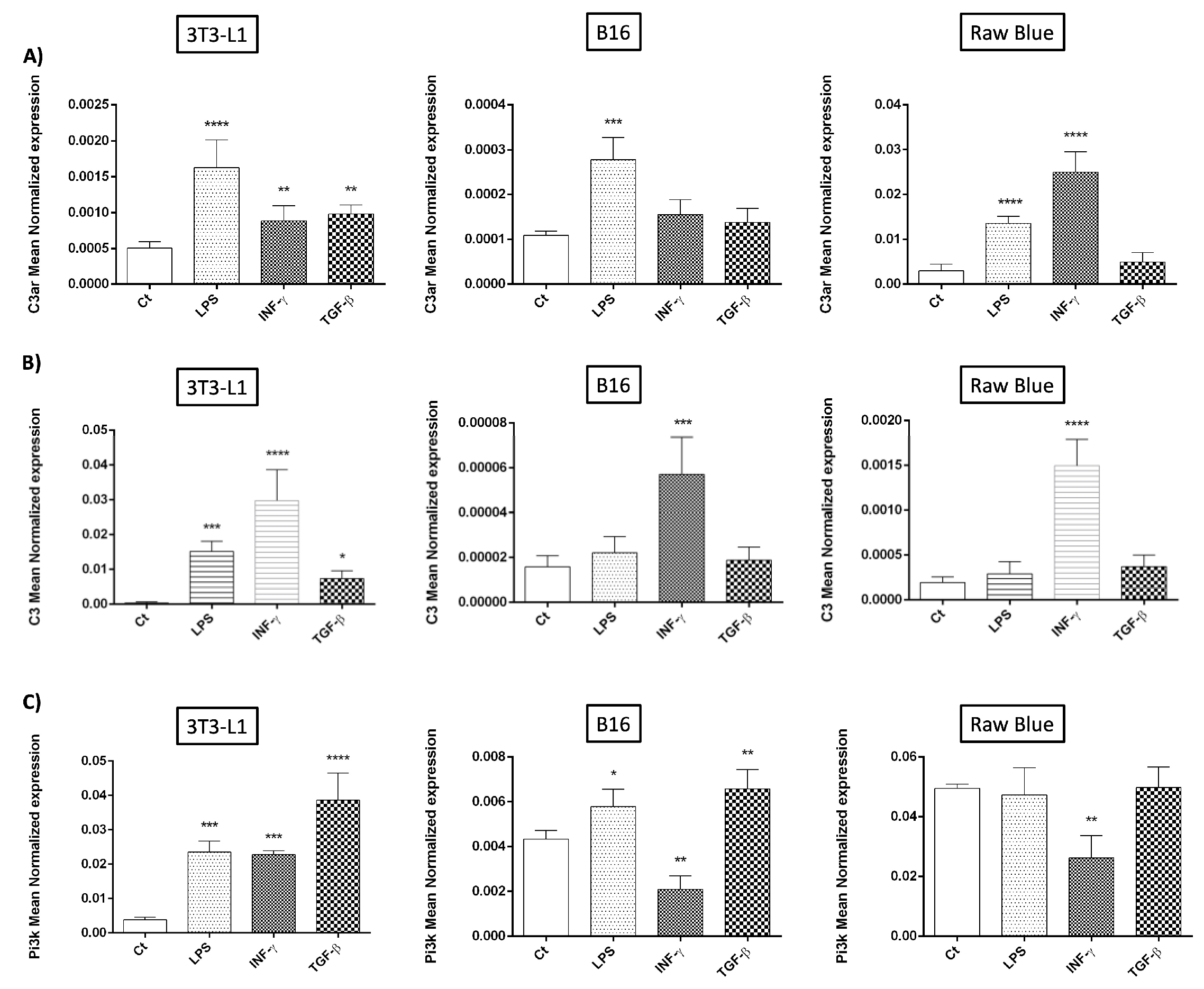
3.2. C3/C3aR Expression
3.3. Recombinant C3a in CHO Expression
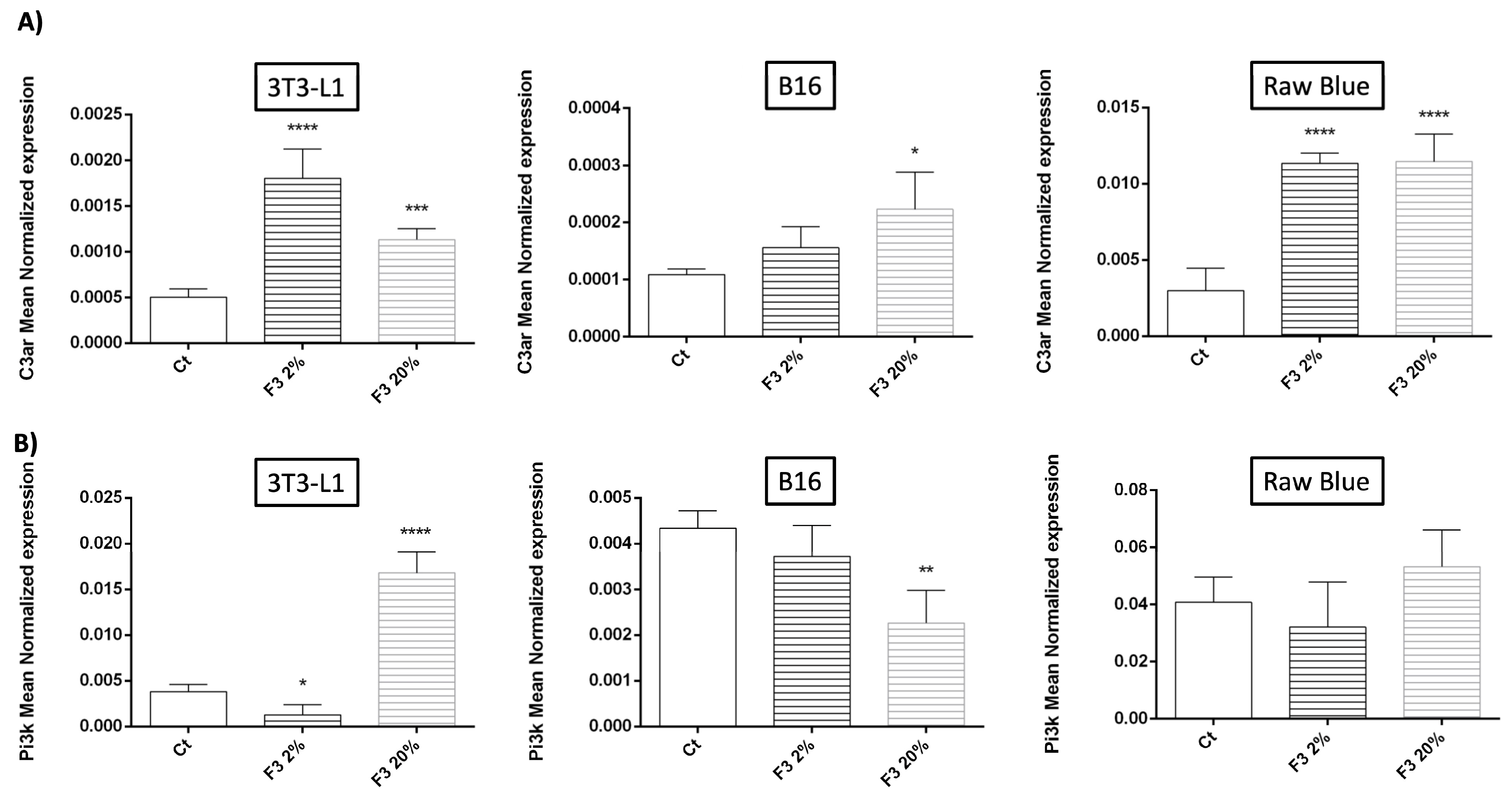
3.4. Modulation of C3aR and PI3K Expression by Recombinant C3a
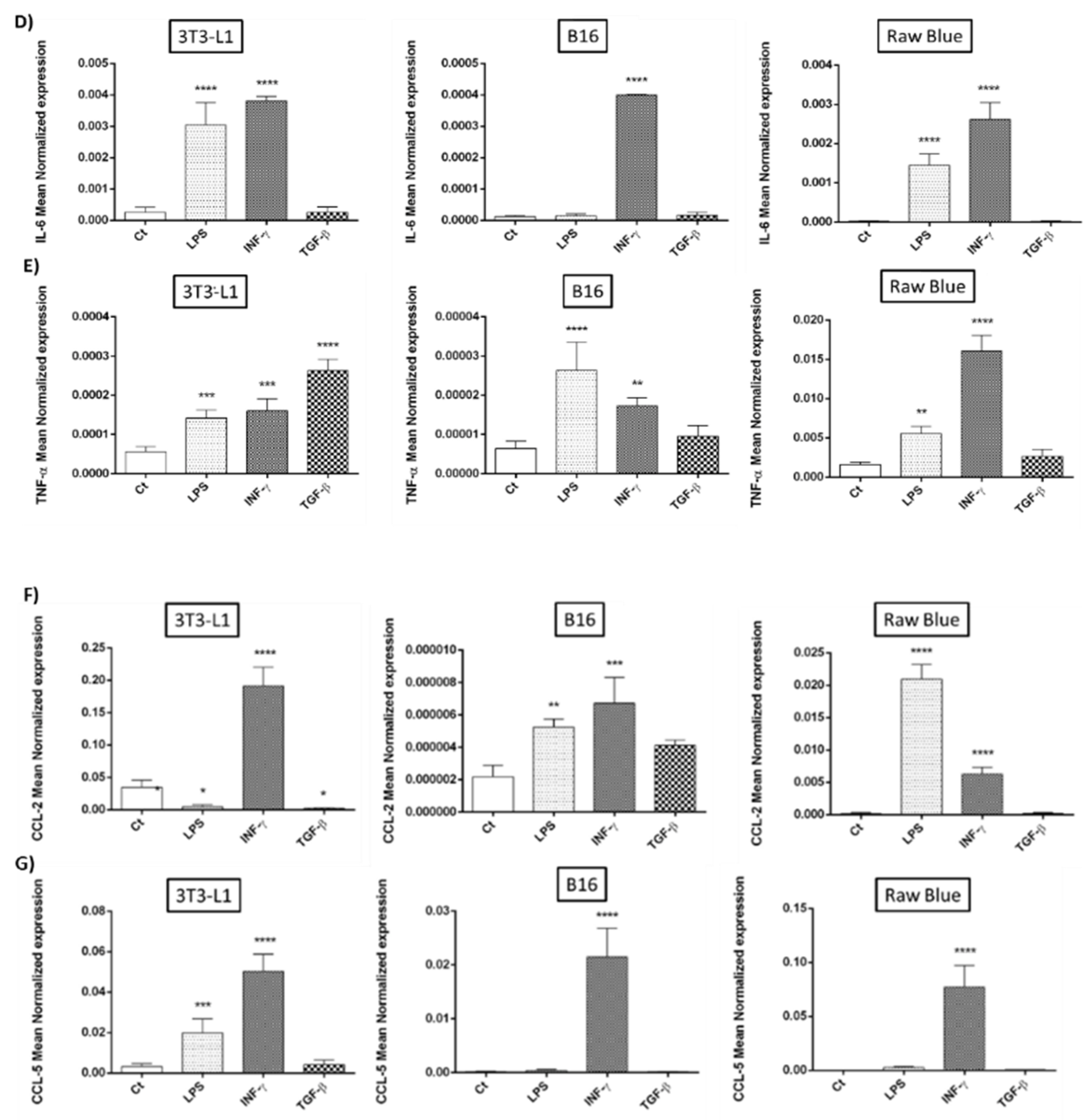
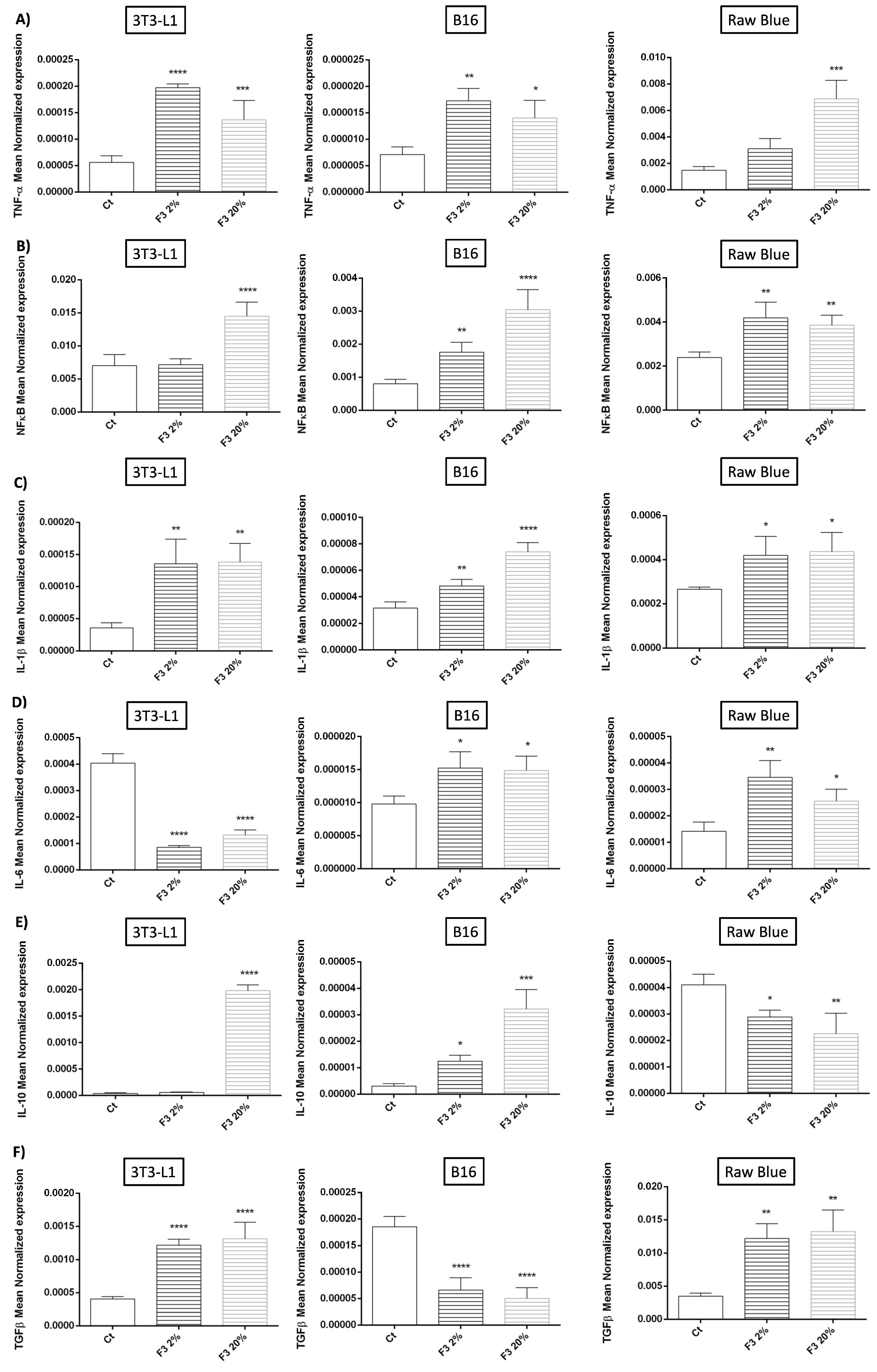
3.5. Modulation of Cytokine Expression by Recombinant C3a
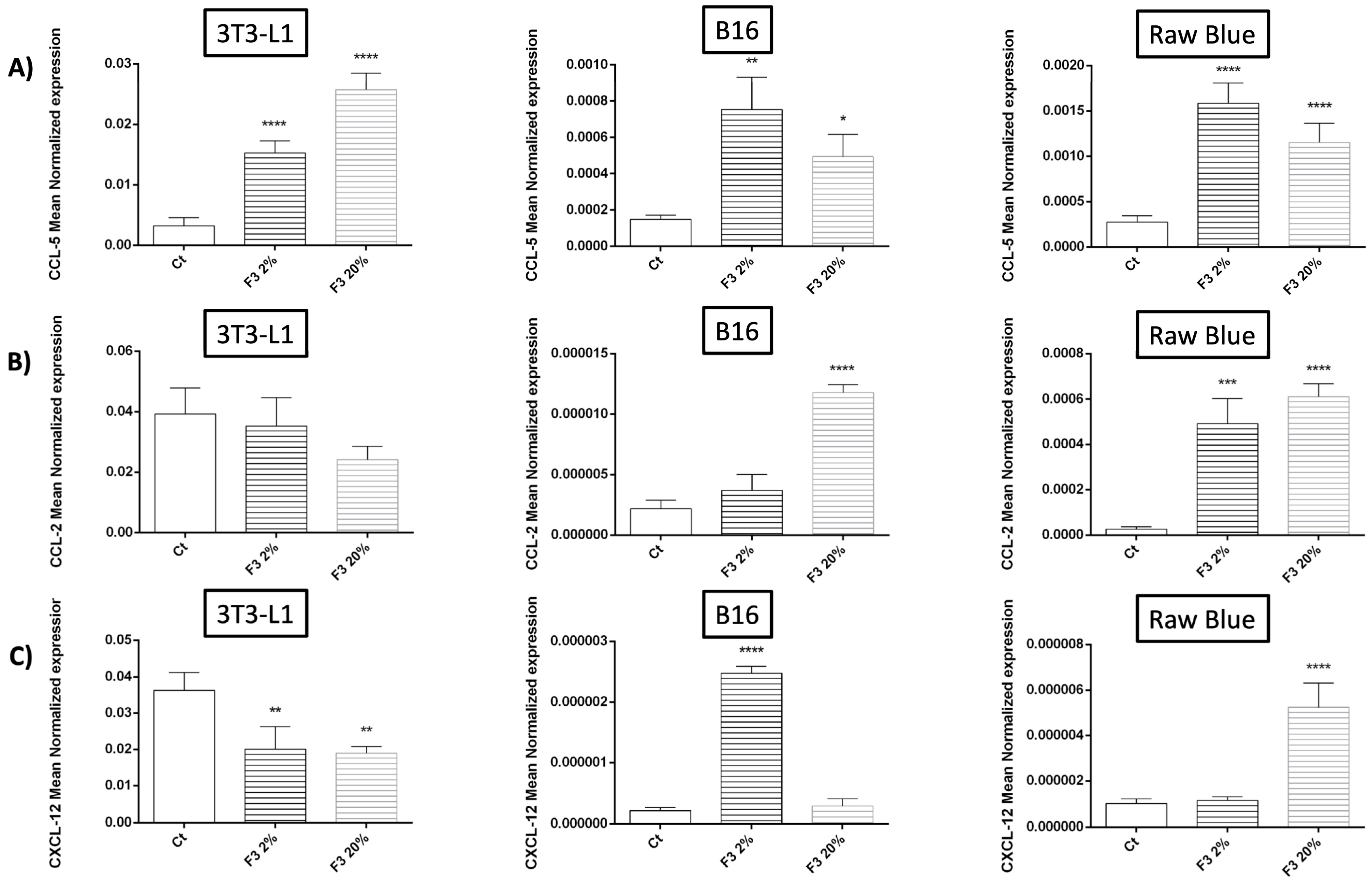
3.6. Modulation of Chemokine Expression by Recombinant C3a
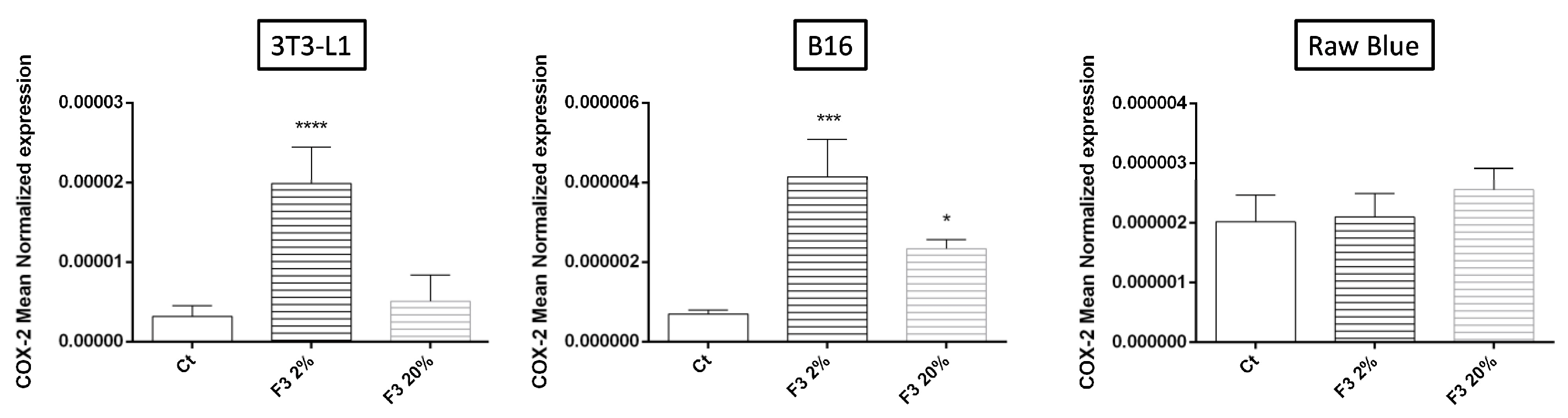
3.7. Regulation of COX-2 Expression Involved in Prostaglandin Biosynthesis by Recombinant C3a
3.8. Modulation of Antioxidant Defense System by Recombinant C3a
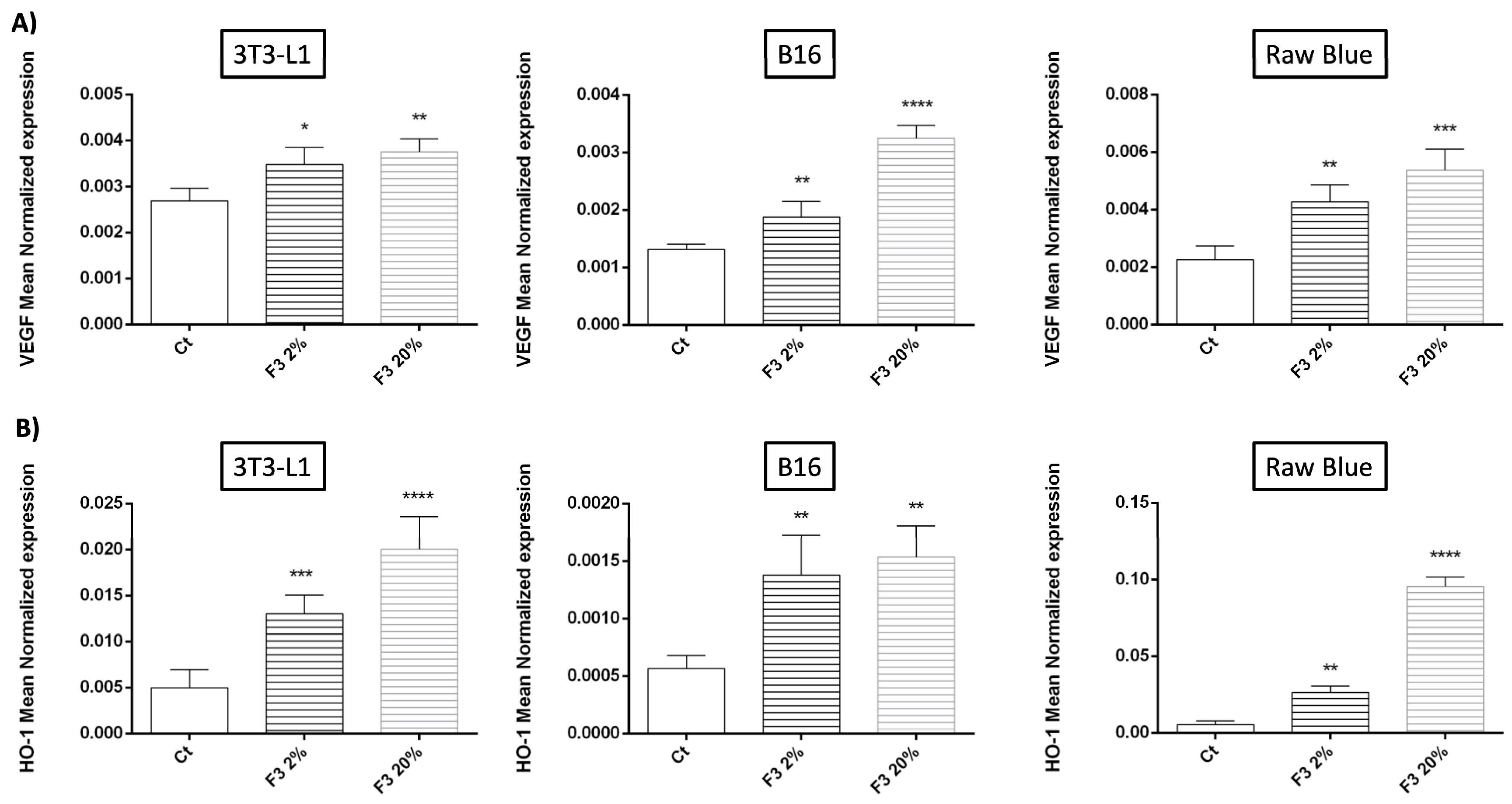
3.9. Pro-Angiogenic Role of C3a
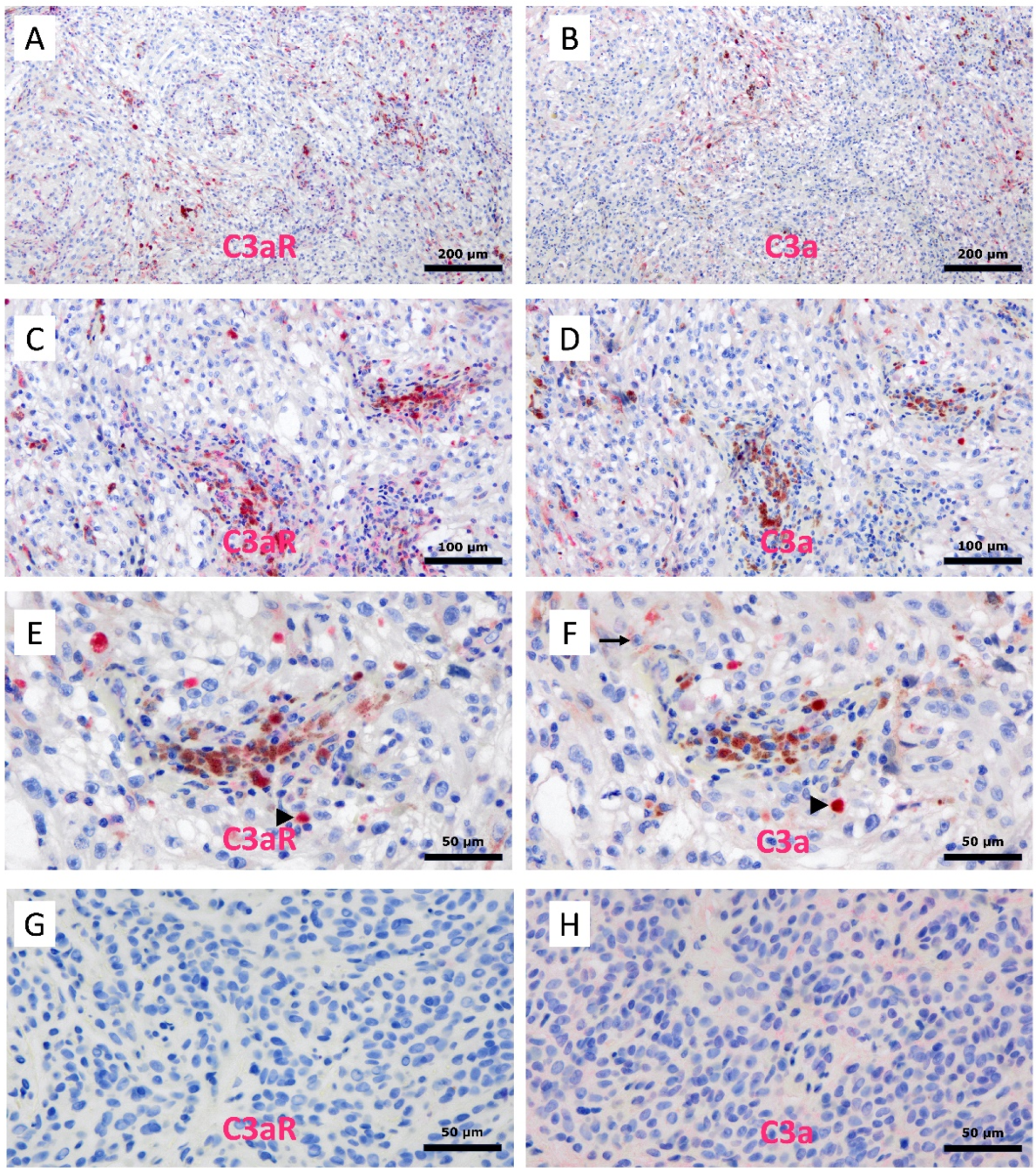
3.10. Expression of C3a and C3aR in Human Melanoma
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walport, M.J. Advances in immunology: Complement (First of two parts). N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P. Complement: A unique innate immune sensor for danger signals. Mol. Immunol. 2004, 41, 1089–1098. [Google Scholar] [CrossRef]
- Heeger, P.S.; Kemper, C. Novel roles of complement in T effector cell regulation. Immunobiology 2012, 217, 216–224. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef]
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Jensenius, J.C.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Structure and activation of C1, the complex initiating the classical pathway of the complement cascade. Proc. Natl. Acad. Sci. USA 2017, 114, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Rus, H.; Cudrici, C.; Niculescu, F. The role of the complement system in innate immunity. Immunol. Res. 2005, 33, 103–112. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Bouwens van der Vlis, T.a.M.; Kros, J.M.; Mustafa, D.a.M.; van Wijck, R.T.A.; Ackermans, L.; van Hagen, P.M.; van der Spek, P.J. The complement system in glioblastoma multiforme. Acta Neuropathol. Commun. 2018, 6, 91. [Google Scholar] [CrossRef]
- Surace, L.; Lysenko, V.; Fontana, A.O.; Cecconi, V.; Janssen, H.; Bicvic, A.; Okoniewski, M.; Pruschy, M.; Dummer, R.; Neefjes, J.; et al. Complement Is a Central Mediator of Radiotherapy-Induced Tumor-Specific Immunity and Clinical Response. Immunity 2015, 42, 767–777. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Cancer and complement. Nat. Biotechnol. 2008, 26, 1348–1349. [Google Scholar] [CrossRef] [PubMed]
- Gelderman, K.A.; Tomlinson, S.; Ross, G.D.; Gorter, A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004, 25, 158–164. [Google Scholar] [CrossRef]
- Zhao, W.-P.; Zhu, B.; Duan, Y.-Z.; Chen, Z.-T. Neutralization of complement regulatory proteins CD55 and CD59 augments therapeutic effect of herceptin against lung carcinoma cells. Oncol. Rep. 2009, 21, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin. Cell Dev. Biol. 2019, 85, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The role of the anaphylatoxins in health and disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef] [PubMed]
- Hugli, T.E. Structure and function of C3a anaphylatoxin. Curr. Top. Microbiol. Immunol. 1990, 153, 181–208. [Google Scholar] [CrossRef]
- Gerard, N.P.; Gerard, C. Complement in allergy and asthma. Curr. Opin. Immunol. 2002, 14, 705–708. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Woodruff, T.M. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef]
- Kildsgaard, J.; Hollmann, T.J.; Matthews, K.W.; Bian, K.; Murad, F.; Wetsel, R.A. Cutting edge: Targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J. Immunol. 2000, 165, 5406–5409. [Google Scholar] [CrossRef]
- Maher, S.G.; McDowell, D.T.; Collins, B.C.; Muldoon, C.; Gallagher, W.M.; Reynolds, J.V. Serum proteomic profiling reveals that pretreatment complement protein levels are predictive of esophageal cancer patient response to neoadjuvant chemoradiation. Ann. Surg. 2011, 254, 809–816, discussion 816–817. [Google Scholar] [CrossRef]
- Ah-Pine, F.; Malaterre-Septembre, A.; Bedoui, Y.; Khettab, M.; Neal, J.W.; Freppel, S.; Gasque, P. Complement Activation and Up-Regulated Expression of Anaphylatoxin C3a/C3aR in Glioblastoma: Deciphering the Links with TGF-β and VEGF. Cancers 2023, 15, 2647. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Zha, H.; Long, H.; Wang, X.; Yang, F.; Gao, J.; Hu, C.; Zhou, L.; Guo, B.; Zhu, B. C3a-C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J. Exp. Clin. Cancer Res. 2020, 39, 11. [Google Scholar] [CrossRef] [PubMed]
- Nataf, S.; Stahel, P.F.; Davoust, N.; Barnum, S.R. Complement anaphylatoxin receptors on neurons: New tricks for old receptors? Trends Neurosci. 1999, 22, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P.; Singhrao, S.K.; Neal, J.W.; Wang, P.; Sayah, S.; Fontaine, M.; Morgan, B.P. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: Analysis in multiple sclerosis and bacterial meningitis. J. Immunol. 1998, 160, 3543–3554. [Google Scholar] [CrossRef]
- Strainic, M.G.; Shevach, E.M.; An, F.; Lin, F.; Medof, M.E. Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat. Immunol. 2013, 14, 162–171. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Discipio, R.G.; Zhao, M.; Khaldoyanidi, S.K. C3a and C5a are chemotactic factors for human mesenchymal stem cells, which cause prolonged ERK1/2 phosphorylation. J. Immunol. 2009, 182, 3827–3836. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, S.-N.; Liu, Q.; Yu, Y.-Y.; Guo, J.; Wang, K.; Xing, B.-C.; Zheng, Q.-F.; Campa, M.J.; Patz, E.F.; et al. Autocrine Complement Inhibits IL10-Dependent T-Cell Mediated Antitumor Immunity to Promote Tumor Progression. Cancer Discov. 2016, 6, 1022–1035. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, S.; Xue, M.; Jing, Y.; Liu, X.; Cai, D.; Zhao, Y.; Bian, Y.; Zhang, Z.; Zhang, L. Aristolochic acid I promotes the invasion and migration of hepatocellular carcinoma cells by activating the C3a/C3aR complement system. Toxicol. Lett. 2020, 378, 51–60. [Google Scholar] [CrossRef]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X.; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Consonni, F.M.; Bleve, A.; Totaro, M.G.; Storto, M.; Kunderfranco, P.; Termanini, A.; Pasqualini, F.; Alì, C.; Pandolfo, C.; Sgambelluri, F.; et al. Heme catabolism by tumor-associated macrophages controls metastasis formation. Nat. Immunol. 2021, 22, 595–606. [Google Scholar] [CrossRef]
- Magrini, E.; Di Marco, S.; Mapelli, S.N.; Perucchini, C.; Pasqualini, F.; Donato, A.; de la Luz Guevara Lopez, M.; Carriero, R.; Ponzetta, A.; Colombo, P.; et al. Complement activation promoted by the lectin pathway mediates C3aR-dependent sarcoma progression and immunosuppression. Nat. Cancer 2021, 2, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.-D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef]
- Sierra-Filardi, E.; Vega, M.A.; Sánchez-Mateos, P.; Corbí, A.L.; Puig-Kröger, A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; He, Y.-W. The Complement Receptors C3aR and C5aR Are a New Class of Immune Checkpoint Receptor in Cancer Immunotherapy. Front. Immunol. 2019, 10, 1574. [Google Scholar] [CrossRef]
- Zha, H.; Wang, X.; Zhu, Y.; Chen, D.; Han, X.; Yang, F.; Gao, J.; Hu, C.; Shu, C.; Feng, Y.; et al. Intracellular Activation of Complement C3 Leads to PD-L1 Antibody Treatment Resistance by Modulating Tumor-Associated Macrophages. Cancer Immunol. Res. 2019, 7, 193–207. [Google Scholar] [CrossRef]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chung, K.-J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.A.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Sequence | Reverse Sequence | Detection |
|---|---|---|---|
| GAPDH | CGACTTCAACAGCAACTCCCACTCTTCC | TGGGTGGTCCAGGGTTTCTTACTCCTT | Sybergreen |
| C3aR | GATTTGTTGGTGGCTCGCAG | GAAACAGAGGCCGTGAGTGT | Sybergreen |
| VEGF | CTCCACCATGCCAAGTGGTC | GTCCACCAGGGTCTCAATCG | Sybergreen |
| NFκB-p50 | CTCTGCATCAGTGACGGTAAAC | TTGTTGTTCTTCAGCCGTGC | Sybergreen |
| TNF-α | GATCGGTCCCCAAAGGGATG | CCACTTGGTGGTTTGTGAGTG | Sybergreen |
| CCL-2 | CTTCTGGGCCTGCTGTTCAC | CTTGAGCTTGGTGACAAAAACTAC | Sybergreen |
| CCL-5 | TGCCCTCACCATCATCCTCA | TTCCTTCGAGTGACAAACACGA | Sybergreen |
| IL-6 | TGATGGATGCTACCAAACTGGA | TGTGACTCCAGCTTATCTCTTGG | Sybergreen |
| IL-10 | GTAGAAGTGATGCCCCAGGC | CACCTTGGTCTTGGAGCTTATT | Sybergreen |
| HO-1 | CACAGGGTGACAGAAGAGGC | CTGCAGGGGCAGTATCTTG | Sybergreen |
| TGFβ-1 | CTGCTGACCCCCACTGATAC | GGGGCTGATCCCGTTGATT | Sybergreen |
| COX-2 | GACACGACTTCGGAGGAGAG | AGACTTTGTCAGAAGTTCTTTTTGT | Sybergreen |
| SOD-1 | CATGGCGATGAAAGCGGTG | GCACTGGTACAGCCTTGTGTA | Sybergreen |
| SOD-2 | CACCGAGGAGAAGTACCACG | CTCCAGCAACTCTCCTTTGGG | Sybergreen |
| Nrf2 (Nfe2l2) | CCAGACAGACACCAGTGGAT | ATATCCAGGGCAAGCGACTCA | Sybergreen |
| Catalase | GTGCATGCATGACAACCAGG | GTGCATGCATGACAACCAGG | Sybergreen |
| PI3K (PRKAA1) | ACCATGGAGGAGAACCCTTATG | ACGGACAGTGCTCCTCCTTA | Sybergreen |
| CXCL-12 | TGCTATGCTGCCTGCTCTTAC | CACAACACTTCCGAAGACCCT | Sybergreen |
| m-IL-10sp m C3a | GTCAAAATTTGCAACTATGTGGGG | GTTTGCAAAGCCAACCACCA | Sybergreen |
| IL1-β | TGCCACCTTTTGACAGTGATG | TGATGTGCTGCTGCGAGATT | Sybergreen |
| C3 | GACAACAACCTACTGCCCGT | TTCCACTGCCCGATGTTGAC | Sybergreen |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanna, J.; Ah-Pine, F.; Boina, C.; Bedoui, Y.; Gasque, P.; Septembre-Malaterre, A. Deciphering the Role of the Anaphylatoxin C3a: A Key Function in Modulating the Tumor Microenvironment. Cancers 2023, 15, 2986. https://doi.org/10.3390/cancers15112986
Hanna J, Ah-Pine F, Boina C, Bedoui Y, Gasque P, Septembre-Malaterre A. Deciphering the Role of the Anaphylatoxin C3a: A Key Function in Modulating the Tumor Microenvironment. Cancers. 2023; 15(11):2986. https://doi.org/10.3390/cancers15112986
Chicago/Turabian StyleHanna, Jolimar, Franck Ah-Pine, Chailas Boina, Yosra Bedoui, Philippe Gasque, and Axelle Septembre-Malaterre. 2023. "Deciphering the Role of the Anaphylatoxin C3a: A Key Function in Modulating the Tumor Microenvironment" Cancers 15, no. 11: 2986. https://doi.org/10.3390/cancers15112986
APA StyleHanna, J., Ah-Pine, F., Boina, C., Bedoui, Y., Gasque, P., & Septembre-Malaterre, A. (2023). Deciphering the Role of the Anaphylatoxin C3a: A Key Function in Modulating the Tumor Microenvironment. Cancers, 15(11), 2986. https://doi.org/10.3390/cancers15112986