
Decursinol Angelate Inhibits Glutamate Dehydrogenase 1 Activity and Induces Intrinsic Apoptosis in MDR-CRC Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Antibodies and Reagents
2.2. Establishment of MultiDrug-Resistant HCT-116 Colorectal Cancer Cell Lines
2.3. Cell Culture and In Vitro Assays
2.4. MTT Assay and Morphological Assessment
2.5. Lactate Dehydrogenase Assay
2.6. ATPase Activity Assay
2.7. Intracellular Metabolite Measurements
2.8. Clonogenic Assay
2.9. Determination of Morphological Changes and Apoptosis by Fluorescence Staining
2.10. Immunofluorescence Staining
2.11. Flow Cytometry Analysis
2.12. Isolation of Proteins
2.13. Western Blotting
2.14. siRNA Transfection
2.15. Statistical Analysis
3. Results
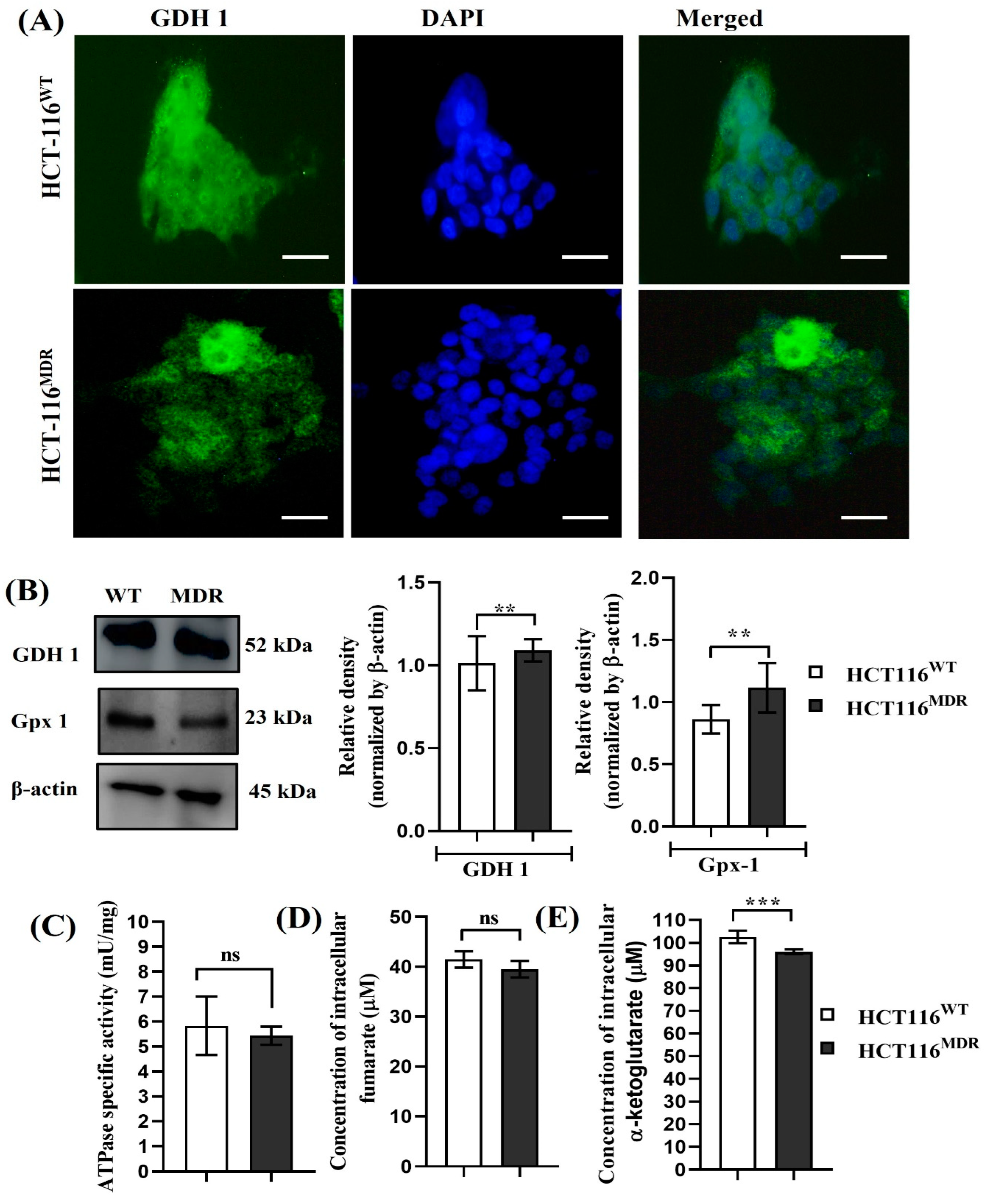
3.1. Phenotypic and Molecular Differences between Parental HCT-116WT and HCT-116MDR Cells
3.2. The Expression of Glutamate Dehydrogenase 1 in Parental HCT-116WT and HCT-116MDR CRC Cells
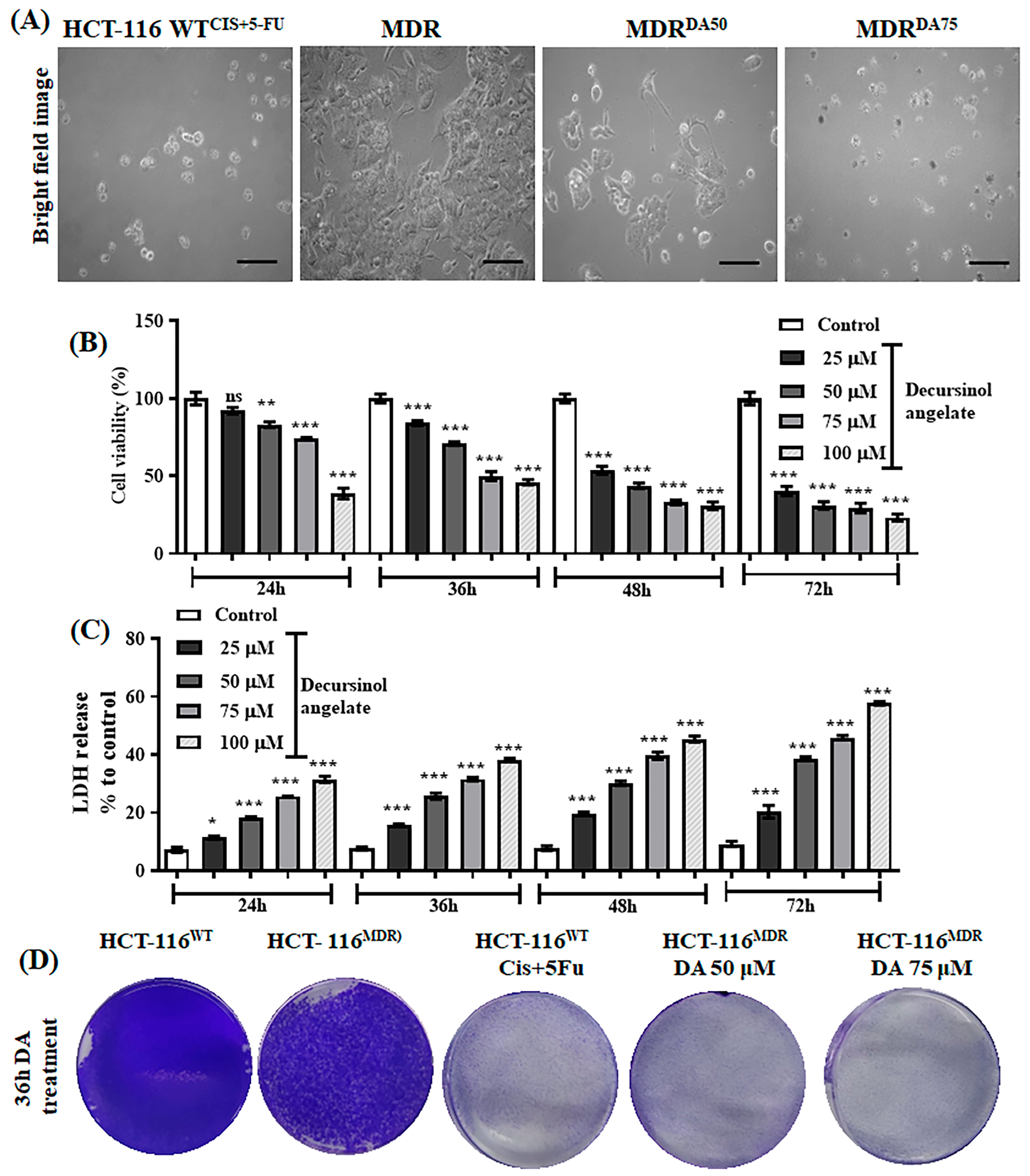
3.3. The Effect of Decursinol Angelate-Induced Cytotoxicity in HCT-116MDR CRC Cells
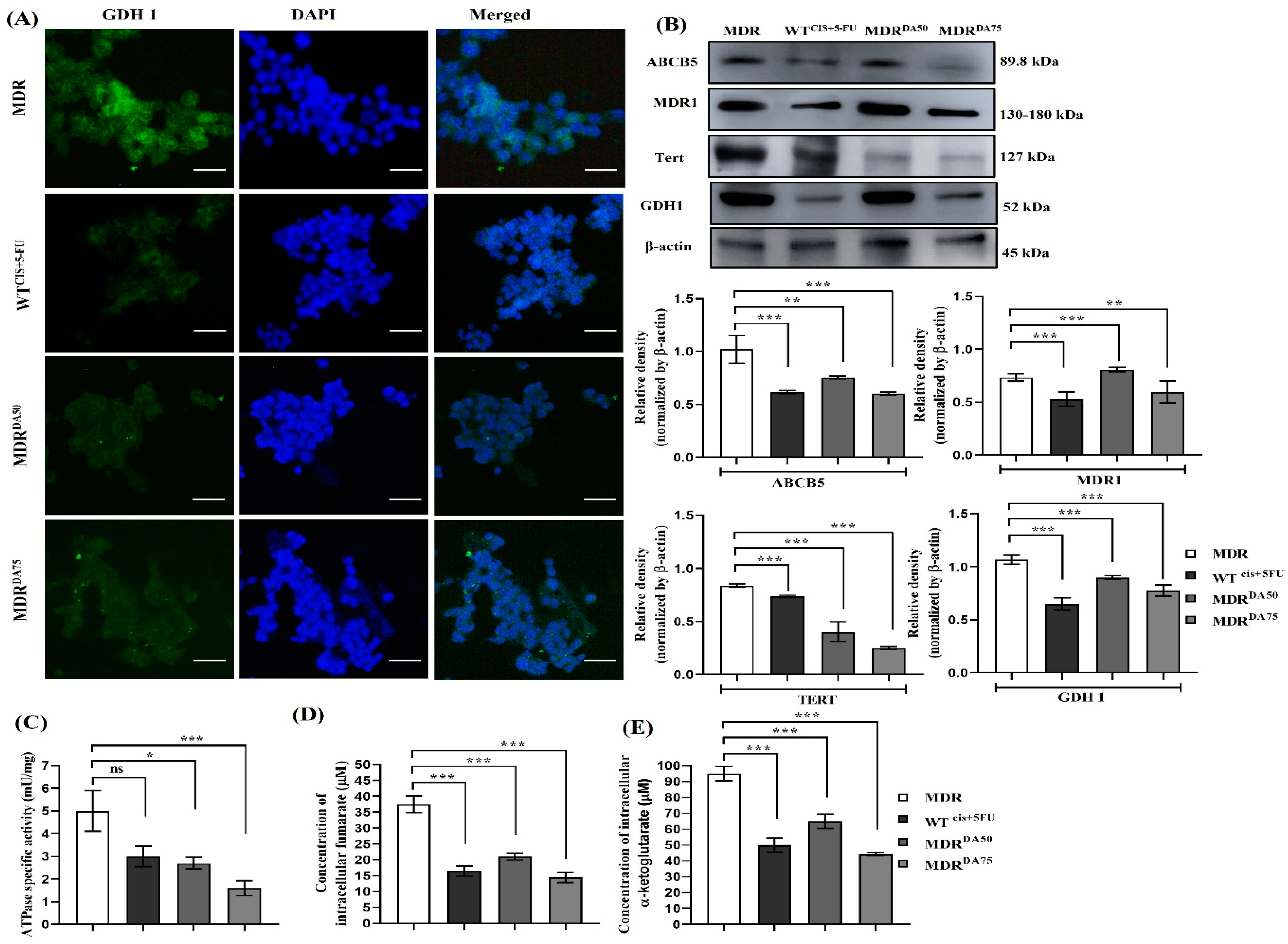
3.4. DA Downregulated MDR Phenotype, DNA Damage Repair, and Inhibited GDH1 Activity
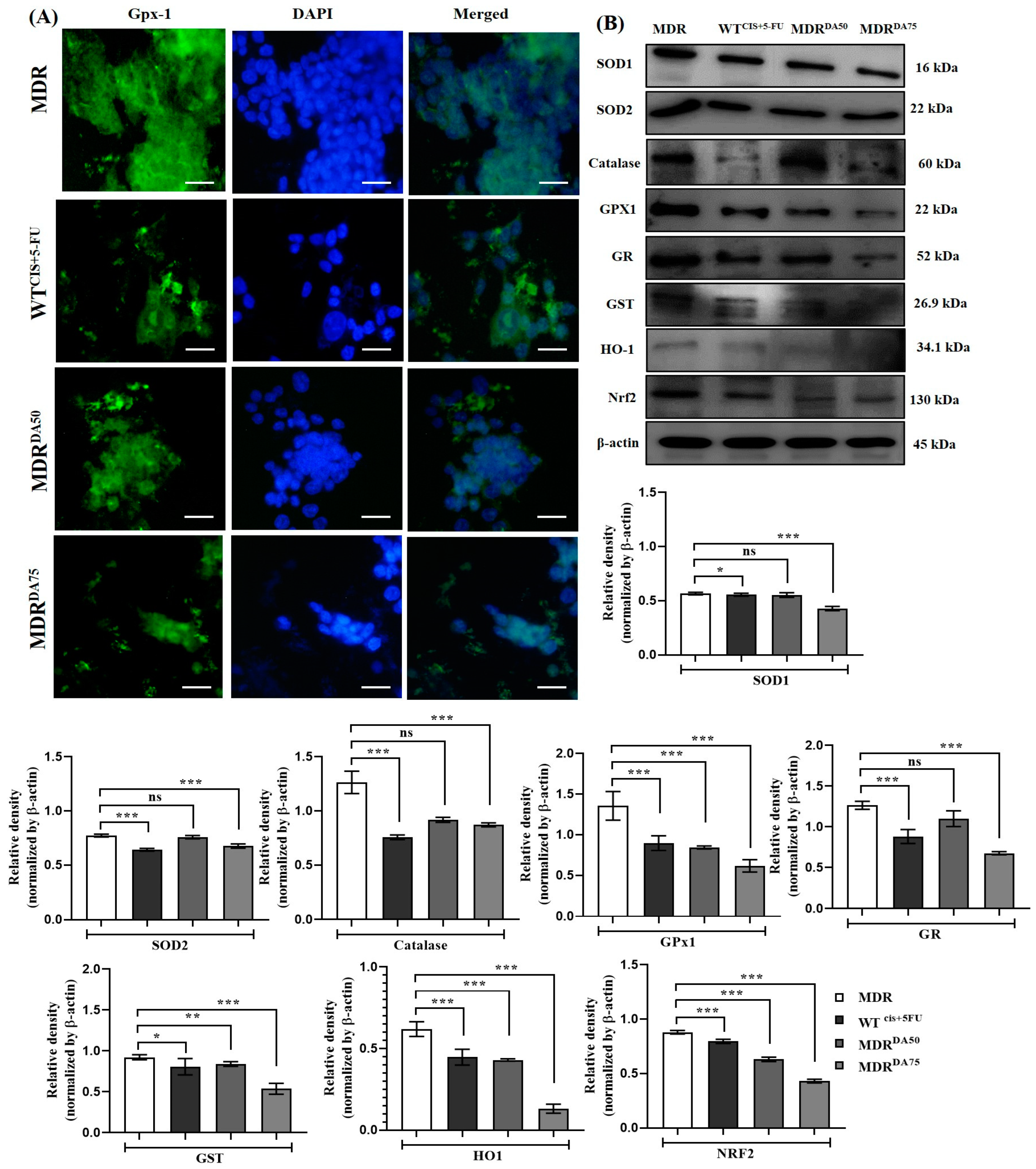
3.5. DA Destabilized Redox Homeostasis Regulated by GDH1 in HCT-116MDR CRC Cells
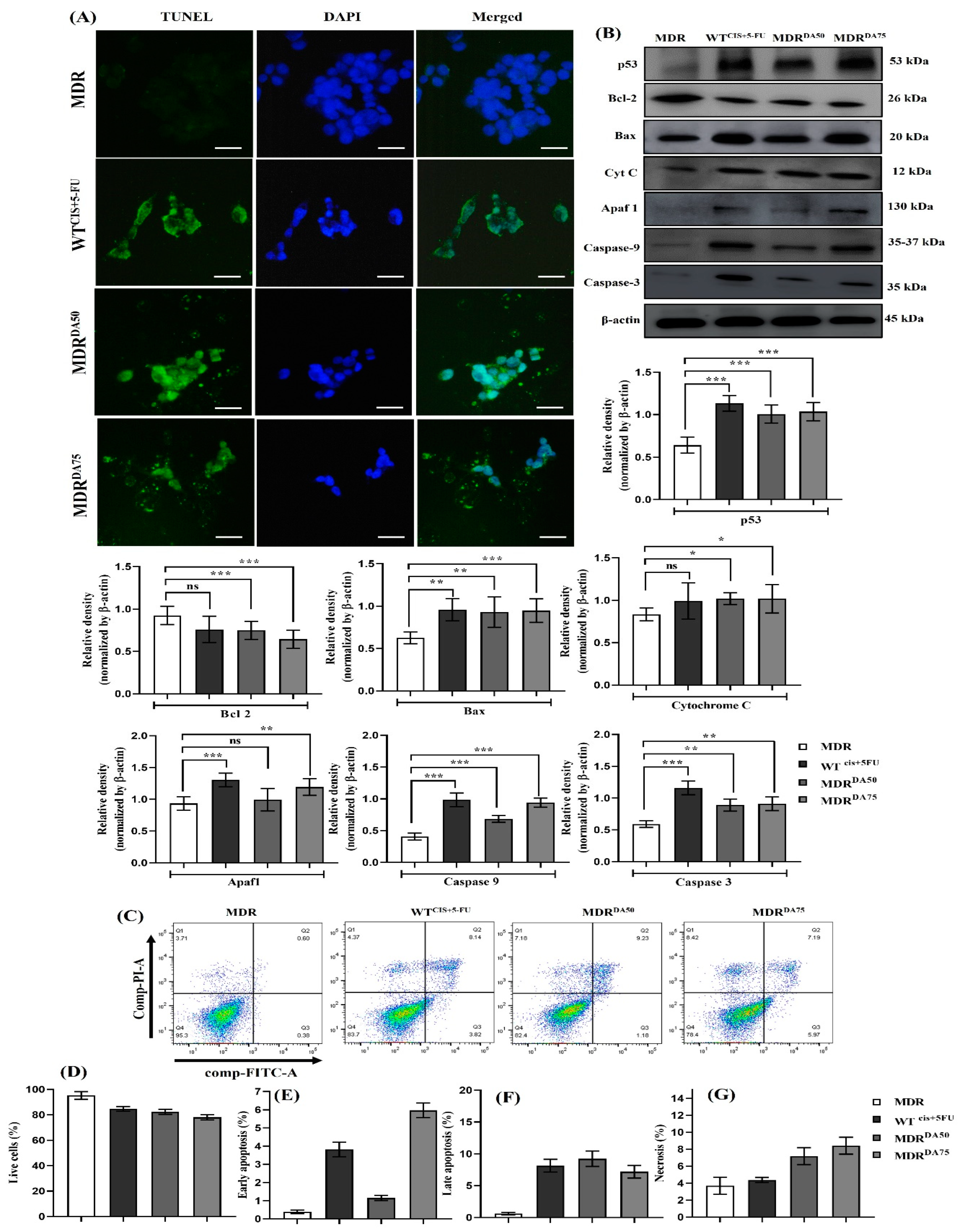
3.6. DA Triggered Intrinsic Apoptosis in HCT-116MDR CRC Cells
3.7. NAC Attenuated DA-Induced Alteration of Antioxidant, DNA Repair and Apoptosis Pathways in HCT-116MDR CRC Cells
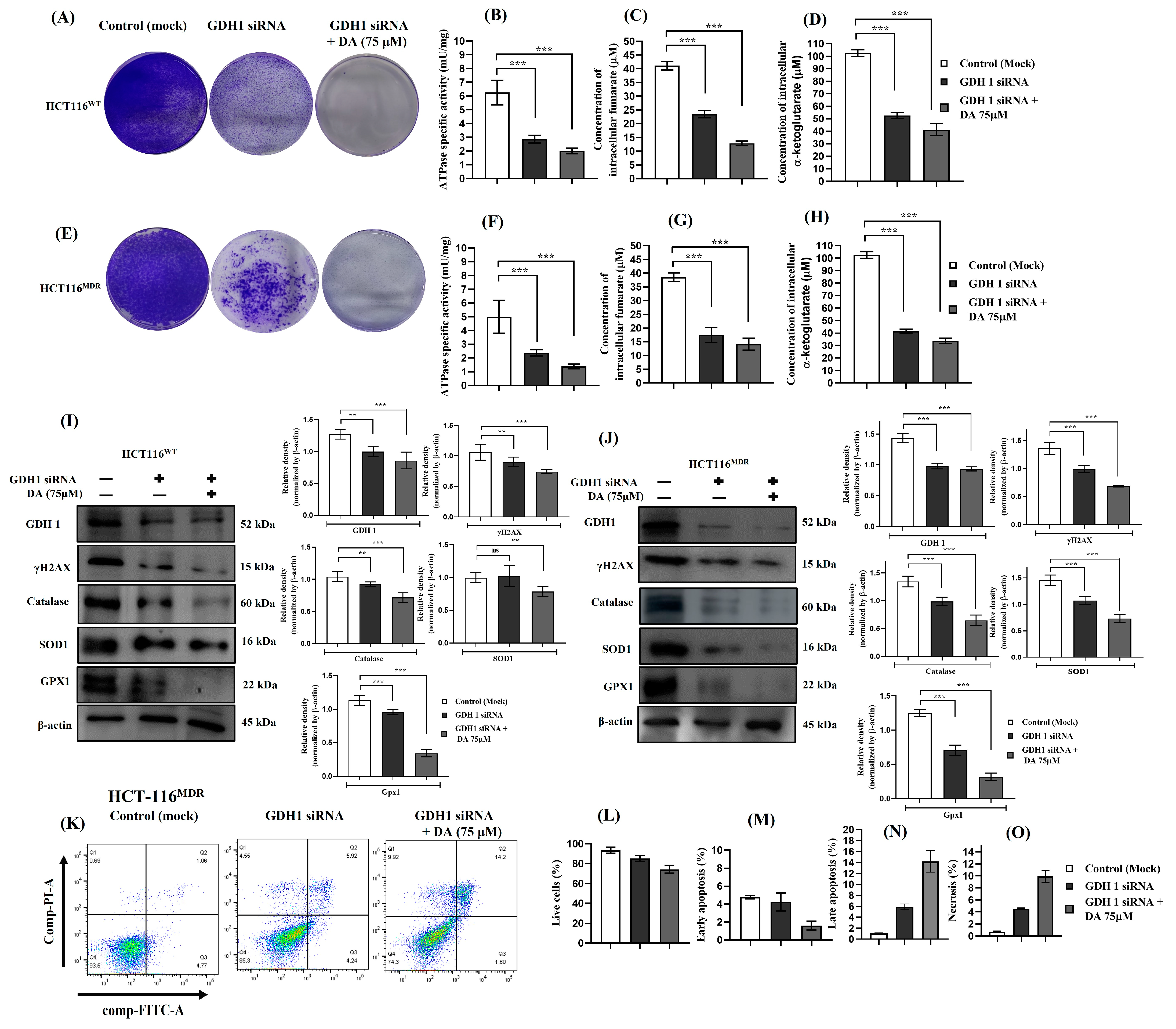
3.8. Silencing GDH1 Impaired Metabolic Activity and Cell Proliferation and Increased Cell Death in HCT-116WT and HCT-116MDR CRC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society Colorectal Cancer Facts & Figures 2020–2022. Atlanta Am. Cancer Soc. 2020.
- International Agency for Research on Cancer, WHO. Global Cancer Observatory. Available online: https://gco.iarc.fr (accessed on 11 January 2021).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.N.; Keretsu, S.; Kang, S.C. Evaluation of Decursin and Its Isomer Decursinol Angelate as Potential Inhibitors of Human Glutamate Dehydrogenase Activity through in Silico and Enzymatic Assay Screening. Comput. Biol. Med. 2022, 151, 106287. [Google Scholar] [CrossRef] [PubMed]
- Chau, I.; Cunningham, D. Adjuvant Therapy in Colon Cancer—What, When and How? Ann. Oncol. 2006, 17, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Alsayadnasser, M.; Vaida, F.; Veerapong, J.; Baumgartner, J.M.; Patel, S.; Ahmad, S.; Barone, R.; Lowy, A.M. Does Primary Tumor Side Matter in Patients with Metastatic Colon Cancer Treated with Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy? Ann. Surg. Oncol. 2019, 26, 1421–1427. [Google Scholar] [CrossRef]
- Loree, J.M.; Pereira, A.A.L.; Lam, M.; Willauer, A.N.; Raghav, K.; Dasari, A.; Van Morris, K.; Advani, S.; Menter, D.G.; Eng, C.; et al. Classifying Colorectal Cancer by Tumor Location Rather than Sidedness Highlights a Continuum in Mutation Profiles and Consensus Molecular Subtypes. Clin. Cancer Res. 2018, 24, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paldino, E.; Tesori, V.; Casalbore, P.; Gasbarrini, A.; Puglisi, M.A. Tumor Initiating Cells and Chemoresistance: Which Is the Best Strategy to Target Colon Cancer Stem Cells? Biomed. Res. Int. 2014, 2014, 859871. [Google Scholar] [CrossRef]
- Crea, F.; Duhagon, M.A.; Farrar, W.L.; Danesi, R. Pharmacogenomics and Cancer Stem Cells: A Changing Landscape? Trends Pharmacol. Sci. 2011, 8, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Kartalou, M.; Essigmann, J.M. Mechanisms of Resistance to Cisplatin. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil Resistance Mechanisms in Colorectal Cancer: From Classical Pathways to Promising Processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer Cell Metabolism: Warburg and Beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbaszadeh, Z.; Çeşmeli, S.; Biray Avcı, Ç. Crucial Players in Glycolysis: Cancer Progress. Gene 2020, 726, 144158. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic Shifts toward Glutamine Regulate Tumor Growth, Invasion and Bioenergetics in Ovarian Cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sim, W.S.; Lee, I.K.; Seu, Y.B.; Kim, I.H. Decursinol Angelate: A Cytotoxic and Protein Kinase C Activating Agent from the Root of Angelica Gigas. Planta Med. 1997, 63, 360–361. [Google Scholar] [CrossRef]
- Shehzad, A.; Parveen, S.; Qureshi, M.; Subhan, F.; Lee, Y.S. Decursin and Decursinol Angelate: Molecular Mechanism and Therapeutic Potential in Inflammatory Diseases. Inflamm. Res. 2018, 67, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.N.; Khan, I.; Kim, C.G.; Park, S.M.; Choi, D.K.; Lee, H.; Hwang, B.S.; Kang, S.C.; Park, J.G. Decursinol Angelate Arrest Melanoma Cell Proliferation by Initiating Cell Death and Tumor Shrinkage via Induction of Apoptosis. Int. J. Mol. Sci. 2021, 22, 4096. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, M.; Cho, H.J.; Paul, S.; Jakhar, R.; Khan, I.; Lee, S.J.; Kim, B.Y.; Krishnan, M.; Khaket, T.P.; Lee, H.G.; et al. Vitexin Induces Apoptosis by Suppressing Autophagy in Multidrug Resistant Colorectal Cancer Cells. Oncotarget 2018, 9, 3278–3291. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.P.; Cho, H.J.; Kim, J.T.; Baek, K.E.; Lee, H.G.; Kang, S.C. Morin Hydrate Reverses Cisplatin Resistance by Impairing Parp1/Hmgb1-Dependent Autophagy in Hepatocellular Carcinoma. Cancers 2019, 11, 986. [Google Scholar] [CrossRef] [Green Version]
- Dey, D.K.; Chang, S.N.; Gu, J.Y.; Kim, K.M.; Lee, J.J.; Kim, T.H.; Kang, S.C. Ultraviolet B-Irradiated Mushroom Supplementation Increased the Ca++ Uptake and Ameliorated the LPS-Induced Inflammatory Responses in Zebrafish Larvae. J. Food Biochem. 2021, 45, e13742. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Bahuguna, A.; Kumar, P.; Bajpai, V.K.; Kang, S.C. In Vitro and in Vivo Antitumor Potential of Carvacrol Nanoemulsion against Human Lung Adenocarcinoma A549 Cells via Mitochondrial Mediated Apoptosis. Sci. Rep. 2018, 8, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.N.; Kim, S.H.; Dey, D.K.; Park, S.M.; Nasif, O.; Bajpai, V.K.; Kang, S.C.; Lee, J.T.; Park, J.G. 5-o-Demethylnobiletin Alleviates Ccl4-Induced Acute Liver Injury by Equilibrating Ros-Mediated Apoptosis and Autophagy Induction. Int. J. Mol. Sci. 2021, 22, 1083. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.N.; Kim, H.J.; Kang, S.C. Quercetin Mitigates Oxidative Stress, Developmental Toxicity and Teratogenic Effects Induced by High-Dose Vitamin D2 in Zebrafish Embryos. Turk. J. Fish. Aquat. Sci. 2022, 22, TRJFAS20269. [Google Scholar] [CrossRef]
- Kim, C.G.; Chang, S.N.; Park, S.M.; Hwang, B.S.; Kang, S.A.; Kim, K.S.; Park, J.G. Moringa Oleifera Mitigates Ethanol-Induced Oxidative Stress, Fatty Degeneration and Hepatic Steatosis by Promoting Nrf2 in Mice. Phytomedicine 2022, 100, 154037. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.N.; Dey, D.K.; Oh, S.T.; Kong, W.H.; Cho, K.H.; Al-Olayan, E.M.; Hwang, B.S.; Kang, S.C.; Park, J.G. Phorbol 12-Myristate 13-Acetate Induced Toxicity Study and the Role of Tangeretin in Abrogating Hif-1α-Nf-Κb Crosstalk in Vitro and in Vivo. Int. J. Mol. Sci. 2020, 21, 9261. [Google Scholar] [CrossRef]
- Chang, S.N.; Park, J.G.; Kang, S.C. Therapeutic Propensity of Ginsenosides Rg1 and Rg3 in Rhabdomyolysis-Induced Acute Kidney Injury and Renohepatic Crosstalk in Rats. Int. Immunopharmacol. 2023, 115, 109602. [Google Scholar] [CrossRef]
- Chang, S.N.; Haroon, M.; Dey, D.K.; Kang, S.C. Rhabdomyolysis-Induced Acute Kidney Injury and Concomitant Apoptosis Induction via ROS-Mediated ER Stress Is Efficaciously Counteracted by Epigallocatechin Gallate. J. Nutr. Biochem. 2022, 110, 109134. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Bahuguna, A.; Bhardwaj, M.; Pal Khaket, T.; Kang, S.C. Carvacrol Nanoemulsion Evokes Cell Cycle Arrest, Apoptosis Induction and Autophagy Inhibition in Doxorubicin Resistant-A549 Cell Line. Artif. Cell. Nanomed. Biotechnol. 2018, 46, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel Strategies to Prevent the Development of Multidrug Resistance (MDR) in Cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [Green Version]
- Danø, K. Active Outward Transport of Daunomycin in Resistant Ehrlich Ascites Tumor Cells. BBA—Biomembr. 1973, 323, 466–483. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. BBA—Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal Duplication and Homology with Bacterial Transport Proteins in the Mdr1 (P-Glycoprotein) Gene from Multidrug-Resistant Human Cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Volpicelli, E.R.; Lezcano, C.; Zhan, Q.; Girouard, S.D.; Kindelberger, D.W.; Frank, M.H.; Frank, N.Y.; Crum, C.P.; Murphy, G.F. The Multidrug-Resistance Transporter Abcb5 Is Expressed in Human Placenta. Int. J. Gynecol. Pathol. 2014, 33, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethy, C.; Kundu, C.N. 5-Fluorouracil (5-FU) Resistance and the New Strategy to Enhance the Sensitivity against Cancer: Implication of DNA Repair Inhibition. Biomed. Pharmacother. 2021, 137, 111285. [Google Scholar] [CrossRef]
- Xu, D.; Tian, W.; Shen, H. Curcumin Prevents Induced Drug Resistance: A Novel Function? Chin. J. Cancer Res. 2011, 23, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Tian, W.; Shen, H. P-Gp Upregulation May Be Blocked by Natural Curcuminoids, a Novel Class of Chemoresistance-Preventing Agent. Mol. Med. Rep. 2013, 7, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Yang, W.; Feng, W.; Cao, L.; Wang, X.; Niu, L.; Li, Y.; Zhou, W.; Zhang, Y.; Liu, J.; et al. Molecular Mechanisms and Clinical Implications of MiRNAs in Drug Resistance of Colorectal Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920947342. [Google Scholar] [CrossRef]
- Cummings, B.S.; Schnellmann, R.G. Measurement of Cell Death in Mammalian Cells. Curr. Protoc. 2021, 1, e210. [Google Scholar] [CrossRef] [PubMed]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; André, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA Repair by ERCC1 in Non–Small-Cell Lung Cancer and Cisplatin-Based Adjuvant Chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Matés, J.M.; Segura, J.A.; Campos-Sandoval, J.A.; Lobo, C.; Alonso, L.; Alonso, F.J.; Márquez, J. Glutamine Homeostasis and Mitochondrial Dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 2051–2061. [Google Scholar] [CrossRef]
- Brunengraber, H.; Roe, C.R. Anaplerotic Molecules: Current and Future. J. Inherit. Metab. Dis. 2006, 29, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate Dehydrogenase 1 Signals through Antioxidant Glutathione Peroxidase 1 to Regulate Redox Homeostasis and Tumor Growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, S.; Yamada, K.; Yamashita, N.; Tada-Oikawa, S.; Kawanishi, S. N-Acetylcysteine, a Cancer Chemopreventive Agent, Causes Oxidative Damage to Cellular and Isolated DNA. Carcinogenesis 1999, 20, 1485–1490. [Google Scholar] [CrossRef] [Green Version]
- De Flora, S.; Izzotti, A.; D’Agostini, F.; Balansky, R.M. Mechanisms of N-Acetylcysteine in the Prevention of DNA Damage and Cancer, with Special Reference to Smoking-Related End-Points. Carcinogenesis 2001, 22, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Balansky, R.; Izzotti, A.; Scatolini, L.; D’Agostini, F.; De Flora, S. Induction by Carcinogens and Chemoprevention by N-Acetylcysteine of Adducts to Mitochondrial DNA in Rat Organs. Cancer Res. 1996, 56, 1642–1647. [Google Scholar] [PubMed]
- Izzotti, A.; Bagnasco, M.; Camoirano, A.; Orlando, M.; De Flora, S. DNA Fragmentation, DNA-Protein Crosslinks, 32P Postlabeled Nucleotidic Modifications, and 8-Hydroxy-2′-Deoxyguanosine in the Lung but Not in the Liver of Rats Receiving Intratracheal Instillations of Chromium(VI). Chemoprevention by Oral N-Acetylcysteine. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 1998, 400, 233–244. [Google Scholar] [CrossRef]
- Izzotti, A.; Balansky, R.; Scatolini, L.; Rovida, A.; De Flora, S. Inhibition by N-Acetylcysteine of Carcinogen-Dna Adducts in the Tracheal Epithelium of Rats Exposed to Cigarette Smoke. Carcinogenesis 1995, 16, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Q.; Han, J.; Feng, J.; Guo, T.; Li, Z.; Min, F.; Jin, R.; Peng, X. N-Acetylcysteine Inhibits Patulin-Induced Apoptosis by Affecting Ros-Mediated Oxidative Damage Pathway. Toxins 2021, 13, 595. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, S.N.; Kang, S.C. Decursinol Angelate Inhibits Glutamate Dehydrogenase 1 Activity and Induces Intrinsic Apoptosis in MDR-CRC Cells. Cancers 2023, 15, 3541. https://doi.org/10.3390/cancers15143541
Chang SN, Kang SC. Decursinol Angelate Inhibits Glutamate Dehydrogenase 1 Activity and Induces Intrinsic Apoptosis in MDR-CRC Cells. Cancers. 2023; 15(14):3541. https://doi.org/10.3390/cancers15143541
Chicago/Turabian StyleChang, Sukkum Ngullie, and Sun Chul Kang. 2023. "Decursinol Angelate Inhibits Glutamate Dehydrogenase 1 Activity and Induces Intrinsic Apoptosis in MDR-CRC Cells" Cancers 15, no. 14: 3541. https://doi.org/10.3390/cancers15143541
APA StyleChang, S. N., & Kang, S. C. (2023). Decursinol Angelate Inhibits Glutamate Dehydrogenase 1 Activity and Induces Intrinsic Apoptosis in MDR-CRC Cells. Cancers, 15(14), 3541. https://doi.org/10.3390/cancers15143541