
Assessment of Colorectal Cancer Risk Factors through the Application of Network-Based Approaches in a Racially Diverse Cohort of Colon Organoid Stem Cells

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Biopsy Collection and Establishment of Colon Organoids in Matrigel
2.3. Stem Cell Enrichment of Colon Organoids
2.4. RNA Isolation from Colon Organoids and RNA Processing
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Mapping Genes to CRC GWAS Loci
2.7. Analysis of Publicly Available Data
2.8. Single-Cell Deconvolution of Bulk RNA-seq Data
2.9. Quantitative PCR (qPCR) of Stem Cell Profiles
3. Results
3.1. Generation of Colon Organoid Dataset
3.2. WGCNA of Stem-Cell-Enriched Colon Organoids
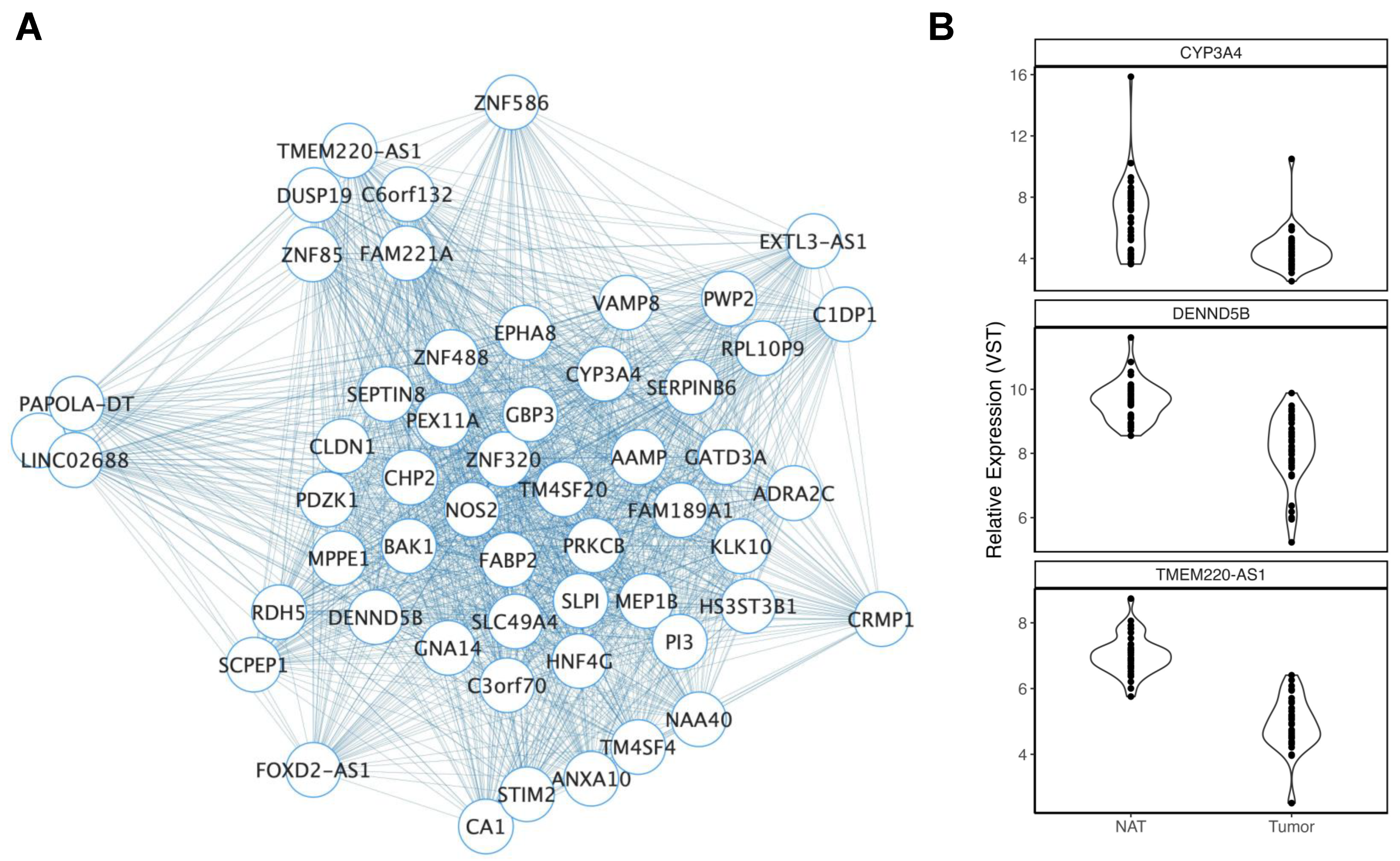
3.2.1. Ancestry
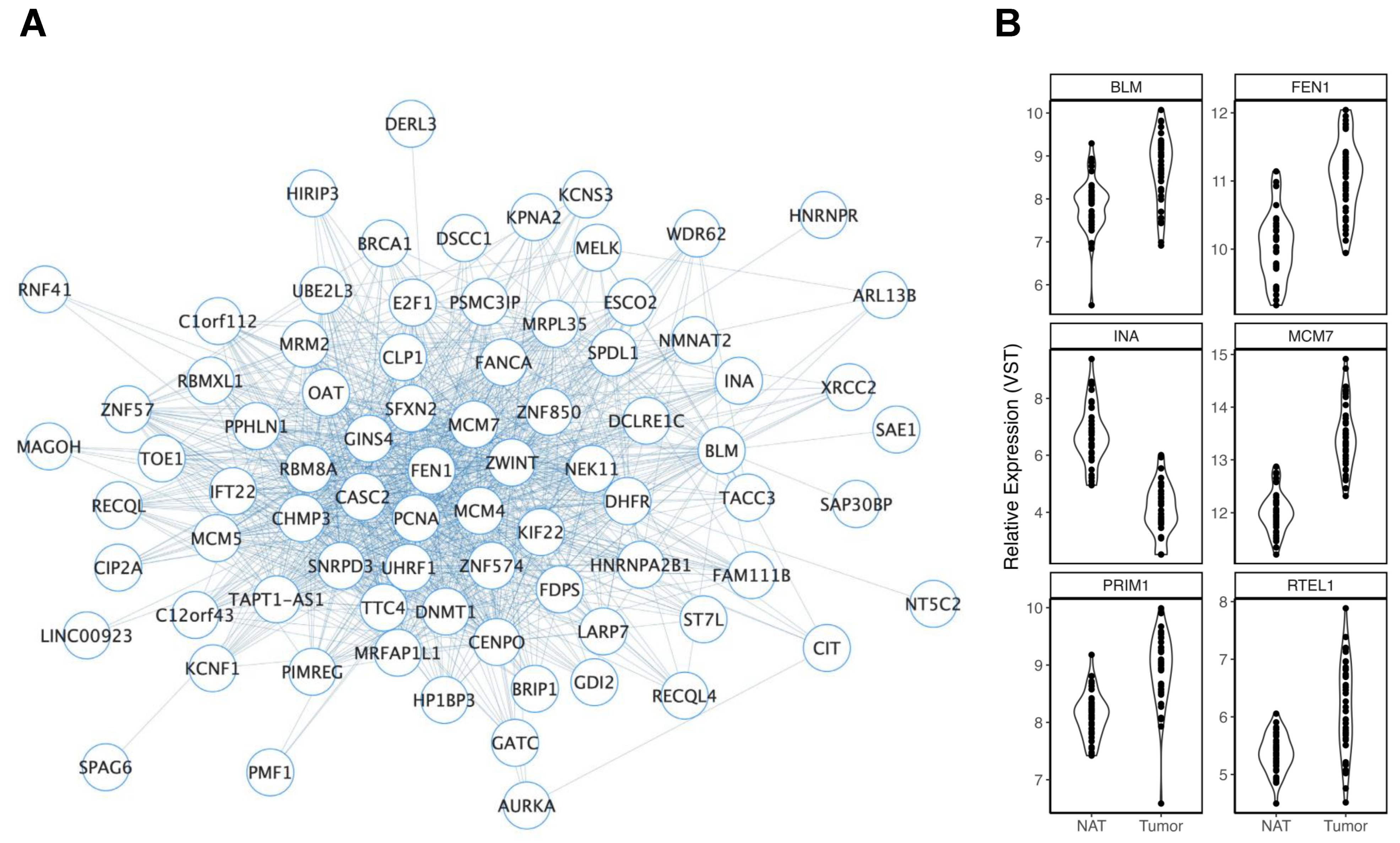
3.2.2. Age
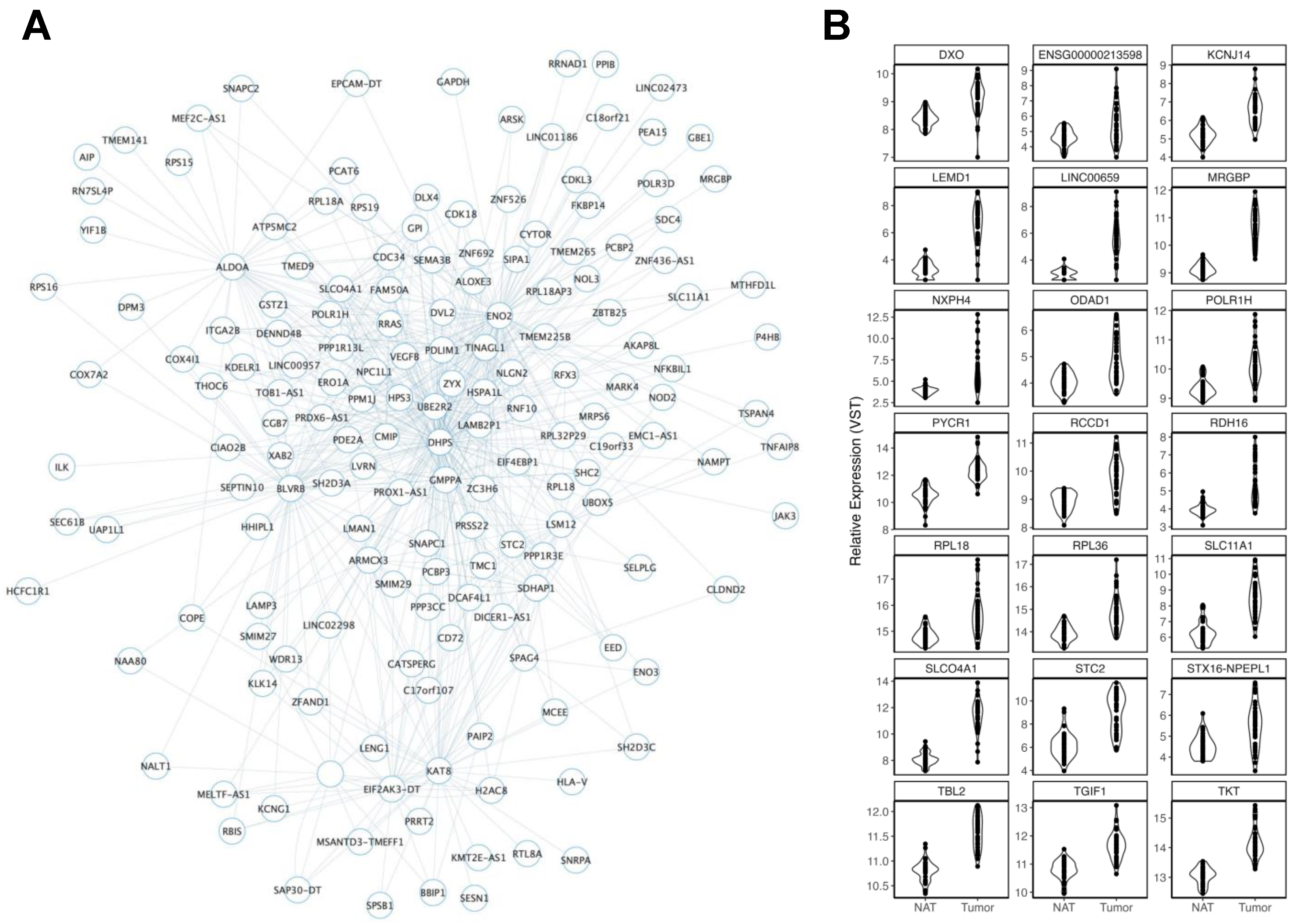
3.2.3. Smoking History
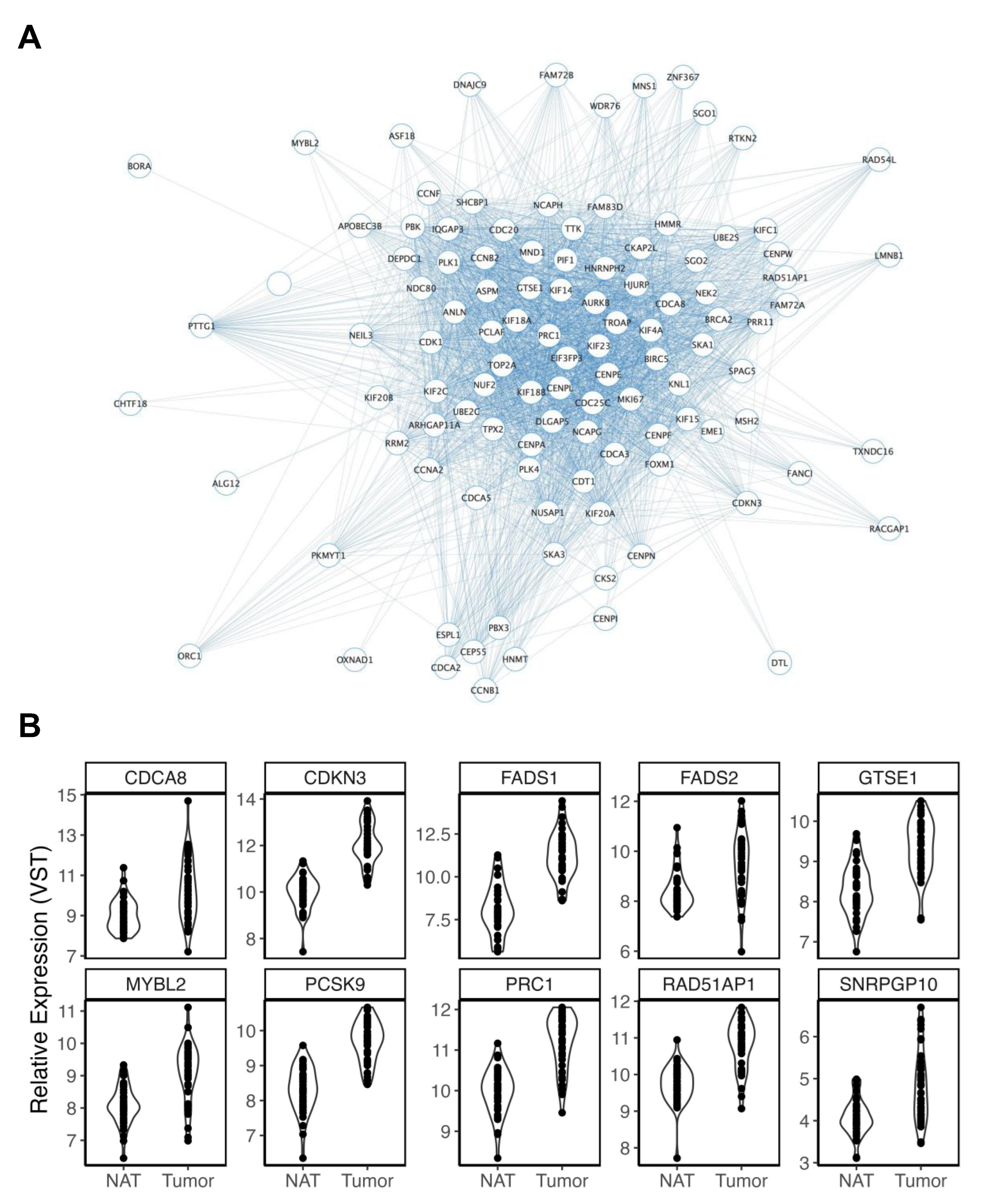
3.2.4. Body Mass Index
3.2.5. Biological Sex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xiong, Z.; He, W.; Xie, K.; Liu, S.; Kong, P.; Jiang, C.; Guo, G.; Xia, L. Proximal shift of colorectal cancer with increasing age in different ethnicities. Cancer Manag. Res. 2018, 10, 2663–2673. [Google Scholar] [CrossRef] [Green Version]
- Petrick, J.L.; Barber, L.E.; Warren Andersen, S.; Florio, A.A.; Palmer, J.R.; Rosenberg, L. Racial Disparities and Sex Differences in Early- and Late-Onset Colorectal Cancer Incidence, 2001–2018. Front. Oncol. 2021, 11, 734998. [Google Scholar] [CrossRef]
- Giaquinto, A.N.; Miller, K.D.; Tossas, K.Y.; Winn, R.A.; Jemal, A.; Siegel, R.L. Cancer statistics for African American/Black People 2022. CA Cancer J. Clin. 2022, 72, 202–229. [Google Scholar] [CrossRef] [PubMed]
- Demb, J.; Earles, A.; Martinez, M.E.; Bustamante, R.; Bryant, A.K.; Murphy, J.D.; Liu, L.; Gupta, S. Risk factors for colorectal cancer significantly vary by anatomic site. BMJ Open Gastroenterol. 2019, 6, e000313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Yang, Y.; Wang, F.; Zhang, P.; Shi, C.; Zou, Y.; Qin, H. Obesity and risk of colorectal cancer: A systematic review of prospective studies. PLoS ONE 2013, 8, e53916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polakis, P. The oncogenic activation of beta-catenin. Curr. Opin. Genet. Dev. 1999, 9, 15–21. [Google Scholar] [CrossRef]
- Oliveira, R.C.; Abrantes, A.M.; Tralhao, J.G.; Botelho, M.F. The role of mouse models in colorectal cancer research—The need and the importance of the orthotopic models. Anim. Model. Exp. Med. 2020, 3, 1–8. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Paredes, J.; Ji, P.; Lacomb, J.F.; Shroyer, K.R.; Martello, L.A.; Williams, J.L. Establishment of three novel cell lines derived from African American patients with colorectal carcinoma: A unique tool for assessing racial health disparity. Int. J. Oncol. 2018, 53, 1516–1528. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; van Es, J.H.; van den Brink, S.; van Houdt, W.J.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Farin, H.F.; van Es, J.H.; Clevers, H.; Langer, R.; Karp, J.M. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat. Methods 2014, 11, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, R.; van Driel, M.S.; Vermeulen, L.; van Neerven, S.M. Intestinal stem cell dynamics in homeostasis and cancer. Trends Cancer 2022, 8, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauss, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Stoney, R.; Robertson, D.L.; Nenadic, G.; Schwartz, J.-M. Mapping biological process relationships and disease perturbations within a pathway network. NPJ Syst. Biol. Appl. 2018, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhou, D.; Qiu, W.; Shi, Y.; Yang, J.-J.; Chen, S.; Wang, Q.; Pan, H. Application of Weighted Gene Co-expression Network Analysis for Data from Paired Design. Sci. Rep. 2018, 8, 622. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.; Lee, I. Single-cell network biology for resolving cellular heterogeneity in human diseases. Exp. Mol. Med. 2020, 52, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.A.M.; Dampier, C.H.; Eaton, S.; Ali, M.W.; Plummer, S.J.; Bryant, J.; Gauderman, W.J.; Peters, U.; Powell, S.M.; Casey, G. Transcriptomic Response to Calcium in Normal Colon Organoids is Impacted by Colon Location and Sex. Cancer Prev. Res. 2022, 15, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Coskun, V.; Liang, A.; Yu, J.; Cheng, L.; Ge, W.; Shi, Z.; Zhang, K.; Li, C.; Cui, Y.; et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell 2015, 161, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devall, M.A.M.; Drew, D.A.; Dampier, C.H.; Plummer, S.J.; Eaton, S.; Bryant, J.; Diez-Obrero, V.; Mo, J.; Kedrin, D.; Zerjav, D.C.; et al. Transcriptome-wide In Vitro Effects of Aspirin on Patient-derived Normal Colon Organoids. Cancer Prev. Res. 2021, 14, 1089–1100. [Google Scholar] [CrossRef]
- Devall, M.; Plummer, S.J.; Bryant, J.; Jennelle, L.T.; Eaton, S.; Dampier, C.H.; Huyghe, J.R.; Peters, U.; Powell, S.M.; Casey, G. Ethanol exposure drives colon location specific cell composition changes in a normal colon crypt 3D organoid model. Sci. Rep. 2021, 11, 432. [Google Scholar] [CrossRef]
- Devall, M.; Dampier, C.H.; Eaton, S.; Ali, M.W.; Díez-Obrero, V.; Moratalla-Navarro, F.; Bryant, J.; Jennelle, L.T.; Moreno, V.; Powell, S.M.; et al. Novel insights into the molecular mechanisms underlying risk of colorectal cancer from smoking and red/processed meat carcinogens by modeling exposure in normal colon organoids. Oncotarget 2021, 12, 1863–1877. [Google Scholar] [CrossRef]
- Devall, M.; Jennelle, L.T.; Bryant, J.; Bien, S.; Peters, U.; Powell, S.; Casey, G. Modeling the effect of prolonged ethanol exposure on global gene expression and chromatin accessibility in normal 3D colon organoids. PLoS ONE 2020, 15, e0227116. [Google Scholar] [CrossRef] [Green Version]
- Devall, M.A.M.; Casey, G. Controlling for cellular heterogeneity using single-cell deconvolution of gene expression reveals novel markers of colorectal tumors exhibiting microsatellite instability. Oncotarget 2021, 12, 767–782. [Google Scholar] [CrossRef]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- Carethers, J.M. Racial and ethnic disparities in colorectal cancer incidence and mortality. Adv. Cancer Res. 2021, 151, 197–229. [Google Scholar] [CrossRef]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef]
- Fernandez-Rozadilla, C.; Timofeeva, M.; Chen, Z.; Law, P.; Thomas, M.; Schmit, S.; Díez-Obrero, V.; Hsu, L.; Fernandez-Tajes, J.; Palles, C.; et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat. Genet. 2023, 55, 89–99. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e722. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Devall, M.; Ali, M.W.; Eaton, S.; Weisenberger, D.J.; Reilley, M.J.; Powell, S.M.; Li, L.; Casey, G. Multi-omic analysis in normal colon organoids highlights MSH4 as a novel marker of defective mismatch repair in Lynch syndrome and microsatellite instability. Cancer Med. 2023, 12, 13551–13572. [Google Scholar] [CrossRef]
- Devall, M.A.; Eaton, S.; Ali, M.W.; Dampier, C.H.; Weisenberger, D.; Powell, S.M.; Li, L.; Casey, G. DNA methylation analysis of normal colon organoids from familial adenomatous polyposis patients reveals novel insight into colon cancer development. Clin. Epigenet. 2022, 14, 104. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Barriga, F.M.; Montagni, E.; Mana, M.; Mendez-Lago, M.; Hernando-Momblona, X.; Sevillano, M.; Guillaumet-Adkins, A.; Rodriguez-Esteban, G.; Buczacki, S.J.A.; Gut, M.; et al. Mex3a Marks a Slowly Dividing Subpopulation of Lgr5+ Intestinal Stem Cells. Cell Stem Cell 2017, 20, 801–816.e807. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE 2009, 4, e5846. [Google Scholar] [CrossRef] [PubMed]
- Hammouz, R.Y.; Kostanek, J.K.; Dudzisz, A.; Witas, P.; Orzechowska, M.; Bednarek, A.K. Differential expression of lung adenocarcinoma transcriptome with signature of tobacco exposure. J. Appl. Genet. 2020, 61, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, K.; Reichmann, R.; Kaaks, R.; Jenab, M.; Bueno-de-Mesquita, H.B.; Dahm, C.C.; Eriksen, A.K.; Tjønneland, A.; Artaud, F.; Boutron-Ruault, M.-C.; et al. Development and validation of a lifestyle-based model for colorectal cancer risk prediction: The LiFeCRC score. BMC Med. 2021, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Freedman, A.N.; Seminara, D.; Gail, M.H.; Hartge, P.; Colditz, G.A.; Ballard-Barbash, R.; Pfeiffer, R.M. Cancer risk prediction models: A workshop on development, evaluation, and application. J. Natl. Cancer Inst. 2005, 97, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.H.; Wilson, B.J.; Gold, J.S.; Frank, N.Y. Clinical Implications of Colorectal Cancer Stem Cells in the Age of Single-Cell Omics and Targeted Therapies. Gastroenterology 2021, 160, 1947–1960. [Google Scholar] [CrossRef]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortini, B.K.; Tring, S.; Devall, M.A.; Ali, M.W.; Plummer, S.J.; Casey, G. SNPs associated with colorectal cancer at 15q13.3 affect risk enhancers that modulate GREM1 gene expression. Hum. Mutat. 2021, 42, 237–245. [Google Scholar] [CrossRef]
- Fortini, B.K.; Tring, S.; Plummer, S.J.; Edlund, C.K.; Moreno, V.; Bresalier, R.S.; Barry, E.L.; Church, T.R.; Figueiredo, J.C.; Casey, G. Multiple functional risk variants in a SMAD7 enhancer implicate a colorectal cancer risk haplotype. PLoS ONE 2014, 9, e111914. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, F.; Sibley, A.B.; Patil, S.A.; Etheridge, A.S.; Jiang, C.; Ou, F.S.; Howell, S.D.; Plummer, S.J.; Casey, G.; Bertagnolli, M.M.; et al. Genomic Analysis of Germline Variation Associated with Survival of Patients with Colorectal Cancer Treated with Chemotherapy Plus Biologics in CALGB/SWOG 80405 (Alliance). Clin. Cancer Res. 2021, 27, 267–275. [Google Scholar] [CrossRef]
- Qiu, X.; Cheng, S.H.; Xu, F.; Yin, J.W.; Wang, L.Y.; Zhang, X.Y. Weighted gene co-expression network analysis identified MYL9 and CNN1 are associated with recurrence in colorectal cancer. J. Cancer 2020, 11, 2348–2359. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zeng, F.; Fan, G.; Dong, Q. Identification of Hub Genes and Construction of a Transcriptional Regulatory Network Associated With Tumor Recurrence in Colorectal Cancer by Weighted Gene Co-expression Network Analysis. Front. Genet. 2021, 12, 649752. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.D.; Fontham, E.T.H.; Church, T.R.; Flowers, C.R.; Guerra, C.E.; LaMonte, S.J.; Etzioni, R.; McKenna, M.T.; Oeffinger, K.C.; Shih, Y.T.; et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 2018, 68, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Chatsirisupachai, K.; Lagger, C.; de Magalhaes, J.P. Age-associated differences in the cancer molecular landscape. Trends Cancer 2022, 8, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.E.; Clendenning, M.; Wong, E.M.; Rosty, C.; Mahmood, K.; Georgeson, P.; Winship, I.M.; Preston, S.G.; Win, A.K.; Dugue, P.A.; et al. DNA Methylation Signatures and the Contribution of Age-Associated Methylomic Drift to Carcinogenesis in Early-Onset Colorectal Cancer. Cancers 2021, 13, 2589. [Google Scholar] [CrossRef]
- Guo, Y.; Ayers, J.L.; Carter, K.T.; Wang, T.; Maden, S.K.; Edmond, D.; Newcomb, P.P.; Li, C.; Ulrich, C.; Yu, M.; et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell 2019, 18, e13013. [Google Scholar] [CrossRef] [Green Version]
- Morris, O.; Deng, H.; Tam, C.; Jasper, H. Warburg-like Metabolic Reprogramming in Aging Intestinal Stem Cells Contributes to Tissue Hyperplasia. Cell Rep. 2020, 33, 108423. [Google Scholar] [CrossRef]
- Devall, M.A.; Sun, X.; Eaton, S.; Cooper, G.S.; Willis, J.E.; Weisenberger, D.J.; Casey, G.; Li, L. A Race-Specific, DNA Methylation Analysis of Aging in Normal Rectum: Implications for the Biology of Aging and Its Relationship to Rectal Cancer. Cancers 2022, 15, 45. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef]
- Sobocinska, J.; Nowakowska, J.; Molenda, S.; Olechnowicz, A.; Guglas, K.; Kozlowska-Maslon, J.; Kazimierczak, U.; Machnik, M.; Oleksiewicz, U.; Teresiak, A.; et al. Zinc Finger Proteins in Head and Neck Squamous Cell Carcinomas: ZNF540 May Serve as a Biomarker. Curr. Oncol. 2022, 29, 9896–9915. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; He, Z.; Wang, C.; Zhou, Y.; Li, F.; Pu, W.; Zhang, X.; Feng, X.; Zhang, M.; Yecheng, X.; et al. Epigenetic silencing of ZNF132 mediated by methylation-sensitive Sp1 binding promotes cancer progression in esophageal squamous cell carcinoma. Cell Death Dis. 2018, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Hakimi, P.; Kao, C.; Kao, A.; Liu, R.; Janocha, A.; Boyd-Tressler, A.; Hang, X.; Alhoraibi, H.; Slater, E.; et al. Reciprocal Changes in Phosphoenolpyruvate Carboxykinase and Pyruvate Kinase with Age Are a Determinant of Aging in Caenorhabditis elegans. J. Biol. Chem. 2016, 291, 1307–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, S.; Seu, P.; Gao, F.Q.; Gros, P.; Goss, J.A. Disruption of the Nramp1 (also known as Slc11a1) gene in Kupffer cells attenuates early-phase, warm ischemia-reperfusion injury in the mouse liver. J. Leukoc. Biol. 2002, 72, 885–897. [Google Scholar] [CrossRef]
- Yu, B.; Zhang, F.; Liu, L.; Liang, Y.; Tang, X.; Peng, Y.; Cai, F.; Zeng, D.; Yuan, X.; Li, J.; et al. The novel prognostic risk factor STC2 can regulate the occurrence and progression of osteosarcoma via the glycolytic pathway. Biochem. Biophys. Res. Commun. 2021, 554, 25–32. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, P.; Liu, Z.; Xing, Y.; Xiao, Y. NXPH4 Promotes Gemcitabine Resistance in Bladder Cancer by Enhancing Reactive Oxygen Species and Glycolysis Activation through Modulating NDUFA4L2. Cancers 2022, 14, 3782. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, X.; Yong, H.; Xu, J.; Qu, P.; Qiao, S.; Hou, P.; Li, Z.; Chu, S.; Zheng, J.; et al. Transketolase promotes colorectal cancer metastasis through regulating AKT phosphorylation. Cell Death Dis. 2022, 13, 99. [Google Scholar] [CrossRef]
- Tejedor, G.; Contreras-Lopez, R.; Barthelaix, A.; Ruiz, M.; Noel, D.; De Ceuninck, F.; Pastoureau, P.; Luz-Crawford, P.; Jorgensen, C.; Djouad, F. Pyrroline-5-Carboxylate Reductase 1 Directs the Cartilage Protective and Regenerative Potential of Murphy Roths Large Mouse Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2021, 9, 604756. [Google Scholar] [CrossRef]
- Cheng, J.; Chen, Y.; Wang, X.; Wang, J.; Yan, Z.; Gong, G.; Li, G.; Li, C. Meta-analysis of prospective cohort studies of cigarette smoking and the incidence of colon and rectal cancers. Eur. J. Cancer Prev. 2015, 24, 6–15. [Google Scholar] [CrossRef]
- Qi, G.; Zhang, C.; Ma, H.; Li, Y.; Peng, J.; Chen, J.; Kong, B. CDCA8, targeted by MYBL2, promotes malignant progression and olaparib insensitivity in ovarian cancer. Am. J. Cancer Res. 2021, 11, 389–415. [Google Scholar]
- Wang, J.; Che, W.; Wang, W.; Su, G.; Zhen, T.; Jiang, Z. CDKN3 promotes tumor progression and confers cisplatin resistance via RAD51 in esophageal cancer. Cancer Manag. Res. 2019, 11, 3253–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heravi, G.; Jang, H.; Wang, X.; Long, Z.; Peng, Z.; Kim, S.; Liu, W. Fatty acid desaturase 1 (FADS1) is a cancer marker for patient survival and a potential novel target for precision cancer treatment. Front. Oncol. 2022, 12, 942798. [Google Scholar] [CrossRef] [PubMed]
- Molendijk, J.; Kolka, C.M.; Cairns, H.; Brosda, S.; Mohamed, A.; Shah, A.K.; Brown, I.; Hodson, M.P.; Hennessy, T.; Liu, G.; et al. Elevation of fatty acid desaturase 2 in esophageal adenocarcinoma increases polyunsaturated lipids and may exacerbate bile acid-induced DNA damage. Clin. Transl. Med. 2022, 12, e810. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.B.; Wages, N.A.; Grant, P.A.; Stukenberg, P.T.; Gentzler, R.D.; Hall, R.D.; Akerley, W.L.; Varghese, T.K.; Arnold, S.M.; Williams, T.M.; et al. MYBL2-Driven Transcriptional Programs Link Replication Stress and Error-prone DNA Repair with Genomic Instability in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 585551. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Liu, S.; Brickell, A.N.; Zhang, J.; Wu, Z.; Zhou, S.; Ding, Z. PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia. Basic Res. Cardiol. 2020, 115, 66. [Google Scholar] [CrossRef]
- Fitieh, A.; Locke, A.J.; Motamedi, M.; Ismail, I.H. The Role of Polycomb Group Protein BMI1 in DNA Repair and Genomic Stability. Int. J. Mol. Sci. 2021, 22, 2976. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Wu, C.C.; Li, P.P.; Fu, Y.M.; Xie, L.H.; Sun, S.S.; Zhou, Y.Y.; Zhu, B.L. The role of MYB proto-oncogene like 2 in tamoxifen resistance in breast cancer. J. Mol. Histol. 2021, 52, 21–30. [Google Scholar] [CrossRef]
- O’Sullivan, D.E.; Sutherland, R.L.; Town, S.; Chow, K.; Fan, J.; Forbes, N.; Heitman, S.J.; Hilsden, R.J.; Brenner, D.R. Risk Factors for Early-Onset Colorectal Cancer: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 1229–1240.e1225. [Google Scholar] [CrossRef]
- Dawson, M.I.; Xia, Z. The retinoid X receptors and their ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef] [Green Version]
- Ogilvie, K.M.; Saladin, R.; Nagy, T.R.; Urcan, M.S.; Heyman, R.A.; Leibowitz, M.D. Activation of the retinoid X receptor suppresses appetite in the rat. Endocrinology 2004, 145, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The Missing Diversity in Human Genetic Studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geneviève, L.D.; Martani, A.; Shaw, D.; Elger, B.S.; Wangmo, T. Structural racism in precision medicine: Leaving no one behind. BMC Med. Ethics 2020, 21, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, C.M.; May, F.P.; Coronado, G.D.; Pujol, T.A.; Thomas, E.G.; Cabreros, I. Racism Is a Modifiable Risk Factor: Relationships Among Race, Ethnicity, and Colorectal Cancer Outcomes. Gastroenterology 2022, 162, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Pew Research Center. One-inTen Black People Living in the U.S. Are Immigrants; Pew Research Center: Washington, DC, USA, 2022. [Google Scholar]
- Pinheiro, P.S.; Medina, H.; Callahan, K.E.; Kwon, D.; Ragin, C.; Sherman, R.; Kobetz, E.N.; Jemal, A. Cancer mortality among US blacks: Variability between African Americans, Afro-Caribbeans, and Africans. Cancer Epidemiol. 2020, 66, 101709. [Google Scholar] [CrossRef]
- Leek, J.T. Svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014, 42, e161. [Google Scholar] [CrossRef] [PubMed]
- Co, J.Y.; Klein, J.A.; Kang, S.; Homan, K.A. Toward Inclusivity in Preclinical Drug Development: A Proposition to Start with Intestinal Organoids. Adv. Biol. 2023, e2200333. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deidentified ID | Sex | Ancestry | Age | Smoking History | BMI |
|---|---|---|---|---|---|
| P1055R | F | EA | 22 | 0 | 26.4 |
| P1081R | M | EA | 66 | 0 | 22.96 |
| P1096R | M | EA | 53 | 0 | 50.4 |
| P1099R | F | AA | 56 | 0 | 42.4 |
| P1112R | F | EA | 50 | 0 | 21.6 |
| P1121R | F | AA | 52 | 0 | 27.72 |
| P1148R | F | EA | 59 | 1 | 25.52 |
| P1176R | M | AA | 69 | 0 | 23.9 |
| P1229R | F | EA | 53 | 0 | 27.79 |
| P1239R | M | AA | 50 | 0 | 32.32 |
| P1246R | F | AA | 54 | 1 | 47.77 |
| P1252R | F | AA | 31 | 1 | 33.78 |
| P1263R | F | EA | 62 | 0 | 49.86 |
| P1268R | F | AA | 58 | 0 | 39.6 |
| P1270R | F | AA | 60 | 0 | 28.1 |
| Trait | Module | GS vs. MM r | GS vs. MM P | r | p | FDR |
|---|---|---|---|---|---|---|
| Age | skyblue4 | 0.49 | 5.30 × 104 | −0.74 | 1.46 × 103 | 0.04 |
| Age | lightpink3 | 0.69 | <2.20 × 1016 | −0.74 | 1.50 × 103 | 0.04 |
| Age | magenta4 | 0.49 | 1.90 × 104 | 0.73 | 1.94 × 103 | 0.04 |
| Age | coral3 | 0.4 | 1.40E × 106 | 0.64 | 0.01 | 0.14 |
| Age | lightsteelblue | 0.49 | <2.20 × 1016 | 0.63 | 0.01 | 0.14 |
| Age | firebrick4 | 0.42 | 8.60 × 104 | −0.61 | 0.02 | 0.15 |
| Age | darkviolet | 0.42 | 1.20 × 103 | −0.57 | 0.03 | 0.21 |
| BMI | darkolivegreen2 | 0.34 | 1.10 × 104 | −0.58 | 0.02 | 0.63 |
| BMI | darkred | 0.39 | 1.90 × 1015 | −0.53 | 0.04 | 0.63 |
| Ancestry | palevioletred2 | 0.55 | 1.20 × 105 | 0.84 | 1.07 × 104 | 5.99 × 103 |
| Ancestry | blue2 | 0.51 | 2.60 × 105 | −0.66 | 6.91 × 103 | 0.18 |
| Smoking History | purple | 0.6 | <2.20 × 1016 | 0.67 | 6.00 × 103 | 0.34 |
| Smoking History | salmon2 | 0.29 | 0.03 | −0.53 | 0.04 | 0.56 |
| Sex | lightcoral | 0.45 | 2.50 × 104 | −0.71 | 3.01 × 103 | 0.17 |
| Sex | plum3 | 0.29 | 0.03 | 0.57 | 0.03 | 0.54 |
| Sex | lightcyan1 | 0.35 | 1.80 × 107 | 0.56 | 0.03 | 0.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devall, M.; Eaton, S.; Yoshida, C.; Powell, S.M.; Casey, G.; Li, L. Assessment of Colorectal Cancer Risk Factors through the Application of Network-Based Approaches in a Racially Diverse Cohort of Colon Organoid Stem Cells. Cancers 2023, 15, 3550. https://doi.org/10.3390/cancers15143550
Devall M, Eaton S, Yoshida C, Powell SM, Casey G, Li L. Assessment of Colorectal Cancer Risk Factors through the Application of Network-Based Approaches in a Racially Diverse Cohort of Colon Organoid Stem Cells. Cancers. 2023; 15(14):3550. https://doi.org/10.3390/cancers15143550
Chicago/Turabian StyleDevall, Matthew, Stephen Eaton, Cynthia Yoshida, Steven M. Powell, Graham Casey, and Li Li. 2023. "Assessment of Colorectal Cancer Risk Factors through the Application of Network-Based Approaches in a Racially Diverse Cohort of Colon Organoid Stem Cells" Cancers 15, no. 14: 3550. https://doi.org/10.3390/cancers15143550
APA StyleDevall, M., Eaton, S., Yoshida, C., Powell, S. M., Casey, G., & Li, L. (2023). Assessment of Colorectal Cancer Risk Factors through the Application of Network-Based Approaches in a Racially Diverse Cohort of Colon Organoid Stem Cells. Cancers, 15(14), 3550. https://doi.org/10.3390/cancers15143550