Simple Summary
Cell therapy approaches, including adoptive cell therapy with engineered T cells remains challenging in the clinical setting of solid tumors. Clinical results have not been encouraging so far, with a general lack of significant therapeutic response and presence of on-target off-tumor toxicity. For this reason, novel cell therapy programs are currently exploring (i) advanced therapy medicinal products capable of increasing T cell affinity or avidity; and (ii) cell therapy in combination with other therapeutic agents. Our review will focus on the current clinical research in this setting, which will likely play a role in improving cancer treatment outcomes in the near future.
Abstract
In the past years cancer treatments have drastically changed, mainly due to the development of immune checkpoint inhibitors capable of immune modulation in vivo, thus providing major clinical benefit in a number of malignancies. Simultaneously, considerable technical refinements have opened new prospects for the development of immune cell-based medicinal products and unprecedented success with chimeric antigen receptor (CAR)-T cells targeting B-cell hematologic malignancies has been obtained. However, T cell therapies introduced and performed in the field of solid tumors have produced so far only limited responses in selected patient populations. This standstill is attributable to the difficulty in identifying target antigens which are homogeneously expressed by all tumor cells while absent from normal tissues, and the limited T cell persistence and proliferation in a hostile tumor microenvironment that favors immune escape. Replicating the results observed in hematology is a major scientific challenge in solid tumors, and ongoing translational and clinical research is focused on obtaining insight into the mechanisms of tumor recognition and evasion, and how to improve the efficacy of cellular therapies, also combining them with immune checkpoint inhibitors or other agents targeting either the cancer cell or the tumor environment. This paper provides an overview of current adaptive T cell therapy approaches in solid tumors, the research performed to increase their efficacy and safety, and results from ongoing clinical trials.
1. Introduction
The past decades have seen an increasing activity among different kinds of immunotherapy in cancer patients. The first experiences were reported in the 1980s, and concerned the development of IL-2 for the treatment of melanoma and renal cell carcinoma [1,2].
Following up approaches to immunotherapy other than IL-2 did not have significant success, and adoptive T cell therapy (ACT), despite a number of early phase I-II trials, did not become part of standard care in the management of solid tumors. Multiple cancer mechanisms have been identified with the aim of escaping immune recognition, including the downregulation of tumor antigens, the generation of a microenvironment able to escape the immune system, and the secretion of cytokines and negative immune regulators thought to be able to silence immune effectors [3,4].
Nevertheless, considerable progress has been made in the field of cancer immune evasion and host–tumor interaction, which has developed immune checkpoint inhibitors [5] and antibodies against programmed death (PD) or programmed death-ligand (PD-L), with the aim of increasing the immune response against tumor cells [6].
On the other hand, a new scenario has opened for the development of immune-cell-based therapy from the success of chimeric antigen receptor (CAR)-T cells against hematologic malignancies [7,8,9]. CARs are chimeric immunoglobulin T cell receptor (TCR) molecules derived from transgenes encoding for single-chain variable fragments, which originate from antibodies capable of recognizing tumor-associated antigens (TAA) [10,11]. Activation signals induce CAR-T cells, leading to cytokine release and transcription factor expression, to promote T cell survival and function and cytotoxic activity against cancer cells [12].
Whereas CAR-T trials for the treatment of leukemia and lymphoma have shown durable clinical remission of the disease [9,13], CAR-T therapy targeting solid tumors has been disappointing. Among the hurdles identified has been the difficulty of recognizing target tumor antigens, and of trafficking T cells to the tumor site, avoiding a hostile tumor microenvironment. It has been suggested that control of advanced solid tumors will not be reached with a single therapeutic drug, but rather with combinations of either conventional or immunotherapy treatments [14]. Concerning this, immune checkpoint inhibitors, such as the CTLA4 antibodies and the anti PD1-PDL1, may have a role in increasing the response to ACT.
Here, we discuss ACT in solid tumors in clinical development, and consider the challenges plaguing the field, with the aim of succeeding in immune evasion to pave the way for effective tumor control.
2. Adaptive Cell Therapy with T Cells Generated and Expanded In Vitro
T cells have a critical role in immune surveillance for cancer, and the lymphocytes infiltrating the tumor‘s environment also have a favorable prognostic role.
First attempts were performed for advanced renal cell carcinoma and metastatic melanoma: recombinant human interleukin-2 (rhIL-2) used in these settings, was capable of favoring human T cell growth [1,2]. This encouraged the collection of T cells generated from patients’ lymphocytes and expanded in vitro with high-dose rhIL-2. The authors reported objective clinical responses, and provided the clinical use of T cells derived from in vitro culture.
2.1. Tumor-Infiltrating Lymphocytes (TILs)
Despite the encouraging responses reported in melanoma [1,2,15], T cells isolated from tumor biopsies (TILs) did not obtain the same results in other cancers.
Early TIL trials reported responses in about 50% of melanoma patients [15,16], and pretreatment with lymphodepleting chemotherapy led to improved TIL persistence in vivo, generating an immune environment to expand the T cells infused [16]. After the first trial that had seen the use of cyclophosphamide prior to T cell infusion [1], subsequent trials were designed with the aim of improving results, through the combining of fludarabine with cyclophosphamide, and testing different doses of total body irradiation [16,17].
Interesting data were reported in hepatocellular carcinoma (HCC) patients, who were randomized to receive adoptive immunotherapy (n = 76) or no adjuvant treatment, (n = 74) after surgical resection [18]. Cellular therapy decreased the frequency of recurrence by 18% compared with controls, and reduced the risk of recurrence by 41%. Therefore, it is encouraging that TILs could suppress tumor growth, supporting the issue of the major benefit of T cell therapy in patients with a minor burden of disease. Unfortunately, TILs became exhausted over time, because immune checkpoint inhibitor molecules, such as PD-1, are up-regulated in TILs isolated from HCC specimens [19]. Moreover, the isolation of an adequate number of TILs from HCC tissue, and the lack of immunogenic antigens, add additional barrier to using TILs as an effective therapy for HCC patients [19].
More recently, selected autologous TILs have shown activity in several other epithelial malignancies, such as cholangiocarcinoma, gynecological (both cervical and ovarian), lung, colorectal, and breast cancer [20,21,22,23,24,25]. Despite the growing interest in T cell therapy, the clinical use of TILs in advanced epithelial cancer has demonstrated limited responses [26]. An important factor may be the poor efficacy of TIL induced by the suppressive tumor microenvironment [27]. An alternative strategy for producing effective ACT therapies is to generate neoantigen-specific T cells de novo by means of TCR-engineered T cells [27]. Initial studies for screening neo-antigens utilized autologous tumor cell c-DNA libraries [28,29,30], but these approaches involved a laborious process for identifying neoepitopes. Subsequently, advances in next-generation sequencing allowed for the recognition of immunogenic tumor antigens though in silico analysis. Using a major histocompatibility complex-binding algorithm, putative T cell epitopes were identified and synthesized and then assessed for recognition by TILs [31].
Currently, there are many TIL ACT clinical studies recruiting patients with gynecological cancer (NCT04611126; 0310845), urothelial carcinoma (NCT04383067), breast cancer (NCT04111510), lung cancer (NCT04072263; NCT03853187; NCT02133196), biliary tract cancer (NCT03801083), and mesenchymal tumors (NCT03449108), as reported in Table 1.

Table 1.
Selected ongoing clinical trials of ATC therapy with T cells, recruiting advanced cancer patients other than melanoma (clinicaltrials.gov).
2.2. Tumor-Infiltrating Lymphocytes (TILs) in Combination with Immune Checkpoint Inhibitors (ICIs)
Among immunotherapy in solid tumors, immune checkpoint inhibitors (ICIs) have obtained interesting results. Melanoma is one of the malignancies that is most responsive to ICIs, and the first results concern anti CTLA-4 (Ipilimumab), which can break peripheral tolerance to self-tissues and induce an antitumor response, reporting a significant improvement in overall survival with a risk reduction of 32–35% [32].
PD-1 and PD-L1 stand out amongst the cancer immunotherapies that have been approved for clinical use, for different diseases [6,33,34,35]. The interaction between programmed death ligand-1 (PD-L1) and its receptor PD-1 has a role in immune self-tolerance and it is typical for the tumor micro-environment to be enriched in PD-L1 in order to promote tumor tolerance by the immune T-cells. Overexpression of PD-L1 has been demonstrated to inhibit the T cell-mediated antitumor response, favoring tumor escape [27]. The combination of different immune therapies has been the following step to improve the clinical response. In this way, the combination of Nivolumab and Ipilimumab in melanoma patients reported a significant increase in clinical efficacy, with a median progression-free survival of 11.5 months versus 6.9 in favor of the combo regimen [33]. While Ipilimumab can activate T cells, the anti-PD-1 antibodies block PD-1 on the activated T cells that are inhibited by PD-L1 expression of cancer cells [36].
In the tumor microenvironment, the high levels of PD-L1 are able to inhibit the activity of transferred TILs, blocking the PD-1 receptor. Therefore, one hypothesis for overcoming these hurdles is to merge TILs with anti PD-1 therapy. One interesting study in this setting concerned the combination of ICIs with TILs in ovarian cancer [37]. Although a modest clinical benefit was observed, interesting immunological observations were reported. The preconditioning with the anti-CTLA-4 antibody Ipilimumab was employed to increase tumor infiltration of tumor-reactive lymphocytes before tumor tissue harvest for TIL production, and data reported an increase in the success rate of ex vivo TIL expansion and an improved quality of the TIL product [37]. These findings confirmed previous animal data, suggesting that, if there is an endogenous antitumor immune response in the animals after tumor implantation, CTLA4 blockade could enhance that endogenous response, and induce tumor regression [36].
Also, the association between TIL and the PD-1 blockade could represent an efficacy scenario for testing TIL. Some experiences have documented a clinical response to TIL after relapse following anti PD-1 therapy, as reported for lung cancer [38]. PD-1 regulates effector T cells within the tissue and tumor microenvironment, enhancing the activity of effector T cells and NK cells. In addition, chronic antigen exposure, appearing in chronic disease as cancer, can manage high levels of PD-1 expression, which can promote a state of exhaustion [36]. As reported, PD-1 is also expressed in a large proportion of TILs from different tumor types [39], and increased levels of PD-1 expression may reflect an exhausted state. On the other hand, PD-1 ligands are commonly upregulated on the tumor cell surface of many different tumors. These findings provide the basis for a combination of TILs and anti-PD-1 immunotherapy, with the aim of enhancing antitumor functions in the tumor microenvironment. Combined TIL and anti-PD-1 therapy reported clinical benefit in the treatment of chemotherapy-resistant advanced osteosarcoma [40,41]. Notably, this strategy displayed an interesting antitumor effect and objective response, with 22 out of the 60 (36.6%) patients enrolled [40], displaying clinical tumor regression and reporting a median overall survival (mOS) of 13.6 months [40], whereas the mOS of patients with refractory disease receiving chemotherapy is no more than 6 months [42]. Similar results were obtained in 30 patients receiving TILs plus nivolumab, and reporting a 33% response rate [41]. To date, several studies have analyzed the use of single anti-PD-1 therapy against osteosarcoma, with an overall response rate of 5% [41,43]. These findings suggest that single-agent anti-PD-1 therapy may not be an effective treatment strategy, while the combination therapy may be a potential strategy for improving treatment of metastatic osteosarcoma. Moreover, a subpopulation of PD-1+T lymphocytes was noticed in the cultured TILs [40,41], suggesting that a PD-1 blockade may boost the cytotoxicity of TILs.
Up to the present date, several studies are ongoing with the aim of exploring TILs in combination with immune checkpoint inhibitors (Table 1).
2.3. Virus-Specific T Cells as ACT
The immune system has developed to recognize viral antigens; therefore, virally infected tumors such as cancers associated with the human papillomavirus (HPV) or Epstein–Barr virus (EBV) can trigger T cell immunity [44].
EBV-specific cytotoxic T cells (CTLs) expanded from the hematopoietic stem cell donor have been shown to prevent and treat EBV-associated lymphoproliferative disease [45,46,47]. Subsequently, autologous EBV-specific CTLs have been successfully employed in the treatment of EBV-associated cancers, such as nasopharyngeal carcinoma (NPC) [48,49,50,51]. These trials have so far documented an encouraging 20% objective response rate, with a 10% rate of complete response. With the aim of increasing clinical response and enhancing T cell survival, lymphodepleting chemotherapy has been administered, based on previous melanoma TILs therapy, without improving outcomes [50,51]. Authors hypothesized that the profound lymphopenia may have also depleted those factors able to promote the T cell effector function or expansion [51], and the presence of CD4+ or other immune T cells are also important for promoting and enhancing T cell effector activity against tumor cells [21,27].
HPV-TILs have also been documented as having a role in the regression of HPV-related cancers [20,52]. Among 29 patients treated with HPV-TILs, 25% reported an objective tumor response; moreover, 5 out of 18 patients (28%) in the cervical cancer cohort and 2 out of 11 (18%) in the non-cervical cancer cohort reported the same [52]. Interestingly, patients recording the highest frequency of HPV-reactive T cells in their peripheral blood (PB) after therapy reported objective tumor responses [20]. Moreover, responding patients who recorded elevated PB T cell reactivity against E6 and/or E7 reported prolonged repopulation with the infused HPV-reactive T cells [20].
Albeit the response rate reported is modest, it may reflect in part the mechanism of tumor escape within immunotherapy and the immunodominant role of nonviral tumor antigens targeted by T cells. To avoid this, a strategy may be to administer peripheral blood T cells that are genetically engineered ex vivo, to target an HPV oncoprotein (E5, E6) with a T cell receptor [52].
ICIs have also demonstrated their potential efficacy in cervical cancer, reporting an ORR in recurrent/advanced disease ranging from 12.2 to 26% [53]. Whereas increased expression of PD-L1 on cervical tumors is associated with a better response to ICIs [54], the response rate in patients with PD-L1-negative is relatively poor [55]. Therefore, anti-PD-1 therapy alone may not be an effective treatment in metastatic cervical cancer patients who had low expression of PD-L1. As is known, the effects of antiPD-1 therapy are mediated by TILs in the tumor microenvironment, and therefore a combination of anti-PD-1 therapy and TILs may provide major antitumor effects. An interesting study reported the results of 80 PD-L1-negative cervical cancer patients treated with a TIL and anti-PD-1 combination therapy; the objective response was observed in 20 patients (25%), and the authors speculated that TILs penetrated the tumor microenvironment and secreted factors, which induced the expression of PD-L1 in the tumor cells [55].
Table 2 summarizes the selected ongoing clinical trials of ACT in virus-associated cancers.
Table 2.
Selected ongoing clinical trials of ACT therapy in virus-associated cancers (clinicaltrials.gov).
2.4. T Cells Targeting Tumor Neoantigens
Unselected TILs also contain non-specific cells. Therefore, not all TIL products have potent antitumor effect, and patients with low levels of tumor antigens could have low levels of tumor-specific TILs. The identification and expansion of T cells with a specificity towards tumor antigens is an encouraging strategy for enhancing the chances of success of ACT. One of the most promising ways is to identify and expand the T cells with TCRs which are specific against tumor antigens. The genetic sequencing approach has permitted the recognition of neoantigens generated by gene rearrangements or mutations [56]. These neoantigens are expressed by all cells within a tumor, as well as across different tumor etiologies, and the identification of TCRs targeting these neoantigens may enable them to be utilized as specific reagents for TCR therapy [24]. Furthermore, neoantigens derived from somatic tumor mutations are likely to stimulate endogenous antitumor immunity, playing a critical role in maintaining durable responses after ACT [57]. Interesting results concerning the in vivo efficacy of T cells targeting tumor neoantigens have been provided in the setting of solid tumors. In particular, metastatic colon cancer patients, treated with TILs containing approximately 75% of CD8+ cells able to recognize a neoantigen derived from a hotspot mutation (G12D) in the KRAS gene, reported a regression of all metastatic lung lesions for almost 9 months [24]. TCRs against KRASG12D mutations restricted to HLA are now being tested in phase I–II of a clinical trial (NCT03745326).
Neoantigens have emerged as a major determinant of long-lasting tumor regressions in different immunotherapy trials; however, durable clinical responses are reported in only a minority of patients. One of the aspects that may negatively affect the treatment efficacy is the poor and heterogeneous expression of the antigens in the tumor load. Some strategies have been utilized to overcome these limitations: neoantigens should be identified in the patient’s tumor by tumor RNA sequencing. Secondly, bioinformatical approaches should be used to recognize the presence of the antigens in all cancer cells, and finally, targeting different neoantigens could minimize the tumor heterogeneity and also the chances of tumor resistance, due to the antigen loss or loss of specific MHC molecules [58].
3. Tumor Mutational Burden
Mutated genes can produce mutated proteins that can be identified by the immune system and presented on the cell surface by MHC molecules [58,59,60]. The tumor mutational burden (TMB) differs among distinct cancer types, and tumors with high TMB have high neoantigen expression, and therefore are predicted to be more immunogenic [61]. Previous experience has demonstrated that patients having a high TMB were more likely to respond to anti-PD1 therapy, in different solid tumor types [62]. Furthermore, tumors with deficiencies in the mismatch repair (MMR) pathway, have high levels of genetic mutations, and therefore high expression of neoantigens [63]. Clinical studies have reported that intestinal cancer patients with MMR deficiency are more likely to respond to ICIs as compared to patients with MMR-proficient tumors [64,65].
Tumors with high TMB and a consequently high expression of neoantigen have high levels of TIL clones among cancer cells, ensuring a sufficient number of tumor-reactive TILs in the final product. It is noteworthy that melanoma is well known for having a high TMB, and it has provided interesting results in TIL ACT.
Previous experiences have reported that TMB could be a biomarker of the response to immunotherapy in patients with different tumor etiologies treated with various types of immunotherapy [54]. Patients with a high TMB had a significantly higher response rate and longer progression-free survival than those with a lower TMB [54].
The association of improved anti-PD-1 and anti-PD-L1 clinical responses with high TMB strongly suggests also that neoantigens are an important target for antitumor immunity by PD-1 inhibition. Moreover, in the peripheral blood of advanced lung cancer patients who received anti-PD-1 therapy, there were detected neoantigen-specific CD8+ T cells, confirming the strong correlation between TMB and neoantigens [38].
4. Adaptive Cell Therapy with CAR-T: Progress and Pitfalls
Whilst TCR therapy requires HLA-restricted specificity and clinical efficacy depends on TCR affinity and the expression of MHC-antigen complexes on the tumor cells, CAR recognition of tumor target cells is HLA-unrestricted. CAR-T cells consist of an antigen-binding domain, derived from a single-chain variable fragment of a monoclonal antibody directed against a tumor-specific antigen, a hinge region, a trans-membrane domain, and the TCR intracellular domain, able to activate T cells [4]. The encouraging results obtained in hematological malignancies [66,67,68] have paved the way for the application of this approach in solid tumors, unfortunately with unsatisfactory results to date.
Despite ameliorating CAR construction, poor in vivo expansion and duration were reported in solid tumors [69,70,71,72]. Second- and third-generation CARs have included one and two costimulatory domains, respectively, aiming to increase cytotoxicity and cytokine production, and improve the proliferation and persistence of CAR-T cells [73,74]. More recently, fourth-generation CAR-T, which incorporate additional stimulatory domains, have been reported [75]. Fourth-generation CAR-T cells share the same construct as second-generation CARs, but include a protein such as interleukin-12 (IL-12) that is constitutively or inducibly expressed upon CAR activation, whilst fifth-generation CAR-T cells are based on the second generation of CARs, with the addition of a truncated cytoplasmic IL-2 receptor β-chain domain with a binding site for the transcription factor STAT3 [76].
Notwithstanding the poor evidence of the efficacy of CAR-T cells in solid tumors, interesting results have been published recently in the treatment of diffuse midline glioma with high expression of GD2 [77]. Along the same lines, a few weeks ago, the authors of [78] described noteworthy results in the treatment of high-risk advanced neuroblastoma with third-generation GD2-CAR-T cells expressing the inducible caspase 9 suicide gene (GD2-CART01). Among 27 heavily pretreated children, 9 patients reported a complete response and 8 patients had a partial response, with an overall response rate of 63%, and a 3-year overall survival and event-free survival of 60% and 36%, respectively [78]. This longer time for disease progression may be a consequence of a persistence of CAR-T cells in vivo, due to the incorporation of two costimulatory domains in the construct of GD2-CART01. The interesting findings reported in this study represent a challenge for immunotherapy in the treatment of solid tumors.
With the aim of increasing the efficacy of CAR-T, some authors have documented the fact that the spatial distance between CAR-T and the target antigens may be equally important for T cell signaling: CAR-T with a shortened extracellular spacer conferred superior recognition of tumor cells compared to the same scFv with a longer spacer [79]. Thus, the design of CAR-T for novel targets should factor in the location of the target epitope and customize the spacer length to optimize CAR-T cell signaling [79].
4.1. CAR-T Interactions with Tumor Microenvironment (TME)
The tumor microenvironment (TME) is a complex network that comprehends the extracellular matrix and several nonmalignant cells. Mechanisms involved in the immunosuppressive nature of TME include (i) physical barriers to tumor invasion by immune cells, (ii) upregulated checkpoint ligands, (iii) a pro-tumor stromal niche, (iv) immunosuppressive soluble factors, and (v) the selected expression of chemokines to recruit immunosuppressive cells such as regulatory T lymphocytes, M2 macrophages, myeloid-derived suppressor cells (MDSCs) and cancer-associated fibroblast (CAFs) (Figure 1) [75].
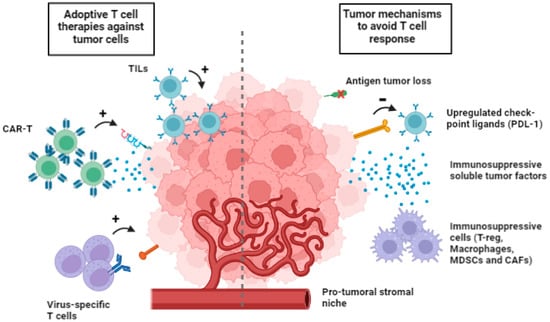
Figure 1.
Multiple cell types available for adoptive cell therapies (on the left) and tumor mechanism to avoid T cell response (on the right). Tumor evasion includes loss of tumor neoantigens, upregulation of checkpoint ligands, immunosuppressive soluble tumor factors, immunosuppressive cells, such as T-reg, macrophages, myeloid-derived suppressor cells (MDSCs) and cancer-associated fibroblast (CAFs).
The efficacy of CAR-T cell therapy in solid cancers is drastically hampered by poor immune cell infiltration [80]. Tumor cells are able to regulate chemokines, contributing to the poor recruitment of CAR-T cells. Engineering CAR-T cells with receptors for chemokines that are overexpressed in the TME has been a challenge. Previous studies reported CAR-T cells engineered to co-express chemokine receptors (such as CCL2 or CCR4) able to enhance T cell homing and improve migration toward tumor cells [81,82]. Once CAR-T cells reach the tumor site, the high density of the extracellular matrix (ECM) associated with solid tumors is a hurdle to overcome. Because of this, some authors have engineered CAR-T cells to express heparanase, which is able to degrade ECM [83].
MDSCs contribute to creating an immunosuppressive environment enhancing the expansion and the activation of T-regs, CD4+ T lymphocytes that can inhibit the function of tumor-specific T lymphocytes producing immunosuppressive cytokines (IL-10 and TGF-β), expressing negative costimulatory molecules (CTLA-4, PD-1 and PD-L1) and consuming IL-2, a cytokine essential for the proliferation of cytotoxic T lymphocytes [84,85].
Lymphodepleting therapy, preceding CAR-T cell infusion, aims to eliminate the recipient’s lymphocyte pool, including T-regs, resulting in increased proliferation and expansion of reinfused T-lymphocytes and inhibition of the tumor immunotolerance mechanism [86].
CAFs are a great part of the tumor stroma, and CXCL12 secreted by CAFs plays an important role in ‘protecting’ tumor cells from immune attack and keeping T cells out of the tumor microenvironment. The fibroblast activation protein (FAP) is highly expressed in CAFs in over 90% of solid tumors, making this protein a potential therapeutic target. Encouraging results have emerged from several studies conducted in vitro and in vivo on CAR-T cells, directed against FAP [87]. Moreover, the aberrant tumor vasculature causes interstitial hypertension that prevents extravasation and a hypoxic microenvironment, especially in the central part of the tumor. Thus, normalizing the tumor vasculature may be beneficial, and anti-vascular endothelial growth factor receptor (VEGFR) CAR-T cells may play a role in the inhibition of the tumor growth [88]. Considering the metabolic barriers, it is worth noticing that the particular anatomical structure of solid tumors generates hostile hypoxia, able to hamper the expansion of CAR-T cells. Strategies to favor CAR-T response in the hypoxic TME are under investigation [89].
Other strategies are studied to improve the efficacy of CAR-T cells. Some cytokines play a role in enhancing CAR-T cell proliferation, survival, and effector function in the immunosuppressive TME. Most cytokines studied in this setting are IL2, IL7, IL15 or IL21, and their association with CAR-T cells has been described in vivo [90].
4.2. CAR-T: How to Increase Endogenous Immune Response
It is well known that tumor cells are able to escape subsets of leukocytes, including CAR-T cells, and to induce immunosuppressive phenotypes [91]. Recently, T cells engineered to secrete Fms-like tyrosine kinase 3 ligand (Flt3L), a hematopoietic cell growth factor, have been able to promote intratumoral proliferation of dendritic cells (DCs), and particularly to induce immunity against tumor cells [92].
Furthermore, CAR-T cells have also been engineered to express different immune-modulatory proteins. For example, CD40L is expressed on T cells, and its interaction with its receptor can lead to the activation of APCs and the apoptosis of CD40+ tumor cells [75]. CD40L expression on CAR-T cells resulted in elevated surface expression of costimulatory molecules, adhesion molecules, HLA molecules and Fas death receptor on CD40+ tumor cells, thus increasing their immunogenicity [92]. These T cells also induced the secretion of proinflammatory IL-12 and showed enhanced antitumor activity in vitro and in vivo [93]. Finally, CAR-T cells can be engineered in molecules able to engage tumor cells by endogenous, non-engineered T cells [94].
“Exhaustion” is a state of functional hyporesponsiveness of T cells during chronic infections, which arises when pathogens are not quickly eliminated, but rather persist. It has been suggested that T cells, and also CAR- and TCR-T cells, exhibit an “exhausted” state in the contest of solid tumors, similar to chronic infections, due to the high antigen load and the immunosuppressive factors in the TME. Recent experiences have documented an enrichment of exhaustion genes and TCR pathways, suggesting that the T cell unresponsiveness is also driven by antigen chronicity and duration of TCR stimulation [95]. Thus, tumor-induced T cell dysfunction is driven by the presence and persistence of tumor antigens, and although microenvironmental factors may contribute, they are not sufficient to induce the dysfunctional phenotype [95,96]. Potential transcription factors able to control the dysregulated gene expression signature are also involved [96]. Therefore, strategies to overcome this antigen-driven dysfunction may be required to improve cancer immunotherapy.
Table 3 summarizes selected ongoing clinical trials of CAR-T therapy.
Table 3.
Selected ongoing clinical trials of CAR-T therapy in solid tumors (clinicaltrials.gov).
5. Conclusions
Recent cell engineering strategies have made great strides in ameliorating the antitumor function of T cells, increasing tumor-targeting specificity, preventing tumor escape, and modifying the TME. Furthermore, the progress in next-generation sequencing and other analyses could enhance our ability to understand these complex interactions and to create the next generation of T cell therapy.
Author Contributions
Conceptualization: S.S., C.C., D.A., P.P. and P.C.; methodology: S.S., C.C., D.A., M.M., G.G., S.B. (Sabrina Borgetto) and S.B. (Sabrina Basso); validation: S.S., C.C., D.A., M.M., G.G., S.B. (Sabrina Borgetto), J.B., C.P., S.B. (Sabrina Basso), P.C. and P.P.; formal analysis: S.S., C.C., S.B. (Sabrina Basso), J.B., C.P., P.C. and P.P.; investigation: S.S., C.C., D.A., M.M., S.B. (Sabrina Borgetto), J.B., C.P. and S.B. (Sabrina Basso); data curation: C.C., D.A., M.M., G.G., S.B. (Sabrina Borgetto) and S.B. (Sabrina Basso); writing—original draft preparation: S.S., D.A., M.M., S.B. (Sabrina Borgetto), C.P. and S.B. (Sabrina Basso); writing—review and editing: S.S., P.C. and P.P.; supervision: P.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukine-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Rosenberg, S.A. Successful immunotherapy of natural killer-resistant established pulmonary melanoma metastases by intravenous adoptive transfer of syngeneic lymphocytes activated in vitro by interleukin 2. J. Exp. Med. 1984, 159, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 2015, 5, e1086862. [Google Scholar] [CrossRef] [PubMed]
- Comoli, P.; Chabannon, C.; Koehl, U.; Lanza, F.; Urbano-Ispizua, A.; Hudecek, M.; Ruggeri, A.; Secondino, S.; Bonini, C.; Pedrazzoli, P. Development of adaptive immune effector therapies in solid tumors. Ann. Oncol. 2019, 30, 1740–1750. [Google Scholar] [CrossRef]
- Eggermont, A.M.M. Advances in systemic treatment of melanoma. Ann. Oncol. 2010, 21 (Suppl. 7), vi339–vi344. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Topalian, S.L.; Weiner, G.P.; Pardoll, D.M. Cancer immunotherapy comes of age. J. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 317, 1507–1517. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell-receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef]
- Dotti, G.; Savoldo, B.; Brenner, M. Fifteen years of gene therapy based on chimeric antigen receptors: “are we nearly there yet?”. Hum. Gene Ther. 2009, 20, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Apetoh, L.; Ladoire, S.; Coukos, G.; Ghiringhelli, F. Combining immunotherapy and anticancer agents: The right path to achieve cancer cure? Ann. Oncol. 2015, 26, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Sekine, T.; Makuuchi, M.; Yamasaki, S.; Kosuge, T.; Yamamoto, J.; Shimada, K.; Sakamoto, M.; Hirohashi, S.; Ohashi, Y.; et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: A randomized trial. Lancet 2000, 356, 802–807. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, J.; Li, H.Y.; Wang, Z.H.; Wu, J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188441. [Google Scholar] [CrossRef]
- Stevanović, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus targeted tumor-infiltratin T cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based in mutation-specific CD4+T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistent metatstatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.-A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8 (+) T cells in immunotherapy naïve ovarian cancer. Nat. Commun. 2018, 9, 1092. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. Cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell therapy with TILs: Training and taming T cells to fight cancer. Front. Imuunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Huang, J.; El-Gamil, M.; Dudley, M.E.; Li, Y.F.; Rosenberg, S.A.; Robbins, P.F. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppror gene locus and a mutated HLA Class I gene product. J. Immunol. 2004, 172, 6057–6064. [Google Scholar] [CrossRef]
- Zhou, J.; Dudley, M.E.; Rosenberg, S.A.; Robbins, P.F. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother. 2005, 28, 53–62. [Google Scholar] [CrossRef]
- Li, Q.; Din, Z.Y. The ways of isolating neoantigens-specific T cells. Front. Oncol. 2020, 10, 1347. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Lee, S.J.; Hodi, F.S.; Rao, U.N.M.; Cohen, G.I.; Hamid, O.; Hutchins, L.F.; Sosman, J.A.; Kluger, H.M.; Eroglu, Z.; et al. Phase III study of adjuvant Ipilimumab (3 or 10 mg/kg) versus high-dose Interferon alfa-2B for resected high-risk melanoma: North American Itergroup E1609. J. Clin. Oncol. 2010, 38, 567–576. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Five-year survival with combined Nivolumab and Ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Pardol, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- Caushi, J.X.; Zhang, J.; Ji, Z.; Vaghasia, A.; Zhang, B.; Hsiue, E.H.-C.; Mog, B.J.; Hou, W.; Justesen, S.; Blosser, R.; et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 2021, 596, 126–132. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, J.; Duan, C.; Liu, Y. Retrospective analysis of adoptive TIL therapy plus anti-PD1 therapy in patients with chemotherapy-resistant metastatic osteosarcoma. J. Immunol. Res. 2020, 7890985. [Google Scholar] [CrossRef]
- Wang, C.; Li, M.; Wei, R.; Wu, J. Adoptive transfer of TILs plus anti-PD1 therapy: An alternative combination therapy for treating metastatic osteosarcoma. J. Bone Oncol. 2020, 25, 100332. [Google Scholar] [CrossRef]
- Chou, A.J.; Gorlick, R. Chemotherapy resistance in osteosarcoma: Current challenges and future directions. Expert Rev. Anticancer Ther. 2006, 6, 1075–1085. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcomas (SARC028): A multicenter, two-cohort, single arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Gao, P.; Lazare, C.; Cao, C.; Meng, Y.; Wu, P.; Zhi, W.; Lin, S.; Wei, J.; Huang, X.; Xi, L.; et al. Immune checkpoint inhibitors in the treatment of virus-associated cancers. J. Hematol. Oncol. 2019, 12, 58. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of gene-modified virus-specific T lymphocytes Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.-F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–936. [Google Scholar] [CrossRef]
- Comoli, P.; Basso, S.; Zecca, M.; Pagliara, D.; Baldanti, F.; Bernardo, M.E.; Barberi, W.; Moretta, A.; Labirio, M.; Paulli, M.; et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am. J. Transplant. 2007, 7, 1648–1655. [Google Scholar] [CrossRef]
- Straathof, K.C.M.; Bollard, C.M.; Popat, U.; Huls, M.H.; Lopez, T.; Morriss, M.C.; Gresik, M.V.; Gee, A.P.; Russell, H.V.; Brenner, M.K.; et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus-specific T Lymphocytes. Blood 2005, 105, 1898–1905. [Google Scholar] [CrossRef]
- Comoli, P.; Pedrazzoli, P.; Maccario, R.; Basso, S.; Carminati, O.; Labirio, M.; Schiavo, R.; Secondino, S.; Frasson, C.; Perotti, C.; et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein-Barr virus-targeted cytotoxic T lymphocytes. J. Clin. Oncol. 2005, 23, 8942–8949. [Google Scholar] [CrossRef]
- Louis, C.U.; Straathof, K.; Bollard, C.M.; Gerken, C.; Huls, M.H.; Gresik, M.V.; Wu, M.-F.; Weiss, H.L.; Gee, A.P.; Brenner, M.K.; et al. Enhancing the in-vivo expansion of adoptively transferred EBV-specific CTL with lymphodepleting CD45 monoclonal antibodies in NPC patients. Blood 2009, 113, 2422–2450. [Google Scholar] [CrossRef]
- Secondino, S.; Zecca, M.; Licitra, L.; Gurrado, A.; Schiavetto, I.; Bossi, P.; Locati, L.; Schiavo, R.; Basso, S.; Baldanti, F.; et al. T-cell theapy for EBV-associated nasopharyngeal carcinoma: Preparative lymphodepleting chemotherapy does not improve clinical results. Ann. Oncol. 2012, 23, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A phase II study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, L.; Tong, R.; Yang, F.; Yin, L.; Li, M.; You, L.; Xue, J.; Lu, Y. PD-1/PD-L1 inhibitors in cervical cancer. Front. Pharmacol. 2019, 10, 65. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Yin, H.; Guo, W.; Sun, X.; Li, R.; Feng, C.; Tan, Y. TILs and anti-PD1 therapy: An alternative combination therapy for PDL1 negative metastatic cervical cancer. J. Immunol. Res. 2020, 2020, 8345235. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer ant the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumor. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Schumaker, T.N.; Scheper, W.; Kvistbong, P. Cancer neoantigens. Ann. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Schumaker, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumor mutational burden with outcomes in patients with advanced solid tumor treated with Pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch repair pathway, genome instability and cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in microsatellite instability-high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Fielding, A.K.; Richards, S.M.; Chopra, R.; Lazarus, H.M.; Litzow, M.R.; Buck, G.; Durrant, I.J.; Luger, S.M.; Marks, D.I.; Franklin, I.M.; et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 2007, 109, 944–950. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.J.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, 22–24. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, A.; Maus, M.V. Making CAR T cells a solid option for solid tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Filies, D.B. Molecular mechanism of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Haynes, N.M.; Trapani, J.A.; Teng, M.W.L.; Jackson, J.T.; Cerruti, L.; Jane, S.M.; Kershaw, M.H.; Smyth, M.J.; Darcy, P.K. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002, 100, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 2020, 4, 473–488. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Abdela, S.M. CART T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhanced tumor localization and tumor eradication by targeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Muranski, P.; Boni, A.; Wrzesinski, C.; Citrin, D.E.; Rosenberg, S.A.; Childs, R.; Restifo, N.P. Increased intensity lymphodepletion and adoptive immunotherapy—How far can we go? Nat. Clin. Pract. Oncol. 2006, 3, 668–681. [Google Scholar] [CrossRef]
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141. [Google Scholar] [CrossRef]
- Boccalatte, F.; Mina, R.; Aroldi, A.; Leone, S.; Suryadevara, C.M.; Placantonakis, D.G.; Bruno, B. Advances and hurdles in CAR T cell immune therapy for solid tumors. Cancers 2022, 14, 5108. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3-specific CAR T cells co-expressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Morgan, M.A.; Schambach, A. Engineering CAR-T cells for improved function against solid tumors. Front. Immunol. 2018, 29, 2493. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T. ells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; Seinstra, B.A.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting Bites circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program intiated early during tumorigenesis. Immunology 2016, 45, 389–401. [Google Scholar] [CrossRef]
- Good, C.R.; Aznar, M.A.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, A.K.; Ma, Y.; et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021, 184, 6081–6100. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).