
SELENOF Controls Proliferation and Cell Death in Breast-Derived Immortalized and Cancer Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines, Culture Conditions and Treatments
2.3. Cell Viability
2.4. FACS Analysis
2.5. RT-Quantitative PCR (QPCR)
2.6. 3D Acinar Assay
2.7. Western Blot
2.8. Transmission Electron Microscopy (TEM) Imaging
2.9. Statistical Analysis
3. Results
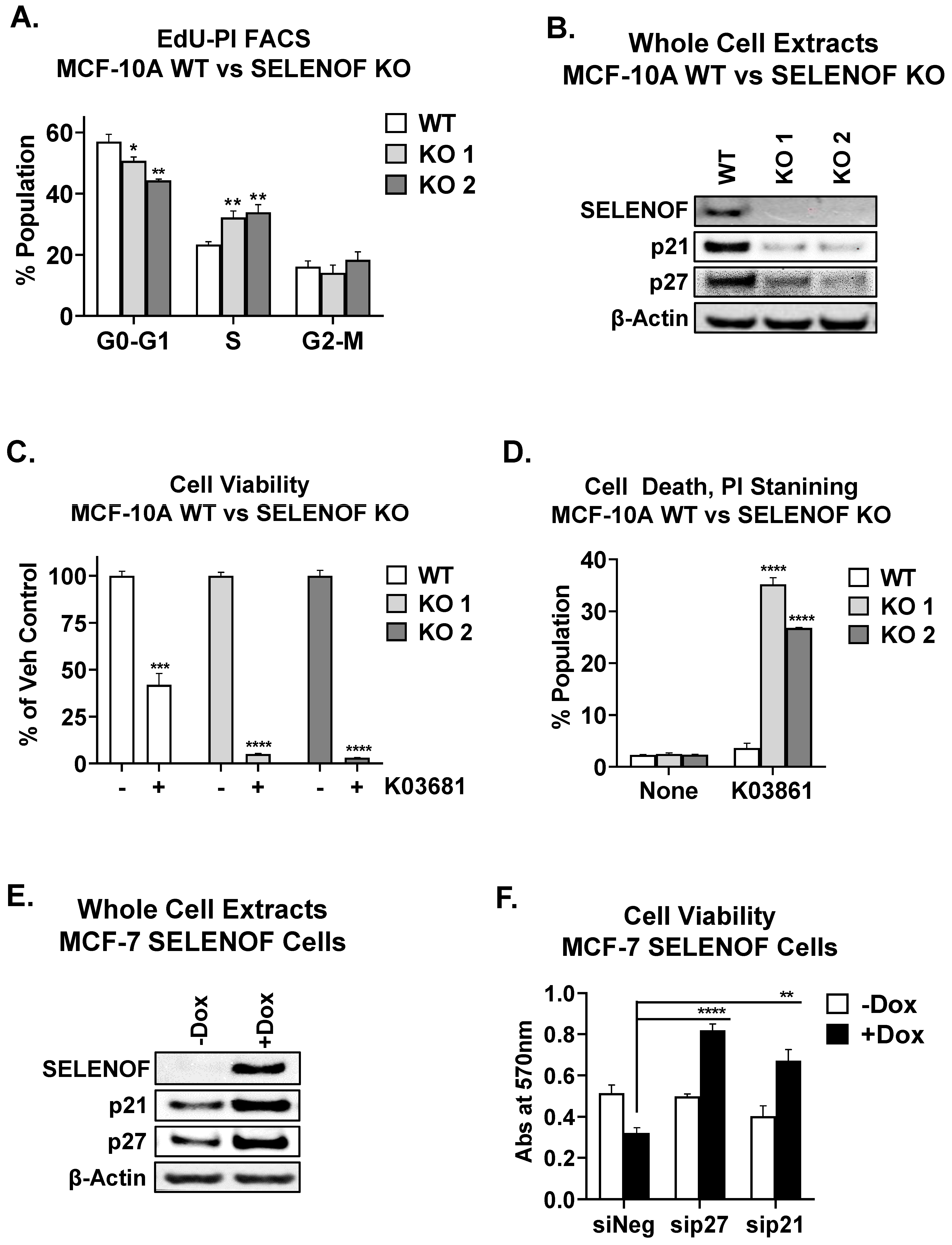
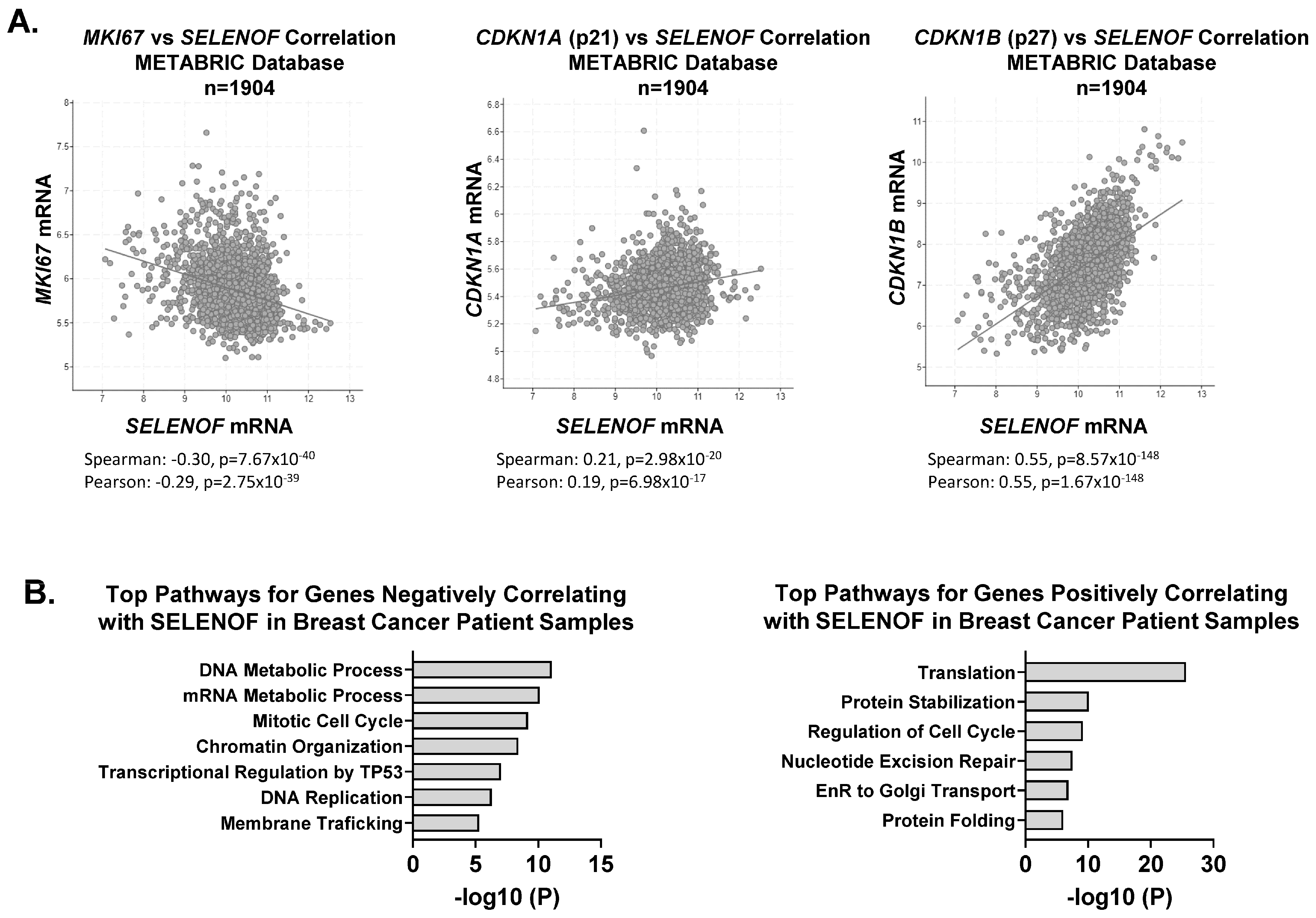
4. SELENOF Controls Proliferation and Cell Death via p21 and p27
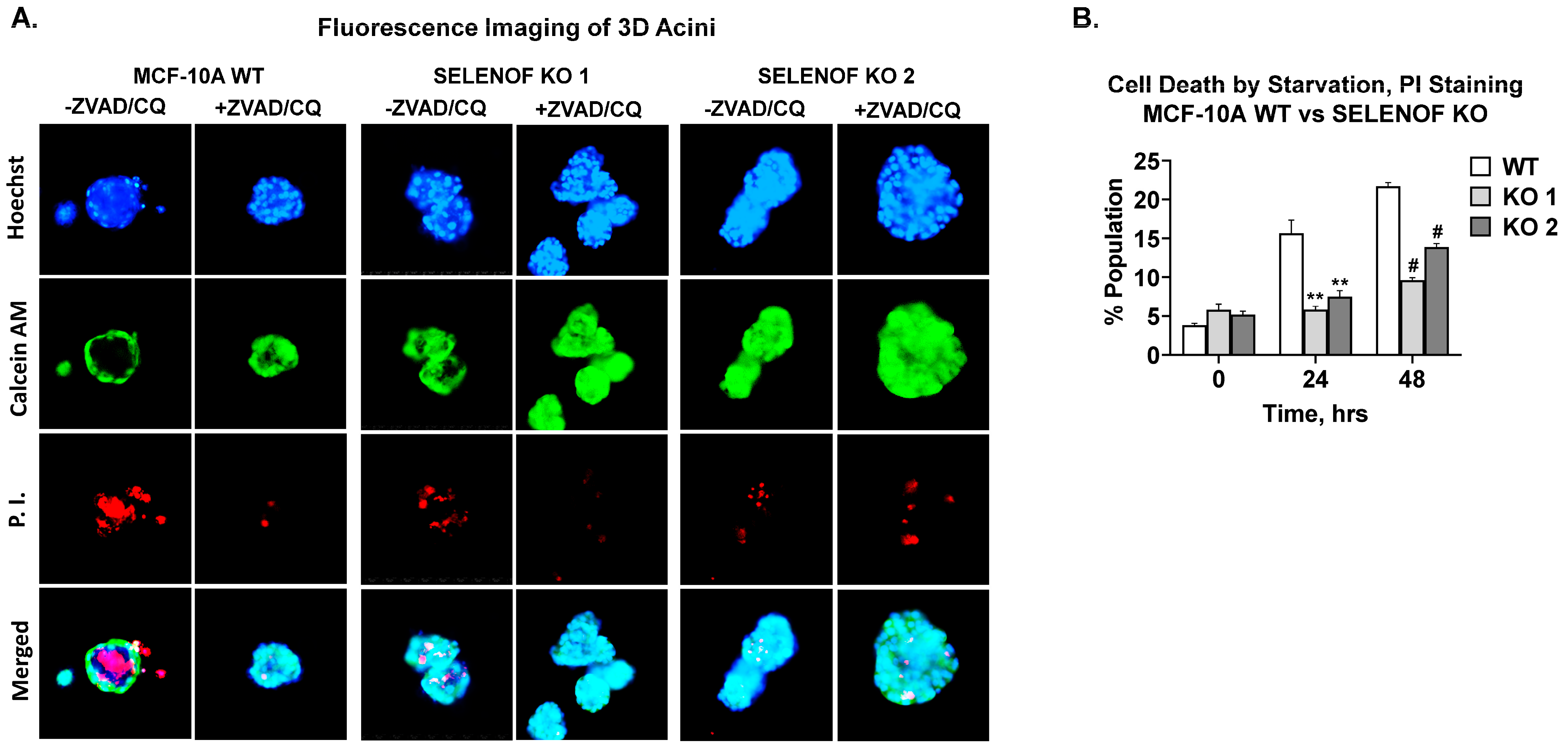
5. SELENOF Is Required for Lumen Formation in 3D Acini
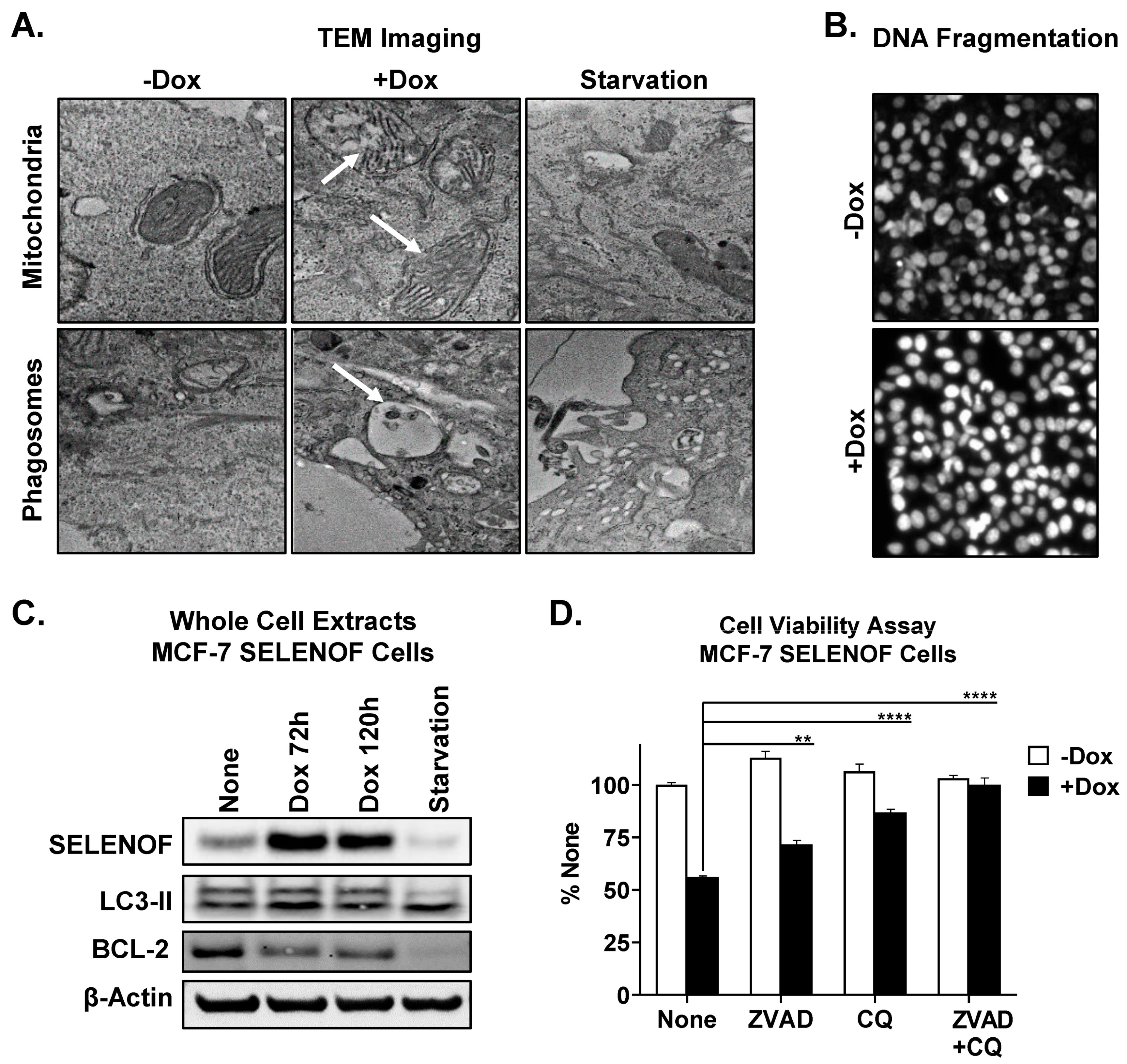
6. SELENOF Overexpression Induces Apoptosis and Autophagy
7. Discussion
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SELENOF (SEP15) | selenoprotein F |
| KO | knockout |
| 2D or 3D | two or three-dimensional |
| Dox | doxycycline |
| EdU | 5-ethynyl-2′-deoxyuridine |
| PI | propidium iodide |
References
- Gladyshev, V.N.; Jeang, K.-T.; Wootton, J.C.; Hatfield, D.L. A new human selenium-containing protein. Purification, characterization and cDNA sequence. J. Biol. Chem. 1998, 273, 8910–8915. [Google Scholar] [CrossRef] [Green Version]
- Flowers, B.; Poles, A.; Kastrati, I. Selenium and breast cancer—An update of clinical and epidemiological data. Arch. Biochem. Biophys. 2022, 732, 109465. [Google Scholar] [CrossRef] [PubMed]
- Diamond, A.M. Selenoproteins of the Human Prostate: Unusual Properties and Role in Cancer Etiology. Biol. Trace Elem. Res. 2019, 192, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.J.; Korotkov, K.V.; Mehta, R.; Hatfield, D.L.; Rotimi, C.N.; Luke, A.; Prewitt, T.E.; Cooper, R.S.; Stock, W.; Vokes, E.E.; et al. Distribution and functional consequences of nucleotide polymorphisms in the 3′-untranslated region of the human Sep15 gene. Cancer Res. 2001, 61, 2307–2310. [Google Scholar]
- Nagai, H.; Negrini, M.; Carter, S.L.; Gillum, D.R.; Rosenberg, A.L.; Schwartz, G.F.; Croce, C.M. Detection and cloning of a common region of loss of heterozygosity at chromosome 1p in breast cancer. Cancer Res. 1995, 55, 1752–1757. [Google Scholar]
- Zigrossi, A.; Hong, L.K.; Ekyalongo, R.C.; Cruz-Alvarez, C.; Gornick, E.; Diamond, A.M.; Kastrati, I. SELENOF is a new tumor suppressor in breast cancer. Oncogene 2022, 41, 1263–1268. [Google Scholar] [CrossRef]
- Korotkov, K.V.; Kumaraswamy, E.; Zhou, Y.; Hatfield, D.L.; Gladyshev, V.N. Association between the 15-kDa selenoprotein and UDP-glucose:glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 2001, 276, 15330–15336. [Google Scholar] [CrossRef] [Green Version]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, A.D.; Labunskyy, V.M.; Fomenko, D.E.; Arac, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar]
- Bang, J.; Huh, J.H.; Na, J.W.; Lu, Q.; Carlson, B.A.; Tobe, R.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L.; Lee, B.J. Cell Proliferation and Motility Are Inhibited by G1 Phase Arrest in 15-kDa Selenoprotein-Deficient Chang Liver Cells. Mol. Cells 2015, 38, 457–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, S.H.; Everley, R.A.; Schildberg, F.A.; Lee, S.G.; Orsi, A.; Barbati, Z.R.; Karatepe, K.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R.; et al. Role of Selenof as a Gatekeeper of Secreted Disulfide-Rich Glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar]
- Hong, L.K.; Kadkol, S.; Sverdlov, M.; Kastrati, I.; Elhodaky, M.; Deaton, R.; Sfanos, K.S.; Wang, H.; Liu, L.; Diamond, A.M. Loss of SELENOF Induces the Transformed Phenotype in Human Immortalized Prostate Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 12040. [Google Scholar] [PubMed]
- Zheng, X.; Ren, B.; Wang, H.; Huang, R.; Zhou, J.; Liu, H.; Tian, J.; Huang, K. Hepatic proteomic analysis of selenoprotein F knockout mice by iTRAQ: An implication for the roles of selenoprotein F in metabolism and diseases. J. Proteom. 2020, 215, 103653. [Google Scholar]
- Zheng, X.; Ren, B.; Li, X.; Yan, H.; Xie, Q.; Liu, H.; Zhou, J.; Tian, J.; Huang, K. Selenoprotein F knockout leads to glucose and lipid metabolism disorders in mice. J. Biol. Inorg. Chem. 2020, 25, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.; Zhou, J.; Liu, H. Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis. Antioxidants 2022, 11, 2105. [Google Scholar] [CrossRef]
- Zhang, D.G.; Zhao, T.; Xu, X.J.; Xu, Y.H.; Wei, X.L.; Jiang, M.; Luo, Z. Selenoprotein F (SELENOF)-mediated AKT1-FOXO3a-PYGL axis contributes to selenium supranutrition-induced glycogenolysis and lipogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194814. [Google Scholar] [CrossRef]
- Flowers, B.; Bochnacka, O.; Poles, A.; Diamond, A.M.; Kastrati, I. Distinct Roles of SELENOF in Different Human Cancers. Biomolecules 2023, 13, 486. [Google Scholar]
- Tang, J.; Wang, F.; Cheng, G.; Si, S.; Sun, X.; Han, J.; Yu, H.; Zhang, W.; Lv, Q.; Wei, J.F.; et al. Wilms’ tumor 1-associating protein promotes renal cell carcinoma proliferation by regulating CDK2 mRNA stability. J. Exp. Clin. Cancer Res. 2018, 37, 40. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.S.; Wang, P.; Yamabe, N.; Fukui, M.; Jay, T.; Zhu, B.T. Docosahexaenoic acid induces apoptosis in MCF-7 cells in vitro and in vivo via reactive oxygen species formation and caspase 8 activation. PLoS ONE 2010, 5, e10296. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar]
- Da Silva, L.; Bray, J.K.; Bulut, G.; Jiang, J.; Schmittgen, T.D. Method for improved integrity of RNA isolated from Matrigel cultures. MethodsX 2020, 7, 100966. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Puleo, J.; Polyak, K. The MCF10 Model of Breast Tumor Progression. Cancer Res. 2021, 81, 4183–4185. [Google Scholar] [CrossRef]
- Petersen, O.W.; Ronnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064–9068. [Google Scholar] [CrossRef]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, K.R.; Reginato, M.; Debnath, J.; Queenan, B.; Brugge, J.S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 3438–3443. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, J.; Ma, C.X. Sequencing Endocrine Therapy for Metastatic Breast Cancer: What Do We Do After Disease Progression on a CDK4/6 Inhibitor? Curr. Oncol. Rep. 2020, 22, 57. [Google Scholar] [CrossRef]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthuswamy, S.K.; Li, D.; Lelievre, S.; Bissell, M.J.; Brugge, J.S. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 2001, 3, 785–792. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekyalongo, R.C.; Flowers, B.; Sharma, T.; Zigrossi, A.; Zhang, A.; Quintanilla-Arteaga, A.; Singh, K.; Kastrati, I. SELENOF Controls Proliferation and Cell Death in Breast-Derived Immortalized and Cancer Cells. Cancers 2023, 15, 3671. https://doi.org/10.3390/cancers15143671
Ekyalongo RC, Flowers B, Sharma T, Zigrossi A, Zhang A, Quintanilla-Arteaga A, Singh K, Kastrati I. SELENOF Controls Proliferation and Cell Death in Breast-Derived Immortalized and Cancer Cells. Cancers. 2023; 15(14):3671. https://doi.org/10.3390/cancers15143671
Chicago/Turabian StyleEkyalongo, Roudy C., Brenna Flowers, Tanu Sharma, Alexandra Zigrossi, An Zhang, Anaisa Quintanilla-Arteaga, Kanishka Singh, and Irida Kastrati. 2023. "SELENOF Controls Proliferation and Cell Death in Breast-Derived Immortalized and Cancer Cells" Cancers 15, no. 14: 3671. https://doi.org/10.3390/cancers15143671
APA StyleEkyalongo, R. C., Flowers, B., Sharma, T., Zigrossi, A., Zhang, A., Quintanilla-Arteaga, A., Singh, K., & Kastrati, I. (2023). SELENOF Controls Proliferation and Cell Death in Breast-Derived Immortalized and Cancer Cells. Cancers, 15(14), 3671. https://doi.org/10.3390/cancers15143671