
Vasculogenic Mimicry Occurs at Low Levels in Primary and Recurrent Glioblastoma
 , and
, and
Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Tissue Samples
2.2. Immunohistochemistry
2.3. Tumour Vessel Quantification
2.4. PSMA Quantification
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics
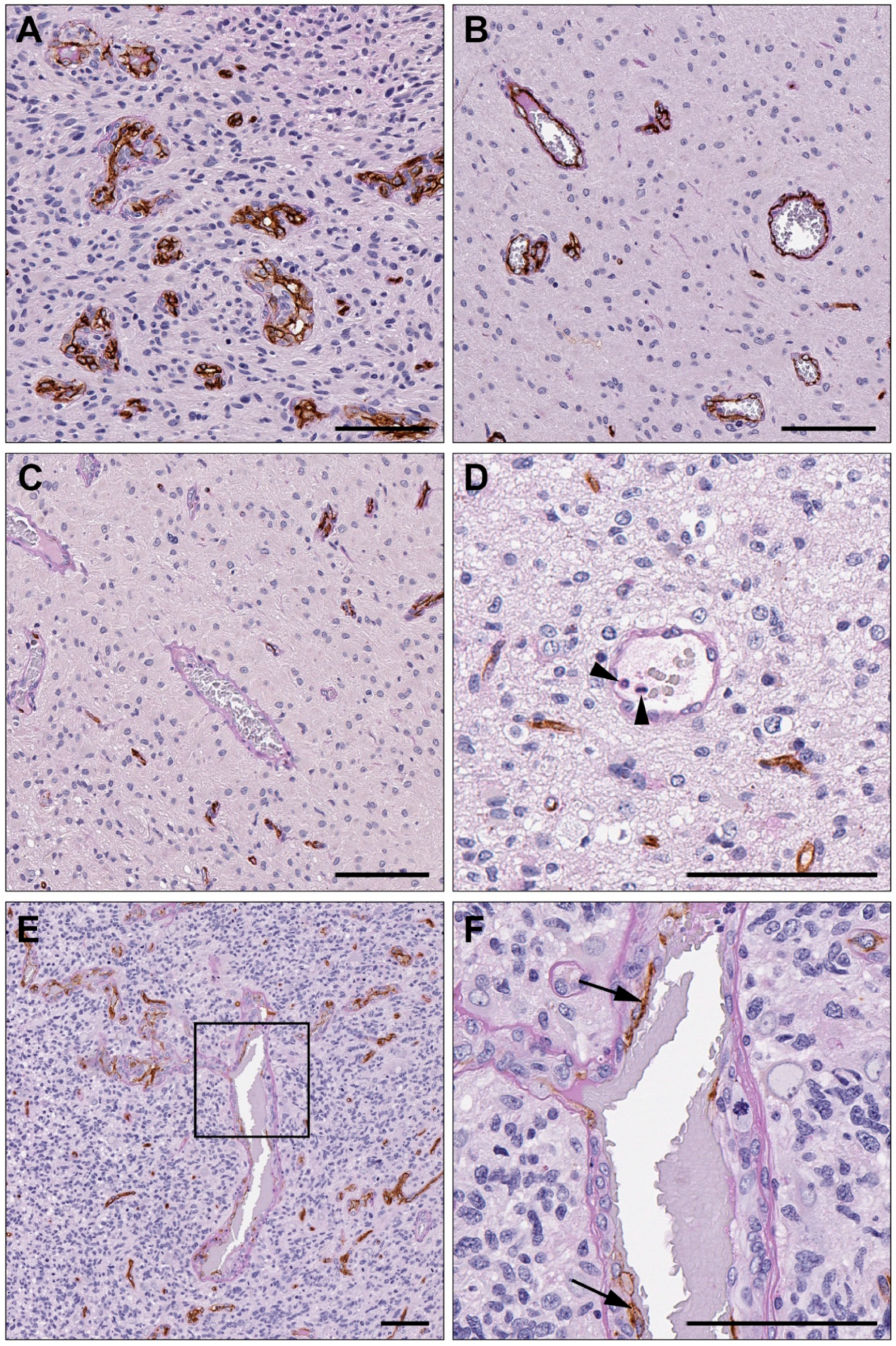
3.2. Vasculogenic Mimicry Is Present in Recurrent Glioblastoma
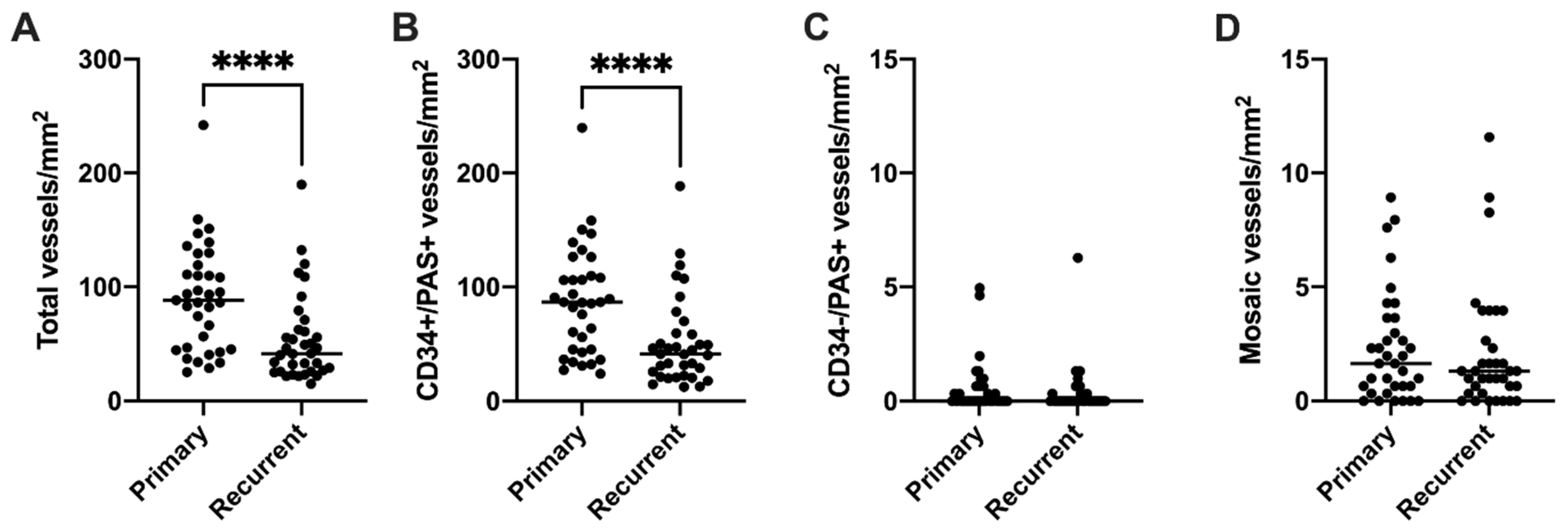
3.3. Endothelial Vessel Density Decreases at Recurrence, While VM Density Does Not Change
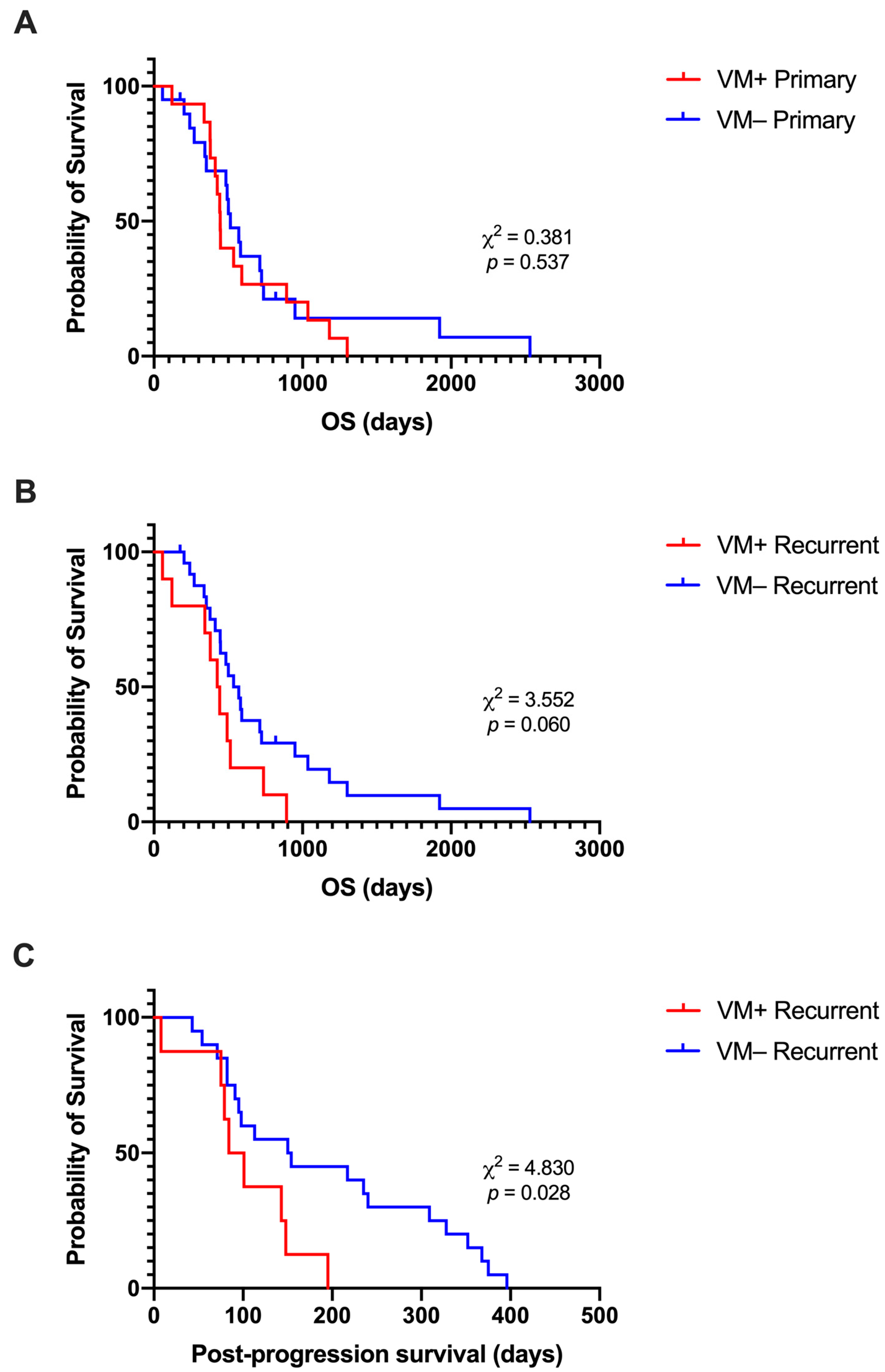
3.4. Presence of VM at Recurrence Is Associated with Shorter Post-Progression Survival
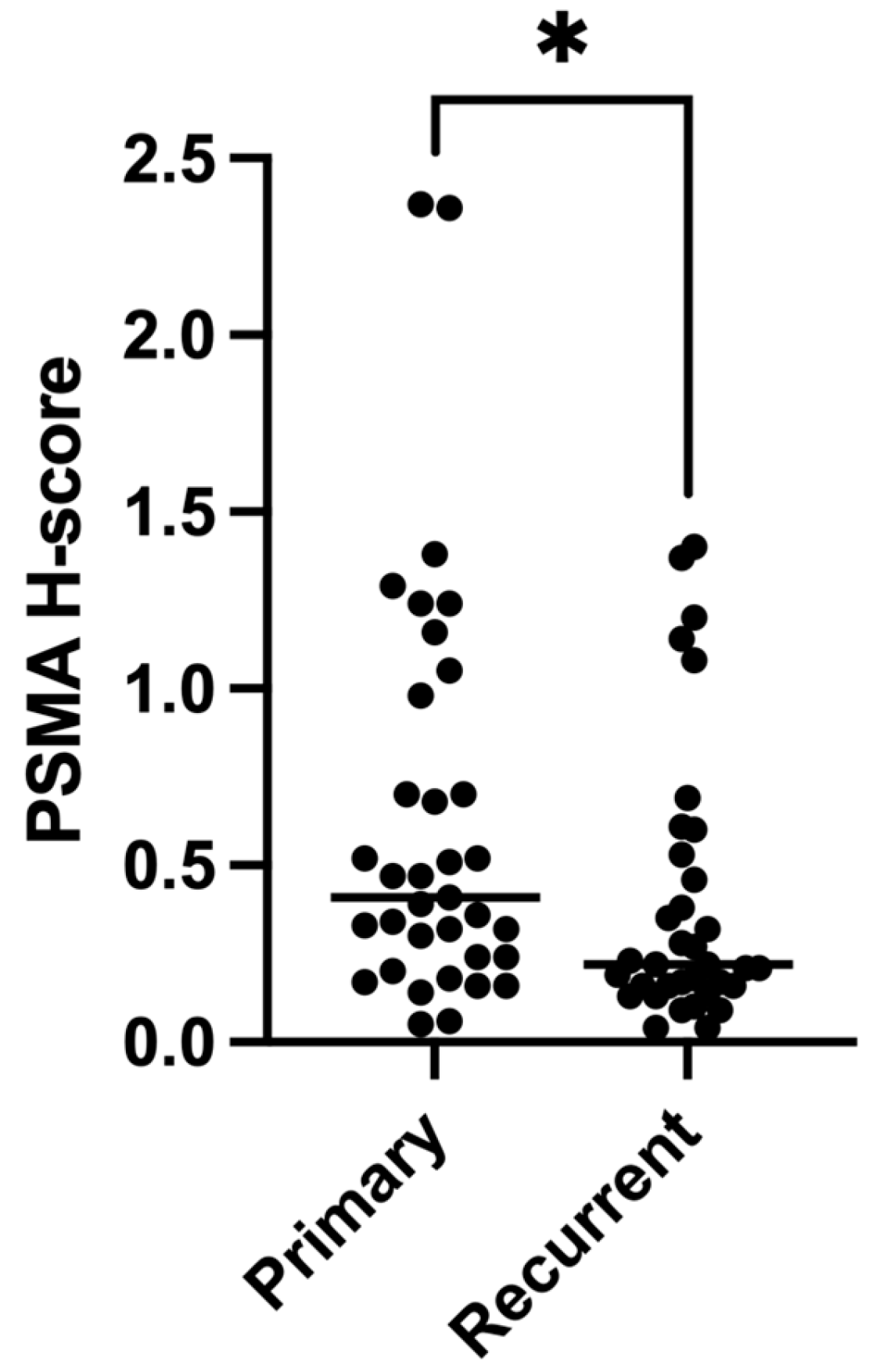
3.5. Expression of PSMA Decreases at Recurrence
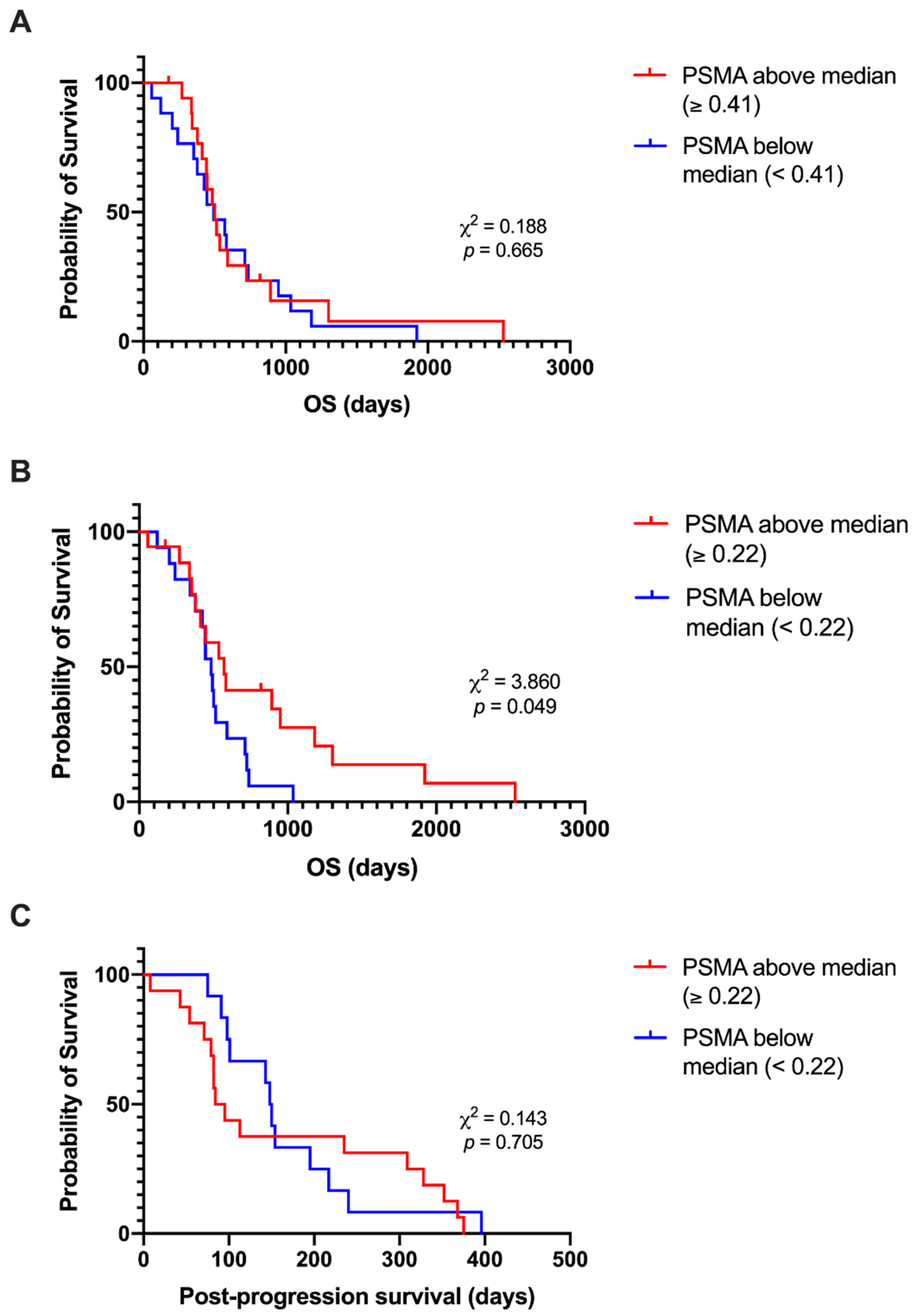
3.6. Lower PSMA Expression at Recurrence Is Associated with Shorter Post-Progression Survival
3.7. Relationships between Abnormal Vasculature and Clinical Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-Term (≥2 Years) Survival in Patients with Glioblastoma in Population-Based Studies Pre- and Post-2005: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef]
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination With Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF Treatment Reduces Blood Supply and Increases Tumor Cell Invasion in Glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seftor, R.E.B.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.G.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J.C. Cooperative Interactions of Laminin 5 Γ2 Chain, Matrix Metalloproteinase-2, and Membrane Type-1-Matrix/Metalloproteinase Are Required for Mimicry of Embryonic Vasculogenesis by Aggressive Melanoma. Cancer Res. 2001, 61, 6322–6327. [Google Scholar]
- Zhang, Z.; Imani, S.; Shasaltaneh, M.D.; Hosseinifard, H.; Zou, L.; Fan, Y.; Wen, Q. The Role of Vascular Mimicry as a Biomarker in Malignant Melanoma: A Systematic Review and Meta-Analysis. BMC Cancer 2019, 19, 1134. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Xu, C.; Li, X.; Yang, X. Twist1 Promotes Astrocytoma Development by Stimulating Vasculogenic Mimicry. Oncol. Lett. 2019, 18, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Q.; Mu, Y.; Zhang, X.; Sai, K.; Pang, J.C.-S.; Ng, H.-K.; Chen, Z. Clinical Significance of Vasculogenic Mimicry in Human Gliomas. J. Neurooncol. 2011, 105, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Mei, X.; Chen, Y.; Zhang, Q.; Chen, F.; Xi, S.; Long, Y.; Zhang, J.; Cai, H.; Ke, C.; Wang, J.; et al. Association between Glioblastoma Cell-derived Vessels and Poor Prognosis of the Patients. Cancer Commun. 2020, 40, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ke, Y.; Lu, G.; Song, Z.; Yu, L.; Xiao, S.; Sun, X.; Jiang, X.; Yang, Z.; Hu, C. Vasculogenic Mimicry Is a Prognostic Factor for Postoperative Survival in Patients with Glioblastoma. J. Neurooncol. 2013, 112, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; O’Keefe, D.S.; Bacich, D.J.; Reuter, V.E.; Heston, W.D.W.; Gaudin, P.B. Prostate-Specific Membrane Antigen Is Produced in Tumor- Associated Neovasculature. Clin. Cancer Res. 1999, 5, 2674–2681. [Google Scholar] [PubMed]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.W.; Condon-Cardo, C. Prostate-Specific Membrane Antigen Expression in Normal and Malignant Human Tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Wernicke, A.G.; Edgar, M.A.; Lavi, E.; Liu, H.; Salerno, P.; Bander, N.H.; Gutin, P.H. Prostate-Specific Membrane Antigen as a Potential Novel Vascular Target for Treatment of Glioblastoma Multiforme. Arch. Pathol. Lab. Med. 2011, 135, 1486–1489. [Google Scholar] [CrossRef] [Green Version]
- Tanjore Ramanathan, J.; Lehtipuro, S.; Sihto, H.; Tóvári, J.; Reiniger, L.; Téglási, V.; Moldvay, J.; Nykter, M.; Haapasalo, H.; Le Joncour, V.; et al. Prostate-specific Membrane Antigen Expression in the Vasculature of Primary Lung Carcinomas Associates with Faster Metastatic Dissemination to the Brain. J. Cell. Mol. Med. 2020, 24, 6916–6927. [Google Scholar] [CrossRef]
- Conway, R.E.; Joiner, K.; Patterson, A.; Bourgeois, D.; Rampp, R.; Hannah, B.C.; McReynolds, S.; Elder, J.M.; Gilfilen, H.; Shapiro, L.H. Prostate Specific Membrane Antigen Produces Pro-Angiogenic Laminin Peptides Downstream of Matrix Metalloprotease-2. Angiogenesis 2013, 16, 847–860. [Google Scholar] [CrossRef]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A New Alternative Mechanism in Glioblastoma Vascularization: Tubular Vasculogenic Mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Wang, J.; Xi, S.; Ni, X.; Chen, Y.; Yu, Y.; Cen, Z.; Yu, Z.; Chen, F.; Guo, C.; et al. Tenascin-c Mediated Vasculogenic Mimicry Formation via Regulation of MMP2/MMP9 in Glioma. Cell Death Dis. 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Hardee, M.E.; Zagzag, D. Mechanisms of Glioma-Associated Neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, Y.; Myskiw, C.; Rommel, A.; Verma, I.M. Mechanisms of Neovascularization and Resistance to Anti-Angiogenic Therapies in Glioblastoma Multiforme. J. Mol. Med. 2013, 91, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, C.L.; Caromile, L.A.; Durrani, K.; Rahman, M.M.; Claffey, K.P.; Fong, G.-H.; Shapiro, L.H. Prostate Specific Membrane Antigen (PSMA) Regulates Angiogenesis Independently of VEGF during Ocular Neovascularization. PLoS ONE 2012, 7, e41285. [Google Scholar] [CrossRef]
- Woo, J.-Y.; Yang, S.H.; Lee, Y.S.; Lee, S.Y.; Kim, J.; Hong, Y.K. Continuous Low-Dose Temozolomide Chemotherapy and Microvessel Density in Recurrent Glioblastoma. J. Korean Neurosurg. Soc. 2015, 58, 426. [Google Scholar] [CrossRef]
- Holzgreve, A.; Biczok, A.; Ruf, V.C.; Liesche-Starnecker, F.; Steiger, K.; Kirchner, M.A.; Unterrainer, M.; Mittlmeier, L.; Herms, J.; Schlegel, J.; et al. PSMA Expression in Glioblastoma as a Basis for Theranostic Approaches: A Retrospective, Correlational Panel Study Including Immunohistochemistry, Clinical Parameters and PET Imaging. Front. Oncol. 2021, 11, 646387. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor Invasion after Treatment of Glioblastoma with Bevacizumab: Radiographic and Pathologic Correlation in Humans and Mice. Neuro-Oncology 2010, 12, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Maddison, K.; Bowden, N.A.; Graves, M.C.; Tooney, P.A. Characteristics of Vasculogenic Mimicry and Tumour to Endothelial Transdifferentiation in Human Glioblastoma: A Systematic Review. BMC Cancer 2023, 23, 185. [Google Scholar] [CrossRef]
- Chen, Y.; Jing, Z.; Luo, C.; Zhuang, M.; Xia, J.; Chen, Z.; Wang, Y. Vasculogenic Mimicry–Potential Target for Glioblastoma Therapy: An in Vitro and in Vivo Study. Med. Oncol. 2012, 29, 324–331. [Google Scholar] [CrossRef]
- Huang, M.; Ke, Y.; Sun, X.; Yu, L.; Yang, Z.; Zhang, Y.; Du, M.; Wang, J.; Liu, X.; Huang, S. Mammalian Target of Rapamycin Signaling Is Involved in the Vasculogenic Mimicry of Glioma via Hypoxia-Inducible Factor-1α. Oncol. Rep. 2014, 32, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, Y.; Guo, C.; Chen, F.; Xi, S.; Zeng, J.; Ke, C.; Chen, Z.; Zhang, Q.; Sharma, H.S. Expression of Twist Associated to Microcirculation Patterns of Human Glioma Correlated with Progression and Survival of the Patient. Int. Rev. Neurobiol. 2020, 151, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Zhao, W.; Ma, Y.; Yang, X. Demonstration of Vasculogenic Mimicry in Astrocytomas and Effects of Endostar on U251 Cells. Pathol. Res. Pract. 2011, 207, 645–651. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.-H.; Huang, M.; Zhang, Y.-H.; Liu, Y.; Yang, Y.-T.; Ding, R.; Ke, Y.-Q.; Li, S.; Li, L.-L. Histone Deacetylase 3 Expression Correlates with Vasculogenic Mimicry through the Phosphoinositide3-Kinase/ERK-MMP-Laminin5gamma2 Signaling Pathway. Cancer Sci. 2015, 106, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Rong, X.; Huang, B.; Li, X.; He, L.; Peng, Y.; Qiu, S. Tumor-Associated Macrophages Induce Vasculogenic Mimicry of Glioblastoma Multiforme through Cyclooxygenase-2 Activation. Oncotarget 2016, 7, 83976–83986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, R.; Akiya, M.; Hashimura, M.; Oguri, Y.; Inukai, M.; Hara, A.; Saegusa, M. ALK Signaling Cascade Confers Multiple Advantages to Glioblastoma Cells through Neovascularization and Cell Proliferation. PLoS ONE 2017, 12, e0183516. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.-Y.; Chen, Z.-P. Does Vasculogenic Mimicry Exist in Astrocytoma? J. Histochem. Cytochem. 2005, 53, 997–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Zhu, Q.; You, W.; Shen, C.; Hu, W.; Chen, X. Silencing of Mig-7 Expression Inhibits in-Vitro Invasiveness and Vasculogenic Mimicry of Human Glioma U87 Cells. NeuroReport 2019, 30, 1135–1142. [Google Scholar] [CrossRef]
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic Blood Vessels in Tumors: Frequency of Cancer Cells in Contact with Flowing Blood. Proc. Natl. Acad. Sci. USA 2000, 97, 14608–14613. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma Stem-like Cells Give Rise to Tumour Endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of Glioblastoma Cells into Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef]
- Pezzolo, A.; Parodi, F.; Marimpietri, D.; Raffaghello, L.; Cocco, C.; Pistorio, A.; Mosconi, M.; Gambini, C.; Cilli, M.; Deaglio, S.; et al. Oct-4+/Tenascin C+ Neuroblastoma Cells Serve as Progenitors of Tumor-Derived Endothelial Cells. Cell Res. 2011, 21, 1470–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Xue, X.; Wang, L.; Zhang, X.; Yan, M.; Tu, Y.; Lin, W.; Jiang, X.; Ren, H.; Zhang, W.; et al. CDH5 Is Specifically Activated in Glioblastoma Stemlike Cells and Contributes to Vasculogenic Mimicry Induced by Hypoxia. Neuro-Oncology 2013, 15, 865–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Gomez, G.A.; Zhao, Y.; Yang, Y.; Cao, D.; Lu, J.; Yang, H.; Lin, S. ETV2 Mediates Endothelial Transdifferentiation of Glioblastoma. Signal Transduct. Target. Ther. 2018, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Zhao, Y.; Huang, Q.; Fei, X.; Diao, Y.; Shen, Y.; Xiao, H.; Zhang, T.; Lan, Q.; Gu, X. Glioma Stem/Progenitor Cells Contribute to Neovascularization via Transdifferentiation. Stem Cell Rev. Rep. 2011, 7, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Porcù, E.; Maule, F.; Boso, D.; Rampazzo, E.; Barbieri, V.; Zuccolotto, G.; Rosato, A.; Frasson, C.; Viola, G.; Della Puppa, A.; et al. BMP9 Counteracts the Tumorigenic and Pro-Angiogenic Potential of Glioblastoma. Cell Death Differ. 2018, 25, 1808–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endothelial Vessel Density, Primary | Endothelial Vessel Density, Recurrent | VM Vessel Density, Primary | VM Vessel Density, Recurrent | Mosaic Vessel Density, Primary |
Mosaic Vessel Density, Recurrent |
Total Vessel Density, Primary |
Total Vessel Density, Recurrent | ||
|---|---|---|---|---|---|---|---|---|---|
| Endothelial Vessel Density, Primary | τb | − | |||||||
| p-value | − | ||||||||
| Endothelial Vessel Density, Recurrent | τb | 0.135 | − | ||||||
| p-value | 0.256 | − | |||||||
| VM Vessel Density, Primary | τb | 0.006 | 0.100 | − | |||||
| p-value | 0.962 | 0.447 | − | ||||||
| VM Vessel Density, Recurrent | τb | 0.024 | −0.273 * | −0.009 | − | ||||
| p-value | 0.858 | 0.043 | 0.952 | − | |||||
| Mosaic Vessel Density, Primary | τb | −0.029 | −0.017 | 0.401 ** | 0.082 | − | |||
| p-value | 0.808 | 0.886 | 0.003 | 0.553 | − | ||||
| Mosaic Vessel Density, Recurrent | τb | 0.028 | −0.095 | 0.163 | 0.483 ** | 0.257 * | − | ||
| p-value | 0.819 | 0.439 | 0.231 | <0.001 | 0.041 | − | |||
| Total Vessel Density, Primary | τb | 0.974 ** | 0.147 | 0.033 | 0.012 | 0.000 | 0.016 | − | |
| p-value | <0.001 | 0.216 | 0.800 | 0.929 | 1.000 | 0.897 | − | ||
| Total Vessel Density, Recurrent | τb | 0.133 | 0.908 ** | 0.131 | −0.189 | 0.019 | 0.005 | 0.145 | − |
| p-value | 0.262 | <0.001 | 0.318 | 0.162 | 0.875 | 0.966 | 0.222 | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maddison, K.; Faulkner, S.; Graves, M.C.; Fay, M.; Bowden, N.A.; Tooney, P.A. Vasculogenic Mimicry Occurs at Low Levels in Primary and Recurrent Glioblastoma. Cancers 2023, 15, 3922. https://doi.org/10.3390/cancers15153922
Maddison K, Faulkner S, Graves MC, Fay M, Bowden NA, Tooney PA. Vasculogenic Mimicry Occurs at Low Levels in Primary and Recurrent Glioblastoma. Cancers. 2023; 15(15):3922. https://doi.org/10.3390/cancers15153922
Chicago/Turabian StyleMaddison, Kelsey, Sam Faulkner, Moira C. Graves, Michael Fay, Nikola A. Bowden, and Paul A. Tooney. 2023. "Vasculogenic Mimicry Occurs at Low Levels in Primary and Recurrent Glioblastoma" Cancers 15, no. 15: 3922. https://doi.org/10.3390/cancers15153922