
Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response


Abstract
Simple Summary
Abstract
1. Introduction
1.1. Breast Cancer Classification
1.2. Nucleosome Organization and Modification Inform Gene Expression
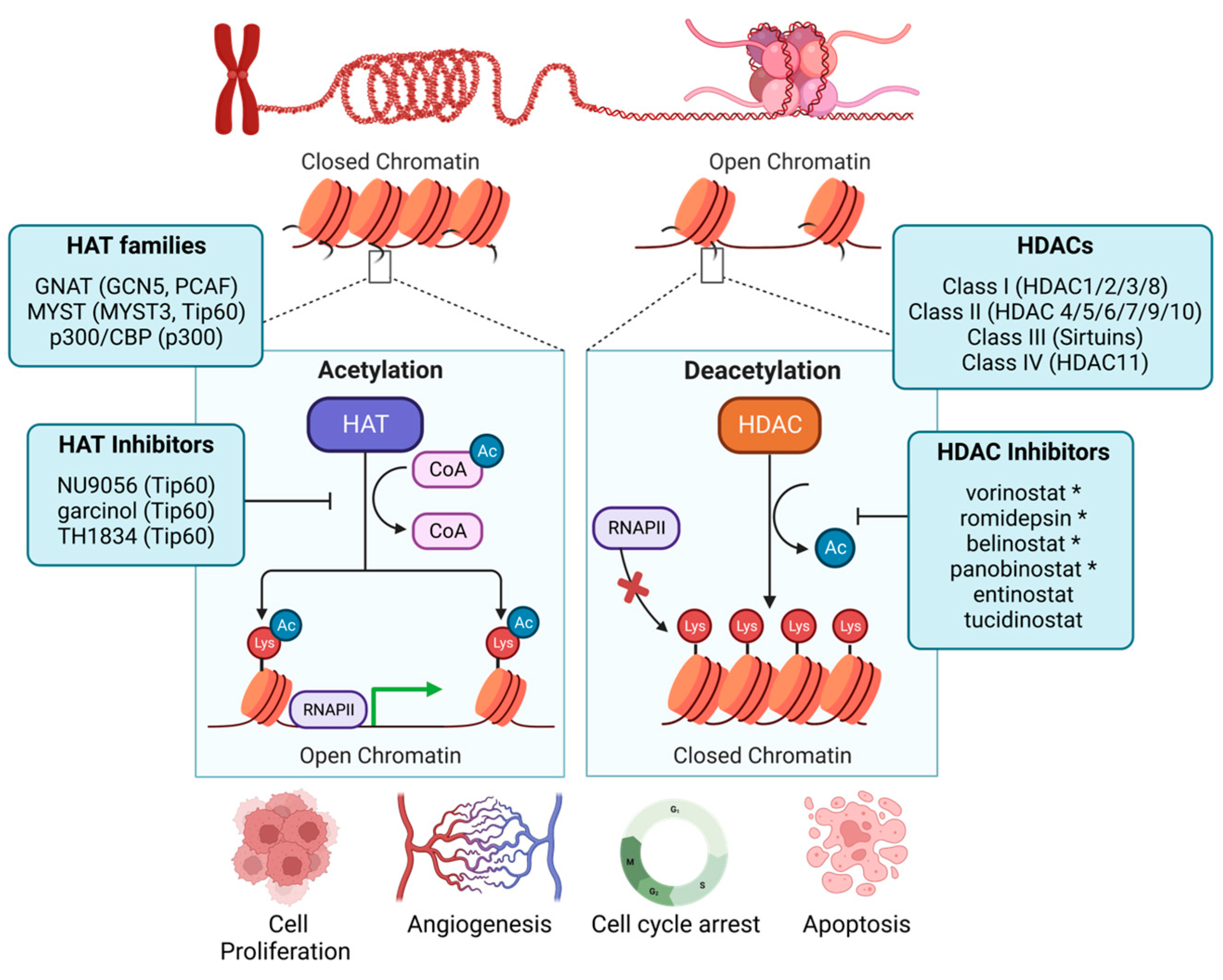
2. Histone-Modifying Complexes
2.1. COMPASS Complex Perturbation and Associated Histone Methylation in Breast Cancer
2.2. Dysregulation of SWI/SNF in Advanced Stage Breast Cancers
3. Histone Acetyltransferases
3.1. Histone Acetylation in Breast Cancer Pathogenesis
3.2. HATs Promote the Transcription of EMT-Specific Markers in Breast Cancer
3.3. HAT-Mediated Regulation of the DNA Damage Response
4. Histone Deacetylases
4.1. HDACs as Prognostic Factors in Breast Cancer Patients
4.2. HDACs Support the Epithelial-to-Mesenchymal Transition (EMT) in Breast Cancer
4.3. HDACs Modulate ER Expression and Signaling
4.4. HDACs as a Therapeutic Target to Overcome Treatment Resistance
4.5. Clinical HDAC Inhibition

{kind=link}
{kind=link}
| Agent | Classification | Target(s) | PTM-Regulation | Approval Indication | Trial Stage in Breast Cancer | Clinical Trial Status |
Reported Adverse Events
≥Grade 3 |
|---|---|---|---|---|---|---|---|
| Vorinostat (SAHA) | Hydroxamic acid | HDAC1, 2, 3, 8 (Class I) and HDAC6 (Class IIb) [144] | H3K14ac and H3K27ac (MCF7); H3K27ac, H3K18ac, H4K5ac (MDA-MB-231) [145]; H3K9ac (TNBC) [146] | Cutaneous T-cell lymphoma (FDA) [147] | I, II | Active; NCT03742245, NCT00616967, NCT04190056, NCT03878524 | Thrombocytopenia, Anemia, Deep vein thrombosis, Dehydration, Pyrexia, Hypotension, Pulmonary embolism, Sepsis [148], Nausea, Fatigue, Vomiting, Asthenia, Constipation, Hypokalemia [149] |
| Romidepsin (FK228) | Cyclic peptide | HDAC1, 2, 3, 8 (Class I) [150] | Unknown | Cutaneous T-cell lymphoma (FDA) [137] | I, II | Active; NCT02393794, NCT01638533 | Anemia, Leucopenia, Neutropenia, Thrombocytopenia, Cardiac disorders, Eye disorders, Gastrointestinal disorders, General disorders and administration site conditions, Immune system disorders, Infections and infest, Metabolism and nutrition disorders, Nervous system disorders, Respiratory, thoracic, and mediastinal disorders, Skin and subcutaneous tissue disorders [151] |
| Belinostat (PXD101) | Hydroxamic acid | Pan-inhibitor for zinc-dependent HDAC [152] | Unknown | Relapsed/Refractory peripheral T-cell lymphoma (FDA) [139] | I | Active; NCT04315233, NCT04703920 | Hypertriglyceridemia, Hemoglobin, Dyspnea, Fatigue, Dehydration, Hypoxia, Nausea, Vomiting, QTc prolonged, Dizziness, Hypercholesterolemia, Allergic reaction, Rash, Diarrhea, Tracheal hemorrhage, Left ventricular dysfunction, Small Bowel Obstruction, Palmar-plantar syndrome [153] |
| Panobinostat (LBH-589) | Hydroxamic acid | Pan-inhibitor for zinc-dependent HDAC [144] | H3K9ac, H4K8ac (TNBC) [154] | Multiple myeloma (FDA) [140] | I | Active; NCT03878524 | Neutropenia, Thrombocytopenia, Diarrhea, Nausea, Infection, Upper Respiratory Tract Infection, Vomiting, Lower Respiratory Tract Infection [155] |
| Entinostat (MS-275) | Benzamide | HDAC1, 2, 3 (Class I), and HDAC9 (Class II) [156] | Unknown | Breakthrough designation (advanced breast cancer) | III | Active; NCT01349959, NCT02115282, NCT02453620, NCT03280563. NCT03538171 (China) | Anorexia, Nausea, Vomiting, Fatigue. Diarrhea, Leukopenia, Neutropenia, Thrombocytopenia, Hypoalbuminemia, Hypocalcemia, Hyponatremia, Hypophosphatemia, ALT [157] |
| Tucidinostat | Benzamide | HDAC1, 2, 3 (Class I), HDAC10 (Class II) [156] | H3K9ac, H3K18ac (TNBC, HR+) [158] | Peripheral T-cell lymphoma (CFDA) | III (China) | Active (China only); NCT05276713, NCT05390476, NCT05632848, NCT05633914, NCT05335473, NCT05411380, NCT04192903, NCT05747313, NCT05186545, NCT05047848, NCT05890287, NCT05749575, NCT05085626, NCT05464173, NCT05400993 | Anemia, Leukopenia, Neutropenia, Thrombocytopenia, Increased alanine aminotransferase, Increased aspartate aminotransferase, Increased transpeptidase, Hypokalemia [159], Diarrhea, Lymphopenia, Decreased Appetite, Blood Alkaline Phosphatase Increased, Gamma-glutamyl Transferase Increased, Weight Decrease [160] |

4.6. Perspectives on HDAC Inhibition in Breast Cancer
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Acheampong, T.; Kehm, R.D.; Terry, M.B.; Argov, E.L.; Tehranifar, P. Incidence Trends of Breast Cancer Molecular Subtypes by Age and Race/Ethnicity in the US From 2010 to 2016. JAMA Netw. Open. 2020, 3, e2013226. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nunez, P.; Acuna-Aguilar, L.E.; Gomez-Valles, F.O.; Ramirez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer; Mayrovitz, H.N., Ed.; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Li, Z.H.; Hu, P.H.; Tu, J.H.; Yu, N.S. Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget 2016, 7, 65024–65033. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Jung, S.J.; Kim, T.H.; Oh, M.K.; Yoon, H.K. Differences in Clinical Outcomes between Luminal A and B Type Breast Cancers according to the St. Gallen Consensus 2013. J. Breast Cancer 2015, 18, 149–159. [Google Scholar] [CrossRef]
- Patel, A.; Unni, N.; Peng, Y. The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer. Cancers 2020, 12, 2081. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jin, J.; Ji, W.; Guan, X. Therapeutic landscape in mutational triple negative breast cancer. Mol. Cancer 2018, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- FitzGerald, M.G.; Marsh, D.J.; Wahrer, D.; Bell, D.; Caron, S.; Shannon, K.E.; Ishioka, C.; Isselbacher, K.J.; Garber, J.E.; Eng, C.; et al. Germline mutations in PTEN are an infrequent cause of genetic predisposition to breast cancer. Oncogene 1998, 17, 727–731. [Google Scholar] [CrossRef][Green Version]
- Pharoah, P.D.; Guilford, P.; Caldas, C.; International Gastric Cancer Linkage, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Simpson, B.; Tupper, C.; Al Aboud, N.M. Genetics, DNA Packaging; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Mersfelder, E.L.; Parthun, M.R. The tale beyond the tail: Histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res. 2006, 34, 2653–2662. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. 2020, 45, 135. [Google Scholar] [CrossRef]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 596. [Google Scholar] [CrossRef] [PubMed]
- Millan-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Tyagi, M.; Imam, N.; Verma, K.; Patel, A.K. Chromatin remodelers: We are the drivers!! Nucleus 2016, 7, 388–404. [Google Scholar] [CrossRef]
- Li, W.; Wu, H.; Sui, S.; Wang, Q.; Xu, S.; Pang, D. Targeting Histone Modifications in Breast Cancer: A Precise Weapon on the Way. Front. Cell Dev. Biol. 2021, 9, 736935. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ennour-Idrissi, K.; Dragic, D.; Durocher, F.; Diorio, C. Epigenome-wide DNA methylation and risk of breast cancer: A systematic review. BMC Cancer 2020, 20, 1048. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; D’Elia, G.; Benincasa, G.; Ferraro, G.; Caliendo, G.; Nicoletti, G.F.; Napoli, C. DNA methylation and breast cancer: A way forward (Review). Int. J. Oncol. 2021, 59, 5278. [Google Scholar] [CrossRef]
- Sher, G.; Salman, N.A.; Khan, A.Q.; Prabhu, K.S.; Raza, A.; Kulinski, M.; Dermime, S.; Haris, M.; Junejo, K.; Uddin, S. Epigenetic and breast cancer therapy: Promising diagnostic and therapeutic applications. Semin. Cancer Biol. 2022, 83, 152–165. [Google Scholar] [CrossRef]
- Trager, M.H.; Sah, B.; Chen, Z.; Liu, L. Control of Breast Cancer Pathogenesis by Histone Methylation and the Hairless Histone Demethylase. Endocrinology 2021, 162, bqab088. [Google Scholar] [CrossRef]
- Cenik, B.K.; Shilatifard, A. COMPASS and SWI/SNF complexes in development and disease. Nat. Rev. Genet. 2021, 22, 38–58. [Google Scholar] [CrossRef]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef]
- Toska, E.; Castel, P.; Chhangawala, S.; Arruabarrena-Aristorena, A.; Chan, C.; Hristidis, V.C.; Cocco, E.; Sallaku, M.; Xu, G.; Park, J.; et al. PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep. 2019, 27, 294–306e295. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Dreijerink, K.M.; Groner, A.C.; Cheng, H.; Ohlson, C.E.; Reyes, J.; Lin, C.Y.; Bradner, J.; Zhao, J.J.; Roberts, T.M.; et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell Rep. 2016, 15, 2692–2704. [Google Scholar] [CrossRef]
- Jones, R.B.; Farhi, J.; Adams, M.; Parwani, K.K.; Cooper, G.W.; Zecevic, M.; Lee, R.S.; Hong, A.L.; Spangle, J.M. Targeting MLL Methyltransferases Enhances the Antitumor Effects of PI3K Inhibition in Hormone Receptor-positive Breast Cancer. Cancer Res. Commun. 2022, 2, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.G.; Couturier, D.L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 2020, 52, 187–197. [Google Scholar] [CrossRef]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Sobczak, M.; Pietrzak, J.; Ploszaj, T.; Robaszkiewicz, A. BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells. Cancers 2020, 12, 349. [Google Scholar] [CrossRef]
- Mehta, G.A.; Angus, S.P.; Khella, C.A.; Tong, K.; Khanna, P.; Dixon, S.A.H.; Verzi, M.P.; Johnson, G.L.; Gatza, M.L. SOX4 and SMARCA4 cooperatively regulate PI3k signaling through transcriptional activation of TGFBR2. NPJ Breast Cancer 2021, 7, 40. [Google Scholar] [CrossRef]
- Do, S.I.; Yoon, G.; Kim, H.S.; Kim, K.; Lee, H.; Do, I.G.; Kim, D.H.; Chae, S.W.; Sohn, J.H. Increased Brahma-related Gene 1 Expression Predicts Distant Metastasis and Shorter Survival in Patients with Invasive Ductal Carcinoma of the Breast. Anticancer. Res. 2016, 36, 4873–4882. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell Biol. 2001, 21, 5979–5991. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes. Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Akhtar, A. The many lives of KATs—Detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 2019, 20, 7–23. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Messier, T.L.; Gordon, J.A.; Boyd, J.R.; Tye, C.E.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef]
- Guillemette, B.; Drogaris, P.; Lin, H.H.; Armstrong, H.; Hiragami-Hamada, K.; Imhof, A.; Bonneil, E.; Thibault, P.; Verreault, A.; Festenstein, R.J. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 2011, 7, e1001354. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef]
- Halasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kalafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells 2019, 8, 485. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yuan, L.; An, J.; Barton, M.C.; Zhang, Q.; Liu, Z. Histone H3 lysine 23 acetylation is associated with oncogene TRIM24 expression and a poor prognosis in breast cancer. Tumour Biol. 2016, 37, 14803–14812. [Google Scholar] [CrossRef]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef]
- Li, Q.L.; Wang, D.Y.; Ju, L.G.; Yao, J.; Gao, C.; Lei, P.J.; Li, L.Y.; Zhao, X.L.; Wu, M. The hyper-activation of transcriptional enhancers in breast cancer. Clin. Epigenetics 2019, 11, 48. [Google Scholar] [CrossRef]
- Bao, C.; Duan, J.; Xie, Y.; Liu, Y.; Li, P.; Li, J.; Zhao, H.; Guo, H.; Men, Y.; Ren, Y.; et al. A novel oncogenic enhancer of estrogen receptor-positive breast cancer. Mol. Ther. Nucleic Acids 2022, 29, 836–851. [Google Scholar] [CrossRef]
- Stejskal, S.; Stepka, K.; Tesarova, L.; Stejskal, K.; Matejkova, M.; Simara, P.; Zdrahal, Z.; Koutna, I. Cell cycle-dependent changes in H3K56ac in human cells. Cell Cycle 2015, 14, 3851–3863. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Pott, S.; Keller, C.; Kamieniarz-Gdula, K.; Caron, M.; Richter, F.; Li, G.; Mittler, G.; Liu, E.T.; Buhler, M.; et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 2013, 152, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, J.; Rousseaux, S.; Tan, M.; Pan, L.; Peng, L.; Wang, S.; Xu, W.; Ren, J.; Liu, Y.; et al. Metabolically controlled histone H4K5 acylation/acetylation ratio drives BRD4 genomic distribution. Cell Rep. 2021, 36, 109460. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Mungamuri, S.K.; Murk, W.; Grumolato, L.; Bernstein, E.; Aaronson, S.A. Chromatin modifications sequentially enhance ErbB2 expression in ErbB2-positive breast cancers. Cell Rep. 2013, 5, 302–313. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Yang, X.; Lu, W.; Chen, Y.; Lin, Y.; Wang, J.; Lin, S.; Yun, J.P. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts. Theranostics 2021, 11, 1473–1492. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, W.; Song, Z.; Wang, M.; Wu, H.; Yang, Y.; Chen, R. A novel miRNA inhibits metastasis of prostate cancer via decreasing CREBBP-mediated histone acetylation. J. Cancer Res. Clin. Oncol. 2021, 147, 469–480. [Google Scholar] [CrossRef]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Lebert, A.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. The Epigenetic Landscape of Promoter Genome-wide Analysis in Breast Cancer. Sci. Rep. 2017, 7, 6597. [Google Scholar] [CrossRef]
- Yu, L.; Liang, Y.; Cao, X.; Wang, X.; Gao, H.; Lin, S.Y.; Schiff, R.; Wang, X.S.; Li, K. Identification of MYST3 as a novel epigenetic activator of ERalpha frequently amplified in breast cancer. Oncogene 2017, 36, 2910–2918. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Frederiks, F.; Tzouros, M.; Oudgenoeg, G.; van Welsem, T.; Fornerod, M.; Krijgsveld, J.; van Leeuwen, F. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol. 2008, 15, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Feng, Q.; Li, Z.; Zhang, Y.; Xu, R.M. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 2003, 112, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Park, J.H.; Choi, H.J.; Park, M.K.; Won, H.Y.; Park, Y.J.; Lee, C.H.; Oh, S.H.; Song, Y.S.; Kim, H.S.; et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun. 2015, 6, 7821. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pang, A.; Li, Y. Function of GCN5 in the TGF-beta1-induced epithelial-to-mesenchymal transition in breast cancer. Oncol. Lett. 2018, 16, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, S.Y.; Choi, J.H.; Woo, H.G.; Xhemalce, B.; Miller, K.M. PCAF-Mediated Histone Acetylation Promotes Replication Fork Degradation by MRE11 and EXO1 in BRCA-Deficient Cells. Mol. Cell 2020, 80, 327–344 e328. [Google Scholar] [CrossRef]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef]
- Marijon, H.; Lee, D.H.; Ding, L.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef]
- Bassi, C.; Li, Y.T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016, 23, 1198–1208. [Google Scholar] [CrossRef]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci. Rep. 2014, 4, 5372. [Google Scholar] [CrossRef]
- Idrissou, M.; Judes, G.; Daures, M.; Sanchez, A.; El Ouardi, D.; Besse, S.; Degoul, F.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. TIP60 Inhibitor TH1834 Reduces Breast Cancer Progression in Xenografts in Mice. OMICS 2019, 23, 457–459. [Google Scholar] [CrossRef]
- Ye, X.; Yuan, L.; Zhang, L.; Zhao, J.; Zhang, C.M.; Deng, H.Y. Garcinol, an acetyltransferase inhibitor, suppresses proliferation of breast cancer cell line MCF-7 promoted by 17beta-estradiol. Asian Pac. J. Cancer Prev. 2014, 15, 5001–5007. [Google Scholar] [CrossRef] [PubMed]
- Chessum, N.; Jones, K.; Pasqua, E.; Tucker, M. Recent advances in cancer therapeutics. Prog. Med. Chem. 2015, 54, 1–63. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, H. Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs). Int. J. Mol. Sci. 2020, 21, 8828. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Robertson, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.; Schwabe, J.W. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun. 2016, 7, 11262. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Florean, C.; Brancolini, C. Class IIa HDACs: From important roles in differentiation to possible implications in tumourigenesis. J. Cell Mol. Med. 2011, 15, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Liu, S.S.; Wu, F.; Jin, Y.M.; Chang, W.Q.; Xu, T.M. HDAC11: A rising star in epigenetics. Biomed. Pharmacother. 2020, 131, 110607. [Google Scholar] [CrossRef]
- Haberland, M.; Johnson, A.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Genetic dissection of histone deacetylase requirement in tumor cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7751–7755. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Huber, E.; Kiefer, F.; Gottlicher, M. Specific and redundant functions of histone deacetylases in regulation of cell cycle and apoptosis. Cell Cycle 2004, 3, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, K.; Yamakawa, M.; Semba, S.; Takeda, H.; Kawata, S.; Kimura, S.; Kimura, W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J. Clin. Pathol. 2007, 60, 1205–1210. [Google Scholar] [CrossRef]
- Choi, J.H.; Kwon, H.J.; Yoon, B.I.; Kim, J.H.; Han, S.U.; Joo, H.J.; Kim, D.Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef]
- Shan, W.; Jiang, Y.; Yu, H.; Huang, Q.; Liu, L.; Guo, X.; Li, L.; Mi, Q.; Zhang, K.; Yang, Z. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am. J. Cancer Res. 2017, 7, 1213–1226. [Google Scholar]
- Krusche, C.A.; Wulfing, P.; Kersting, C.; Vloet, A.; Bocker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and -3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005, 90, 15–23. [Google Scholar] [CrossRef]
- Muller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer--overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef]
- Seo, J.; Min, S.K.; Park, H.R.; Kim, D.H.; Kwon, M.J.; Kim, L.S.; Ju, Y.S. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in Invasive Ductal Carcinomas of the Breast. J. Breast Cancer 2014, 17, 323–331. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Dimitroulis, D.; Kouraklis, G.; Garmpi, A.; Sarantis, P.; Koustas, E.; Patsouras, A.; Psilopatis, I.; Antoniou, E.A.; et al. Clinical Significance of the Histone Deacetylase 2 (HDAC-2) Expression in Human Breast Cancer. J. Pers. Med. 2022, 12, 1672. [Google Scholar] [CrossRef]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 2015, 165, 36–47. [Google Scholar] [CrossRef]
- Manshouri, R.; Coyaud, E.; Kundu, S.T.; Peng, D.H.; Stratton, S.A.; Alton, K.; Bajaj, R.; Fradette, J.J.; Minelli, R.; Peoples, M.D.; et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat. Commun. 2019, 10, 5125. [Google Scholar] [CrossRef]
- Yin, X.; Teng, X.; Ma, T.; Yang, T.; Zhang, J.; Huo, M.; Liu, W.; Yang, Y.; Yuan, B.; Yu, H.; et al. RUNX2 recruits the NuRD(MTA1)/CRL4B complex to promote breast cancer progression and bone metastasis. Cell Death Differ. 2022, 29, 2203–2217. [Google Scholar] [CrossRef]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.J.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 314. [Google Scholar] [CrossRef]
- Jiang, Y.G.; Luo, Y.; He, D.L.; Li, X.; Zhang, L.L.; Peng, T.; Li, M.C.; Lin, Y.H. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int. J. Urol. 2007, 14, 1034–1039. [Google Scholar] [CrossRef]
- Coelho, B.P.; Fernandes, C.F.L.; Boccacino, J.M.; Souza, M.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.R.; et al. Multifaceted WNT Signaling at the Crossroads between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef]
- Chen, H.C.; Zhu, Y.T.; Chen, S.Y.; Tseng, S.C. Wnt signaling induces epithelial-mesenchymal transition with proliferation in ARPE-19 cells upon loss of contact inhibition. Lab. Investig. 2012, 92, 676–687. [Google Scholar] [CrossRef]
- Pantelaiou-Prokaki, G.; Mieczkowska, I.; Schmidt, G.E.; Fritzsche, S.; Prokakis, E.; Gallwas, J.; Wegwitz, F. HDAC8 suppresses the epithelial phenotype and promotes EMT in chemotherapy-treated basal-like breast cancer. Clin. Epigenetics 2022, 14, 7. [Google Scholar] [CrossRef]
- Shah, P.; Gau, Y.; Sabnis, G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res. Treat. 2014, 143, 99–111. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J. Biol. Chem. 2013, 288, 11555–11571. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Rajabi, H.; Takahashi, H.; Yasumizu, Y.; Li, W.; Jin, C.; Long, M.D.; Hu, Q.; Liu, S.; Fushimi, A.; et al. MUC1-C Activates the NuRD Complex to Drive Dedifferentiation of Triple-Negative Breast Cancer Cells. Cancer Res. 2019, 79, 5711–5722. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, L.; He, T.; Xiao, X.; Liu, X.; Wang, L.; Yang, L.; Yang, M.; Zhang, T.; Chen, R.; et al. TWIST represses estrogen receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int. J. Biol. Sci. 2012, 8, 522–532. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, A.; Wang, R.A.; Mishra, S.K.; Adam, L.; Bagheri-Yarmand, R.; Mandal, M.; Vadlamudi, R.K.; Kumar, R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat. Cell Biol. 2001, 3, 30–37. [Google Scholar] [CrossRef]
- Heuer, J.; Heuer, F.; Sturmer, R.; Harder, S.; Schluter, H.; Braga Emidio, N.; Muttenthaler, M.; Jechorek, D.; Meyer, F.; Hoffmann, W. The Tumor Suppressor TFF1 Occurs in Different Forms and Interacts with Multiple Partners in the Human Gastric Mucus Barrier: Indications for Diverse Protective Functions. Int. J. Mol. Sci. 2020, 21, 2508. [Google Scholar] [CrossRef]
- Liu, X.F.; Bagchi, M.K. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J. Biol. Chem. 2004, 279, 15050–15058. [Google Scholar] [CrossRef]
- Criscitiello, C.; Fumagalli, D.; Saini, K.S.; Loi, S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: Overview of clinical use and molecular biomarkers for patient selection. Onco Targets Ther. 2010, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, P.N.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Cornwell, M.; Pun, M.; Buchwalter, G.; Nguyen, M.; Bango, C.; Huang, Y.; Kuang, Y.; Paweletz, C.; Fu, X.; et al. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc. Natl. Acad. Sci. USA 2017, 114, E4482–E4491. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Zhu, L.; Lao, Z.; Zhou, T.; Zang, L.; Ge, W.; Jiang, M.; Xu, J.; Cao, Y.; et al. SOX9 is a critical regulator of TSPAN8-mediated metastasis in pancreatic cancer. Oncogene 2021, 40, 4884–4893. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shepherd, J.; Zhao, D.; Bollu, L.R.; Tahaney, W.M.; Hill, J.; Zhang, Y.; Mazumdar, A.; Brown, P.H. SOX9 Is Essential for Triple-Negative Breast Cancer Cell Survival and Metastasis. Mol. Cancer Res. 2020, 18, 1825–1838. [Google Scholar] [CrossRef]
- Xue, Y.; Lian, W.; Zhi, J.; Yang, W.; Li, Q.; Guo, X.; Gao, J.; Qu, H.; Lin, W.; Li, Z.; et al. HDAC5-mediated deacetylation and nuclear localisation of SOX9 is critical for tamoxifen resistance in breast cancer. Br. J. Cancer 2019, 121, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Yard, B.; Chie, E.K.; Adams, D.J.; Peacock, C.; Abazeed, M.E. Radiotherapy in the Era of Precision Medicine. Semin. Radiat. Oncol. 2015, 25, 227–236. [Google Scholar] [CrossRef]
- Ouellette, M.M.; Zhou, S.; Yan, Y. Cell Signaling Pathways That Promote Radioresistance of Cancer Cells. Diagnostics 2022, 12, 656. [Google Scholar] [CrossRef]
- Tsai, C.L.; Liu, W.L.; Hsu, F.M.; Yang, P.S.; Yen, R.F.; Tzen, K.Y.; Cheng, A.L.; Chen, P.J.; Cheng, J.C. Targeting histone deacetylase 4/ubiquitin-conjugating enzyme 9 impairs DNA repair for radiosensitization of hepatocellular carcinoma cells in mice. Hepatology 2018, 67, 586–599. [Google Scholar] [CrossRef]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Chen, Y.A.; Chiou, Y.S.; Ho, S.Y.; Lin, P.; Wang, Y.J. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS ONE 2013, 8, e76340. [Google Scholar] [CrossRef]
- Baschnagel, A.; Russo, A.; Burgan, W.E.; Carter, D.; Beam, K.; Palmieri, D.; Steeg, P.S.; Tofilon, P.; Camphausen, K. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol. Cancer Ther. 2009, 8, 1589–1595. [Google Scholar] [CrossRef]
- Wang, S.; Song, M.; Zhang, B. Trichostatin A enhances radiosensitivity and radiation-induced DNA damage of esophageal cancer cells. J. Gastrointest. Oncol. 2021, 12, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Sharda, A.; Rashid, M.; Shah, S.G.; Sharma, A.K.; Singh, S.R.; Gera, P.; Chilkapati, M.K.; Gupta, S. Elevated HDAC activity and altered histone phospho-acetylation confer acquired radio-resistant phenotype to breast cancer cells. Clin. Epigenetics 2020, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, J.; Skorupa, M.; Jarkiewicz-Tretyn, J.; Cybulski, C.; Tretyn, A. Harnessing Epigenetics for Breast Cancer Therapy: The Role of DNA Methylation, Histone Modifications, and MicroRNA. Int. J. Mol. Sci. 2023, 24, 7235. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat. Rev. Drug. Discov. 2007, 6, 21–22. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert. Rev. Anticancer. Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Braunstein, Z.; Ruiz, M.; Hanel, W.; Shindiapina, P.; Reneau, J.C.; Brammer, J.E. Recent Advances in the Management of Relapsed and Refractory Peripheral T-Cell Lymphomas. J. Pers. Med. 2022, 12, 964. [Google Scholar] [CrossRef]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug. Benefits 2016, 9, 84–87. [Google Scholar]
- Sabnis, G.J.; Goloubeva, O.G.; Kazi, A.A.; Shah, P.; Brodie, A.H. HDAC inhibitor entinostat restores responsiveness of letrozole-resistant MCF-7Ca xenografts to aromatase inhibitors through modulation of Her-2. Mol. Cancer Ther. 2013, 12, 2804–2816. [Google Scholar] [CrossRef]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J. Clin. Oncol. 2021, 39, 3171–3181. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Chapter 32—Pharmacoepigenomics; Tollefsbol, T.O., Ed.; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Feng, X.; Han, H.; Zou, D.; Zhou, J.; Zhou, W. Suberoylanilide hydroxamic acid-induced specific epigenetic regulation controls Leptin-induced proliferation of breast cancer cell lines. Oncotarget 2017, 8, 3364–3379. [Google Scholar] [CrossRef]
- Palczewski, M.B.; Kuschman, H.P.; Bovee, R.; Hickok, J.R.; Thomas, D.D. Vorinostat exhibits anticancer effects in triple-negative breast cancer cells by preventing nitric oxide-driven histone deacetylation. Biol. Chem. 2021, 402, 501–512. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Petrich, A.; Nabhan, C. Use of class I histone deacetylase inhibitor romidepsin in combination regimens. Leuk. Lymphoma 2016, 57, 1755–1765. [Google Scholar] [CrossRef]
- Chiappella, A.; Dodero, A.; Evangelista, A.; Re, A.; Orsucci, L.; Usai, S.V.; Castellino, C.; Stefoni, V.; Pinto, A.; Zanni, M.; et al. Romidepsin-CHOEP followed by high-dose chemotherapy and stem-cell transplantation in untreated Peripheral T-Cell Lymphoma: Results of the PTCL13 phase Ib/II study. Leukemia 2023, 37, 433–440. [Google Scholar] [CrossRef]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef]
- Luu, T.; Frankel, P.; Beumer, J.H.; Lim, D.; Cristea, M.; Appleman, L.J.; Lenz, H.J.; Gandara, D.R.; Kiesel, B.F.; Piekarz, R.L.; et al. Phase I trial of belinostat in combination with 13-cis-retinoic acid in advanced solid tumor malignancies: A California Cancer Consortium NCI/CTEP sponsored trial. Cancer Chemother. Pharmacol. 2019, 84, 1201–1208. [Google Scholar] [CrossRef]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012, 14, R79. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Kalff, A.; Gartlan, K.H.; Savvidou, I.; Khong, T.; Ramachandran, M.; Cooke, R.E.; Bowen, K.; Hill, G.R.; Reynolds, J.; et al. Phase II trial of single-agent panobinostat consolidation improves responses after sub-optimal transplant outcomes in multiple myeloma. Br. J. Haematol. 2021, 193, 160–170. [Google Scholar] [CrossRef]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 2019, 43, 130–134. [Google Scholar] [CrossRef]
- Ryan, Q.C.; Headlee, D.; Acharya, M.; Sparreboom, A.; Trepel, J.B.; Ye, J.; Figg, W.D.; Hwang, K.; Chung, E.J.; Murgo, A.; et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J. Clin. Oncol. 2005, 23, 3912–3922. [Google Scholar] [CrossRef]
- Cao, L.; Zhao, S.; Yang, Q.; Shi, Z.; Liu, J.; Pan, T.; Zhou, D.; Zhang, J. Chidamide Combined with Doxorubicin Induced p53-Driven Cell Cycle Arrest and Cell Apoptosis Reverse Multidrug Resistance of Breast Cancer. Front. Oncol. 2021, 11, 614458. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, X.; Zhang, H.; Wang, X.; Yuan, Y.; Zhang, S.; Jiang, Z.; Wang, T. Clinical outcomes of tucidinostat-based therapy after prior CDK4/6 inhibitor progression in hormone receptor-positive heavily pretreated metastatic breast cancer. Breast 2022, 66, 255–261. [Google Scholar] [CrossRef]
- Rai, S.; Kim, W.S.; Ando, K.; Choi, I.; Izutsu, K.; Tsukamoto, N.; Yokoyama, M.; Tsukasaki, K.; Kuroda, J.; Ando, J.; et al. Oral HDAC inhibitor tucidinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Phase IIb results. Haematologica 2023, 108, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: Defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003, 2, 151–163. [Google Scholar] [PubMed]
- Valdez, B.C.; Brammer, J.E.; Li, Y.; Murray, D.; Liu, Y.; Hosing, C.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Romidepsin targets multiple survival signaling pathways in malignant T cells. Blood Cancer J. 2015, 5, e357. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Katoh, T.; Shimodaira, H.; Oda, A.; Takahashi, O.; Ishioka, C. Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors. Cancer Sci. 2012, 103, 1994–2001. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Howell, G.M.; Teggart, C.A.; Chowdhury, A.; Person, J.J.; Bowers, D.M.; Brattain, M.G. Histone deacetylase inhibitor belinostat represses survivin expression through reactivation of transforming growth factor beta (TGFbeta) receptor II leading to cancer cell death. J. Biol. Chem. 2011, 286, 30937–30948. [Google Scholar] [CrossRef]
- Qian, X.; Ara, G.; Mills, E.; LaRochelle, W.J.; Lichenstein, H.S.; Jeffers, M. Activity of the histone deacetylase inhibitor belinostat (PXD101) in preclinical models of prostate cancer. Int. J. Cancer 2008, 122, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Asano, T.; Isono, M.; Ito, K.; Asano, T. Panobinostat synergizes with bortezomib to induce endoplasmic reticulum stress and ubiquitinated protein accumulation in renal cancer cells. BMC Urol. 2014, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Idso, J.M.; Lao, S.; Schloemer, N.J.; Knipstein, J.; Burns, R.; Thakar, M.S.; Malarkannan, S. Entinostat augments NK cell functions via epigenetic upregulation of IFIT1-STING-STAT4 pathway. Oncotarget 2020, 11, 1799–1815. [Google Scholar] [CrossRef] [PubMed]
- Truong, A.S.; Zhou, M.; Krishnan, B.; Utsumi, T.; Manocha, U.; Stewart, K.G.; Beck, W.; Rose, T.L.; Milowsky, M.I.; He, X.; et al. Entinostat induces antitumor immune responses through immune editing of tumor neoantigens. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Zhang, P.; Du, Y.; Bai, H.; Wang, Z.; Duan, J.; Wang, X.; Zhong, J.; Wan, R.; Xu, J.; He, X.; et al. Optimized dose selective HDAC inhibitor tucidinostat overcomes anti-PD-L1 antibody resistance in experimental solid tumors. BMC Med. 2022, 20, 435. [Google Scholar] [CrossRef]
- Jiang, L.; Ma, Z.; Ye, X.; Kang, W.; Yu, J. Clinicopathological factors affecting the effect of neoadjuvant chemotherapy in patients with gastric cancer. World J. Surg. Oncol. 2021, 19, 44. [Google Scholar] [CrossRef]
- Sun, L.B.; Zhao, G.J.; Ding, D.Y.; Song, B.; Hou, R.Z.; Li, Y.C. Comparison between better and poorly differentiated locally advanced gastric cancer in preoperative chemotherapy: A retrospective, comparative study at a single tertiary care institute. World J. Surg. Oncol. 2014, 12, 280. [Google Scholar] [CrossRef]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra251. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, R.S.; Sad, K.; Fawwal, D.V.; Spangle, J.M. Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response. Cancers 2023, 15, 4005. https://doi.org/10.3390/cancers15154005
Lee RS, Sad K, Fawwal DV, Spangle JM. Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response. Cancers. 2023; 15(15):4005. https://doi.org/10.3390/cancers15154005
Chicago/Turabian StyleLee, Richard Sean, Kirti Sad, Dorelle V. Fawwal, and Jennifer Marie Spangle. 2023. "Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response" Cancers 15, no. 15: 4005. https://doi.org/10.3390/cancers15154005
APA StyleLee, R. S., Sad, K., Fawwal, D. V., & Spangle, J. M. (2023). Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response. Cancers, 15(15), 4005. https://doi.org/10.3390/cancers15154005