Simple Summary
Cancer cells are dependent on normal cells for their survival and functionality because they can use nanoscale tubes to steal the mitochondria from immune cells. It also highlights the significance of mitochondria in the biology of cancer cells as the key organelles for cellular metabolism and energy generation. Recent research has shown that mitochondria are critical for cancer cell survival in the hostile tumor microenvironments, immune system evasion, acquisition of more aggressive characteristics, and treatment resistance. This article discusses the role of mitochondrial metabolism in cancer biology, customized cancer therapy, and how it affects cancer resistance to chemotherapy, immunotherapy, and radiation. For instance, by scavenging the produced reactive oxygen species, functioning mitochondria might enhance cancer resistance to radiation. According to this hypothesis, targeting mitochondria may improve oncological results. The tumors can respond completely to anticancer therapies or even experience malignant progression while receiving them. As a result, individualized cancer treatment is essential. Up until now, genetic analysis has been the foundation for customized cancer treatment. There is evidence that cancers with a high mitochondrial concentration are more difficult to cure. Evaluation of mitochondrial metabolism before therapy may supplement genetic data and enhance the personalization of oncological interventions.
Abstract
Energy is needed by cancer cells to stay alive and communicate with their surroundings. The primary organelles for cellular metabolism and energy synthesis are mitochondria. Researchers recently proved that cancer cells can steal immune cells’ mitochondria using nanoscale tubes. This finding demonstrates the dependence of cancer cells on normal cells for their living and function. It also denotes the importance of mitochondria in cancer cells’ biology. Emerging evidence has demonstrated how mitochondria are essential for cancer cells to survive in the harsh tumor microenvironments, evade the immune system, obtain more aggressive features, and resist treatments. For instance, functional mitochondria can improve cancer resistance against radiotherapy by scavenging the released reactive oxygen species. Therefore, targeting mitochondria can potentially enhance oncological outcomes, according to this notion. The tumors’ responses to anticancer treatments vary, ranging from a complete response to even cancer progression during treatment. Therefore, personalized cancer treatment is of crucial importance. So far, personalized cancer treatment has been based on genomic analysis. Evidence shows that tumors with high mitochondrial content are more resistant to treatment. This paper illustrates how mitochondrial metabolism can participate in cancer resistance to chemotherapy, immunotherapy, and radiotherapy. Pretreatment evaluation of mitochondrial metabolism can provide additional information to genomic analysis and can help to improve personalized oncological treatments. This article outlines the importance of mitochondrial metabolism in cancer biology and personalized treatments.
1. Introduction
Cancer is a heterogeneous illness made up of various biological entities that require various therapies. Due to this problem, the world is moving away from one-size-fits-all cancer treatment regimens toward ones that are risk-adapted [1]. Recent researchers aim to identify the predictive factors influencing outcomes to personalized therapies and enhance quality of life while preserving efficacy. Predictive indicators for therapy response and toxicity are as important to illness as prognostic factors.
Cancer cells require normal cells for survival and function. By using nanoscale tube-like structures, cancer cells steal mitochondria from immune cells (CD8+ T cells and natural killer [NK] cells) [2]. Aside from providing energy, mitochondria also play a significant role in cancer cell survival and growth. Moreover, mitochondria are critical to the biology of cancer stem cells (CSCs), contributing to their resistance to chemo- and radiotherapy [3].
The purpose of this article is to provide a detailed understanding of mitochondrial function in cancer metabolism and how it is relevant to improving different cancer treatments, particularly radiotherapy (RT). RT is used in over 50% of cancer cases [4], and aims to deliver the maximum dose to the affected area while minimizing harm to healthy tissues. Each RT schedule is determined by several factors, including beam type, total and per fraction doses, treatment length, time between fractions, and dose rate. Personalized radiotherapy aims to optimize the RT schedule—per the specific tumor and host characteristics—to maximize treatment outcomes while minimizing the likelihood of adverse effects [5]. Currently, RT recommendations are mainly based on population averages obtained from studies. This paradigm has two problems: tumors are generally heterogeneous with different genetic and epigenetic signatures, and tumor hosts vary in racial, ethnic, and genetic features, which might affect the treatment outcomes [5]. Emerging evidence reflects the importance of patient characteristics, including age, gender, ethnicity, comorbidities, lifestyle, and intrinsic characteristics of cancer on treatment response [6,7,8]. This strategy has become a discipline in oncology called Personalized Cancer Treatment. To date, personalized oncology has been principally based on genomic analysis, using different testing, for example, next-generation sequencing (NGS) [9]. This paper illustrates how mitochondrial metabolism can serve as a predictive factor of treatment response. This additional information can improve the existing personalized treatment based on genomic analysis. The varied function of mitochondria in cancer metabolism is discussed in the following section, along with how essential healthy mitochondria are used for the survival and development of cancer.
2. The Pivotal Role of Mitochondria in Cancer Cells’ Metabolism
Cancer cells rely on functional mitochondria to survive in the harsh tumor microenvironment (TME), evade the immune system, progress to less differentiated types, and resist different treatment modalities [10], as follows: (Figure 1)
- (A)
- Surviving in the TME via the following mechanism:
- (A1)
- Metabolic switch to glycolysis: cancer cells are reorganized to tolerate the hypoxic, acidic, and hypoglycemic conditions of TME. Hypoxia-inducible factor-1α (HIF-1α) is one of the primary regulators of this metabolic alteration. In the harsh TME, HIF-1α overexpression leads to a metabolic switch from oxidative phosphorylation (OxPhos) into glycolysis. This alteration can maintain the cellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) level in the hypoxic TME. It has been demonstrated that HIF-1α relies on functional mitochondria for a secure continuous function [11]. In 2020, van Gisbergen et al. realized that cancer cells with severe mitochondrial dysfunction showed a decrease in CAIX expression and HIF-1α levels. The authors concluded that functional mitochondria are essential for the stabilization of HIF-1α [11].
- (A2)
- Scavenging reactive oxygen species (ROS): hypoxic condition of TME is associated with increased ROS production in cancer cells. When there is insufficient oxygen availability, the electron transport across the mitochondrial complexes is slowed down. This causes the electrons to leak out of the electron transport chain (ETC) and interact with oxygen, producing ROS. Functional mitochondria can detoxify the released ROS by preserving the cellular NADPH sources. This function is mediated by increased NADH production, representing mitochondrial function [12,13].
- (A3)
- Arresting cell cycle: cancer cells can tolerate the harsh TME by dormancy, which is the mitotic arrest at the G0/G1 cycle phase [14]. Cell cycle progression is regulated by a dedicated system consisting of cyclins and cyclin-dependent kinases (CDK). It has been demonstrated that mitochondria can mediate dormancy in colon cancer cells by HIF-dependent activation of p21 and p27 (two CDK-cyclin inhibitors) [11,15], in prostate cancer cells by activating the MAPK-p38 pathway [16,17], and in leukemic stem cells by activating the mTOR pathway [18,19].
- (A4)
- Maintaining pH homeostasis: In contrast to normal cells, cancer cells can tolerate acidic TME using a dedicated transmembrane glycoprotein called carbonic anhydrase IX (CA IX). This protein preserves intracellular pH by absorbing extracellular bicarbonate and sending out intracellular lactate [20,21]. It has been demonstrated that mitochondria are the upregulators of CA IX [11].
- (A5)
- Mediating autophagy: mitochondria can facilitate autophagy by raising the level of intracellular ROS, which leads to the inactivation of mTORC1 (an autophagy inhibitor) and the activation of NRF2 (an autophagy activator) [22,23,24,25].
- (A6)
- Angiogenesis: secretion of different angiogenic factors (e.g., VEGF, PGF, angiopoietin) in cancer cells is HIF-dependent [26]. Mitochondria conduct angiogenesis by securing HIF function [11].
- (A7)
- Mitochondrial hijacking: cancer cells can steal mitochondria from T cells (and NK cells) via nano-scale tubes. Saha et al. demonstrated that this process is GTP-dependent [2]. Functional mitochondria can secure mitochondria hijacking by providing GTP from their TCA cycle [27].
- (B)
- Immune evasion: completed via facilitating TME acidification, glucose influx, PD-1 upregulation on T cells (by mitochondrial hijacking) [28], recruiting myeloid-derived suppressor cells (MDSCs), PD-L1 overexpression on cancer cells (via STING-IFN pathway), MHC-1 downregulation, and the secretion of immunosuppressants [10]. Additionally, T cells’ mitochondrial hijacking leads to PD-1 upregulation on T-cells and depletes their energy to provide long-term cancer-fighting action [28].
- (C)
- Aggressiveness: mitochondria are crucial for cancer progression via mediating genomic instability, quiescence evasion, and epithelial-to-mesenchymal transition (EMT) [10]. An increase in cellular ROS is the most common promoter of these three processes. Genomic instability is mediated by an increase in ROS levels and damage to nuclear nucleosides and inducing minority MOMP (mitochondrial outer membrane permeabilization) [10]; quiescence evasion is conducted by an increase in cellular ROS and following the activation of the Ras pathway [29,30]; Section 3 summarizes how mitochondria are involved in EMT. ROS is a double-edged sword, destroying cancer cells at high levels and promoting cancer progression at moderate levels. Functional mitochondria help cancer cells to maintain cellular ROS at higher levels (so-called “elevated ROS balance”), facilitating cancer progression without damage to the cellular structures [31].
- (D)
- Treatment resistance: mitochondria can protect cancer cells from chemotherapy and RT by eliminating the released ROS. Additionally, they increase chemotherapy resistance by encouraging the function of efflux pumps (by providing ATP) and inducing cell cycle arrest. Additionally, mitochondrial hijacking from T cells impairs the long-term effects of anti-PD-1 treatment [10].

Figure 1.
Schematic model of how mitochondria contribute to cancer cells’ survival in tumor microenvironment (A), immune evasion (B), progression (C), and resistance to different treatment modalities (D). Section D also demonstrates the importance of mitochondrial metabolism in ‘6Rs’ of radiobiology. EMT indicates epithelial-mesenchymal transition; MDSC, myeloid-derived suppressor cell; MHC-1, major histocompatibility complex class I; PD-1, programmed cell death protein-1; PD-L1, programmed cell death protein-ligand 1; ROS, reactive oxygen species; and TME, tumor microenvironment (retrieved from [10,32]).
3. Mitochondria Individualized Role in Cancer Metastasis
Metastasis happens in a very diverse and individualized pattern [33]. The players in the molecular pathway of metastasis and the therapeutic response to metastasis should also be considered in a personalized and idealized context. In order for cancer cells to spread, they must first undergo EMT, during which they lose intercellular adhesions and obtain high capacity for local migration, vascular invasion, and resistance to apoptotic stimuli. [34]. It has been found that there is a link between EMT and the stemness of cancer cells. These two processes are controlled by common mediators such as HIFs, SNAIL, and SLUG/SOX9 [35,36]. More functional mitochondria can promote EMT through releasing more mitochondrial ROS (mtROS), which activates different pathways, such as MAPK PI3K/Akt/mTOR, and VEGFA–SOX2–SNAI2 pathways [36,37,38]. Moreover, it is essential to acknowledge that mitochondria are directly involved in the cancer cells’ proliferation, invasion, and metastasis by enabling the linkage between β1 integrin and the extracellular matrix [39]. This process is mediated by lysyl oxidase (LOX), which requires HIF-1α for a secured function. Mitochondria can promote this process by promoting HIF-1α stability [11,40]. It is of utmost importance to employ targeted anti-mitochondrial to impede the process of EMT and curb the spread of cancer cells throughout the body. This approach can prove to be instrumental in arresting the progression of cancer and enhancing the effectiveness of treatment. Precisional targeting of cancer-specific mitochondria can reduce their ability to de-differentiate, proliferate, and metastasize, and helps to improve the treatment results and overall prognosis.
4. Targeting Mitochondria: A Practical Strategy for Personalized Cancer Treatment
Thanks to the developments in medical genetics and molecular biology, the function of mitochondria in several cellular functions, including apoptosis, redox balance, macromolecule production, and calcium homeostasis, has been demonstrated [41,42]. In contrast to the ancient Warburg theory, the mitochondria of cancer cells are functional, supporting their survival and function [10]. As noted earlier, mitochondria can contribute to the development, progression, and metastasis of cancer. In addition, it has a crucial role in treatment resistance. As noted in Section 2, functional mitochondria can help cancer cells to overcome chemotherapy effects by scavenging released ROS and activating multidrug resistance pumps [10]. Also, they can improve the resistance against immunotherapy, by inhibiting the immune cells’ entry to the TME by depleting the glucose content of TME, acidifying the TME, and mediating the mitochondria hijacking from immune cells [10,43]. Next, we outline how mitochondria can improve the cancer cells resistance against radiotherapy. In a recent study, Taghizadeh-Hesary et al. demonstrated that mitochondria have a contributing role in tumor response to radiotherapy. They demonstrated that mitochondria are involved in so-called 6Rs of radiobiology [32] (Figure 1). The details of this link were presented as follows:
(a) Repair: DNA damage is the primary cause of RT’s cytotoxic effects. Cancer cells with improved DNA repair mechanisms can counteract this effect. Mitochondria can support ATP-dependent proteins responsible for DNA integrity-related, including PARP-1 [44], XRCC1 [45], ATM [46], and DNA ligases [47], by providing enough ATP molecules.
(b) Repopulation: Mitochondria can support cancer cells proliferation by supplying the building materials, including nucleic acids, amino acids, and lipids through stabilizing HIF-1 and metabolic switching to glycolysis [48].
(c) Reoxygenation: HIF-1 can mediate tissue reoxygenation by promoting the expression of different angiogenic factors and shielding endothelial cells from radiation effects [49]. HIF-1 needs functional mitochondria to function properly [11]. Consequently, healthy mitochondria can aid in the reoxygenation of tumor tissue.
(d) Redistribution: Cyclin-Cdk complexes carefully control the cancer cells’ cell cycle [50]. The radiosensitive phases of the cell cycle are G2 and M and the radioresistant phases are G1 and S [51]. Cell cycle progression depends on dynamic responses of mitochondria during the G1 and S phases, when mitochondria fuse to form a hyperactive network; after that, they undergo fission to ensure proper partitioning between the two daughter cells [50]. In addition, functional mitochondria can help the cell cycle progression by supplying enough energy [52].
(e) Reactivation: Cancer cells have the ability to avoid activated immune cells by using immune inhibitory molecules like programmed death-ligand 1 (PD-L1) [53]. It has been shown that in the hypoxic TME, HIF-1α mediates PD-L1 expression on cancer cells [54]. This process is supported by functioning mitochondria which help to stabilize HIF-1α [11]. On the other hand, Akbari et al. found a direct correlation between T cell mitochondrial capacity and the expression of PD-1. If T cells have limited mitochondrial capacity, they may experience an overexpression of PD-1, ultimately leading to inactivity [28]. The study conducted by Saha et al. conclusively showed that specific nanotubes enable cancer cells to hijack the mitochondria of T and NK cells [2]. Applying this strategy, cancer cells deliberately raise their PD-L1 levels while boosting PD-1 in immune cells to cunningly evade the immune system.
(f) Radiosensitivity: Functional mitochondria can reduce the radiosensitivity of cancer cells by scavenging the released ROS and mediating the removal of damaged mitochondria, a process called mitophagy [32]. Hitherto, numerous biological factors have been linked to the intrinsic radiosensitivity of cancer cells, including p53, transforming growth factor beta (TGF-β), and isocitrate dehydrogenase 1 (IDH1) among others. For instance, p53 can improve radioresistance by enhancing the mitochondrial DNA integrity and PGC-1α (peroxisome proliferator-activated receptor γ coactivator-1α) overexpression [55,56]. For the detailed mechanisms of other corresponding factors, the readers are referred to the study by Taghizadeh-Hesary et al. [32] (Table 1).

Table 1.
The biological factors of radioresistance from the mitochondria perspective.
This section illustrated how functional mitochondria can improve the tumor resistance against the various treatments. Therefore, inhibiting the cancer cells’ mitochondria can potentially improve the treatment results.
5. Enhancing the Normal Cells’ Mitochondria Reduces the Radiotherapy Toxicity
The way normal tissue responds to radiation is mainly influenced by its DNA repair capacity, repopulation, and radiosensitivity [101]. As previously stated, having functional mitochondria is crucial for cells to promote DNA repair and growth and to reduce RT-induced oxidative stress. Inflammation caused by radiation is an important stage in the development of normal tissue damage. It occurs when ROS is released from the damaged cells [102]. Functional mitochondria help to reduce inflammation and prevent tissue damage by effectively eliminating ROSs [103]. Hence, enhanced mitochondrial content of normal cells can effectively mitigate the adverse impact of radiation exposure. In this section, we outline how mitochondrial metabolism is connected to the known factors’ normal tissue radiosensitivity.
- Recruiting genotypic and proteomic data of patients with breast or head and neck cancer, a series of proteins are recognized as a determinant for normal tissue toxicity to radiation; including CHIT1, PDGFB, STIM1, and THPO proteins as improving radiosensitivity, and SERPINC1 and SLC4A as enhancing radioresistance [104]. Mitochondrial metabolism interprets the mechanism of action of STIM1, SERPINC1, and SLC4A. STIM1 (stromal interaction molecule 1) regulates intracellular calcium level [105] and downregulates mitochondrial metabolism as its knockout leads to more metabolically active mitochondria [106]. STIM1 exacerbates radiation toxicity by preventing mitochondrial function from neutralizing the radiation-induced ROSs. Apoptosis and mitochondrial dysfunction are instead encouraged by SERPINC1 knockout because it activates the Bax pathway [107]. In the mitochondrial anti-oxidative system, SLC4 (solute carrier 4) scavenges ROS to improve radioresistance [108]. Hence, SERPINC1 and SLC4 may enhance radioresistance by enhancing mitochondrial metabolism and their capacity to scavenge ROS molecules.
- TGF-β overexpression increases the susceptibility of radiation-induced pulmonary fibrosis [109] and its activation affects mitochondrial respiration via impairing the mitochondrial complex IV in lung epithelial cells [110].
- The JAK/STAT signaling pathway in human cells is thought to provide protection against radiation. The activation of STAT3 enhances the ability of normal cells to withstand radiation by promoting the production of NADPH (which helps maintain a balanced redox state) and ATP (which helps ensure DNA stability); hence, it enhances the mitochondrial ETC in normal cells [111].
- Radiation toxicities are more likely to affect older people. Higher ROS production and decreased antioxidant capability in older people have been blamed for this impact [112]. As people get older, there is mounting evidence that their ability to produce ATP and NADPH is reduced because of an accumulation of mtDNA mutations and ROS damage to the mitochondrial substructures [113]. The cellular redox processes (such as glutathione) and the ATP-dependent enzymes responsible for repairing DNA damage are each impaired, necessitating NAPDH to function [114]. As a result, its relationship with radiation damage may be influenced by aging’s impact on mitochondrial metabolism.
- Several mechanisms have been proposed to explain how smoking during RT may increase the frequency and severity of radiation-induced acute and delayed toxicities [115]. Through endothelial damage and coagulation, it impairs tissue repair and triggers an inflammatory cascade, which increases the rate and severity of acute radiation toxicities and causes late toxicities [116]. Both acute and late radiation toxicities from tobacco smoke affect mitochondria negatively. Smoke exposure alters the mitochondrial membrane potential, which causes the release of ROS from the mitochondria and ultimately results in cellular death. DAMPs are then released into the extracellular matrix, where they connect to toll-like receptors (TLRs) on tissue macrophages and trigger the NF-kB pathway. Inflammatory cytokines are released as a result, which damages healthy tissue and exacerbates acute radiation-induced inflammation [116]. The main cause of delayed radiation toxicities, which manifest at least three months after RT, is the replacement of normal tissues by fibrotic tissues with inadequate blood flow [117]. In order for tissue regeneration and angiogenesis to be mediated by wound macrophages—the key players in wound healing—proper mitochondrial function is a crucial precondition and determining factor in the early stages of wound repair [118]. Therefore, increased radiation toxicity in smokers is justified by mitochondrial damage.
Alcohol intake can also enhance the incidence and severity of tissue fibrosis after radiation exposure, which can aggravate radiation-induced toxicities [119]. In order for macrophages to effectively repair the damaged tissue, as mentioned above, functional mitochondria are necessary [118]. Since ethanol can harm normal cells’ mitochondria by inducing oxidative stress, its detrimental effects on mitochondrial metabolism may contribute to the radiotherapy’s delayed toxicities [120]. As a result, continued use of cigarettes or alcohol during RT may each cause certain radiation-induced toxicities.
6. Immune Cells’ Mitochondria: A Chance to Improve Treatment Results
In addition to immunotherapy, a powerful immune system can improve the treatment results of radiotherapy and chemotherapy [32,121]. To improve the normal cells’ mitochondrial content and activity several strategies can be employed. The mitochondria quality can increase by two strategies; (1) improving the lifestyle by regular exercise [122], specific diets (low-specific dynamic action diet [123], branched-chain amino acid-rich diet [124], and Mediterranean diet [125]); good sleep [126], healthy weight [127], alcohol abstinence [128], and smoking cessation [129]; and (2) mitochondria boosting agents (e.g., coenzyme Q10, activators of adenosine AMPK, acetyl-L-Carnitine; mammalian target of rapamycin [mTOR], PGC-1α, etc.) [130,131]. In addition, the human gut microbiota is another modulator of mitochondrial fitness. It has been demonstrated that microbiota-derived metabolites are necessary for the proper action of mitochondrial metabolisms, including glycolysis, tricarboxylic acid (TCA) cycle, oxidative phosphorylation, as well as amino acid and fatty acid metabolism. The mitochondrial boosting strategies are diverse. Detailed information is presented in the following sources [131,132].
7. Heteroplasmy Provides Unique Profiles in Cancer
Heteroplasmy is the presence of more than one type of organellar genome (mitochondrial DNA or plastid DNA) within a cell. The amount of heteroplasmy is determined during oogenesis and is inherited from the mother. There are variations in the percentage of mutant alleles between oocytes and then between children. Heteroplasmy or the presence of at least two mtDNA variants within the single cell, and its level (the proportion of mutated mtDNA) are frequently seen with and in accompany tumor heterogeneity. One of the major challenges to understanding and elucidating the role of the variations in tumor growth is the heteroplasmy levels of the mtDNA variants. In turn, intratumor genetic heterogeneity affects personalized medicine strategies in a significant way since it can reduce the effectiveness of treatments and result in treatment resistance. It is interesting to note that numerous studies have linked heteroplasmic levels to both cancer risk and survival [133,134,135,136,137]. It would be essential to advance knowledge of the biological mechanisms at play, including proliferation, metastasis, and intratumoral heterogeneity, as well as the clinical implications of heteroplasmy, via recognizing the crucial role of heteroplasmy in cancer. The high mutation rate found in mtDNA, which is between 10 and 17 times higher than that of the nuclear genome, is explained by the lack of histones, effective DNA repair mechanisms, and closeness to reactive oxygen species (ROS) produced by the OxPhos system (mostly from Complex I and III) (nDNA) [138,139,140,141]. In humans, mtDNA is only inherited via the maternal line as a single unit called a haplotype, which may be shared by populations with similar ancestries. Factually, a set of haplotypes or a haplogroup can be used to distinguish across populations or ethnic groupings while certain haplogroups have advantages for environmental adaptation but are also linked to cancer [142,143,144,145,146,147,148,149].
The degree of heteroplasmy varies greatly between different kinds of cancer and individuals. It has been demonstrated that when tumors progressed, heteroplasmy varied amongst tissues. Based on the idea that some heteroplasmic variations are finally able to become dominant or are lost in cancer cells based on their tumor-promoting impact, a likely bottleneck process was proposed. The G1576C and G12009A mutations are the most prevalent in tumor cells compared to normal cells (7.8% versus 0.35% and 68.8% versus 0.35%, respectively) [150].
Although a very limited number of studies have been completed on the mechanisms of heteroplasmy shifting in cancer, there is proof that cell niche and the nucleus-mitochondrial environment regulate the OxPhos system’s energy performance, choosing particular mutant alleles [151]. For instance, it has been demonstrated that fumarate accumulation in renal cancer alters the mitochondrial content by inactivating core components necessary for mtDNA replication [152]. Alterations in DNA polymerase gamma (POLG) and mitochondrial transcription factor A (TFAM) expression, mutations in nDNA-encoded genes involved in mitochondrial biogenesis, nuclear and mitochondrial epigenetic modifications, as well as intrinsic and extrinsic stimuli, may all result in anomalies in mtCNVs [153,154,155]. Examples include the dysregulated expression of nuclear genes such as dynamin 1 (DRP1), mitofusin 1 (MFN1) and 2 (MFN2) mitochondrial fusion and fission proteins, BCL2 inter-acting protein 3 (BNIP3), PTEN-induced kinase 1 (PINK1), and hypoxia inducible factor 1 (HIF1), observed in lung, bladder, and breast cancers [156,157]. The role of the tumor microenvironment in altering the allelic frequencies of mtDNA mutations was also hypothesized based on an investigation of primary tumors and their distant metastasis [158]. Additionally, NOX2-derived redox signaling has been shown to be used by bone marrow stromal cells to transfer functioning mitochondria to acute myeloid leukemia blasts [159,160]. Together, these pathways may be crucial for the emergence of a tumorigenic environment-adaptive, unique response that is represented in the alteration of the allelic frequencies of mtDNA mutations. The nuclear insertions of mitochondrial origin (NumtS), which have been linked to cancer, should be considered in the next investigations on heteroplasmy. NumtS or mtDNA segments integrated into the nucleus during evolution are thought to occur at a rate of ~5 × 10e−6 per germ cell every generation [161].
Currently, methods based on mitochondrial gene editing have been proposed as a treatment choice for reestablishing the OxPhos system in conditions brought on by mtDNA mutations. A possible therapeutic target for cancer has been suggested to include components involved in mitochondrial biogenesis and metabolism [162,163,164]. Overall, in the context of personalized oncology, the importance of heteroplasmy lies in its potential implications for diagnosis, prognosis, and treatment of certain types of cancers. Heteroplasmy analysis can contribute to tracking heterogeneity within a tumor and monitoring the clonal evolution of mtDNA mutations; it may also guide treatment decisions and the development of personalized therapeutic strategies. Heteroplasmy analysis can be performed using non-invasive methods, such as liquid biopsies, which involve analyzing the circulating tumor DNA (ctDNA) or cell-free DNA (cfDNA) shed by tumors into the bloodstream.
8. Practical and Potential Methods to Target Cancer Cells’ Mitochondria
This section outlines the examined and proposed methods to target the cancer cells’ mitochondria. The available methods can be categorized based on their target, as follows: (1) inhibiting mitochondrial metabolism, including isocitrate dehydrogenase inhibitors (IDH1/2) (ivosidenib [IDH1] and enasidenib [IDH2]), lactate dehydrogenase inhibitors (galloflavin), OxPhos inhibitors (venetoclax plus azacytidine [165], gamitrinib [166]), mitochondrial ETC inhibitors (metformin, deguelin, and rotenone [complex I], and oligomycin and gboxin [complex V]) [167], (2) inhibiting mitochondrial upregulators, including mTOR inhibitors (temsirolimus) [168], (3) inhibiting mitochondrial protein translation (tigecycline) [169], and (4) mitochondrial apoptosis inducers, including BH3-mimetics [170]. In this regard, an emerging study (OPTIMUM, NCT04945148) aims to evaluate the impact of adding metformin (an OxPhos inhibitor) in improving the efficacy of available standard treatment in patients with glioblastoma (radiotherapy plus temozolomide). Table 2 outlines the list of clinical trials evaluating the impact of antimitochondrial therapy on radiotherapy efficacy.
Table 2.
Trials on the combination of radiotherapy and therapeutic targeting of mitochondrial metabolism (source http://clinicaltrials.gov).
As noted in Section 2, Saha et al. demonstrated that cancer cells can improve their mitochondrial content by hijacking mitochondria from immune cells [2]. In addition, cancer cells can enhance their neighboring cells’ malignancy by transferring mitochondria between themselves. This behavior has been demonstrated by Lu et al. in bladder cancer cells [171]. Saha et al. demonstrated that the Ras/Rho GTPase signaling is actively implicated in the nanotube formation. They showed that a farnesyltransferase and geranylgeranyltransferase type 1 inhibitor (L-778123) could effectively inhibit the nanotube formation [2]. Therefore, L-778123 (and similar agents) can effectively reduce the mitochondrial content of the cancer cells.
In addition, nanotechnology-based platforms have been applied to target cancer cells’ mitochondria. In comparison with non-mitochondria-targeting approaches, mitochondria-targeting nanomaterials have overcome the limitations of photodynamic therapy (PDT) (e.g., hypoxia) and photothermal therapy (PTT) and have improved the penetration and intra-mitochondrial accumulation of chemotherapeutics [172]. For instance, in 2020, Xu et al. introduced a polypyrrole-silica (Py@Si)-based hybrid nanoparticle to improve doxorubicin accumulation in CD44+ cancer cells [173]. To successfully deliver nanocarriers into the mitochondria, it is crucial to consider the challenges posed by the hydrophobic and double membrane of mitochondria, as well as its highly negative potential [174]. To overcome these barriers, several strategies have been applied in the design of nanocarriers. For example, adding lipophilic cations, peptides, or aptamers to polymeric nanoparticles can enable them to penetrate the mitochondrial matrix [175].
A novel approach to target the cancer cells’ mitochondria is put forward here. Taking a look at the mitochondrial ETC, another mitochondrial-targeting approach can be considered. Based on the electromagnetic principles, electric flux creates a magnetic field surrounding it [176] (Figure 2):
where B is Magnetic field; I is Current; r is Distance from the conductor; and μo is Permeability of free space (=4π × 10−7 N/A2).
B = μoI/2πr
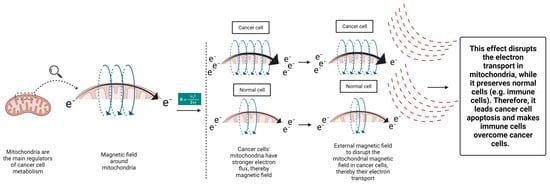
Figure 2.
The proposed method to target cancer cells’ mitochondria using magnetic fields.
Hence, we may envision a magnetic field surrounding a mitochondrion due to its active ETC. Studies have shown that cancer cells’ mitochondria have a stronger electron flux in their ETC compared to normal cells, which aids in responding to their metabolism [177]. This characteristic can serve as an opportunity to target cancer cells’ mitochondria by an extrinsic therapeutic magnetic field, with specific intensity sparing the normal cells’ mitochondria. Electrons generated from different metabolic processes are channeled towards the mitochondrial ETC to support the cellular metabolic pathways. By disrupting the cancer cells’ mitochondria, their ETC become impaired (reverse direction of Formula 1). This effect can eventually turn off the power button of cancer cells’ mitochondria and impede their support on cancer cells’ metabolism and TME. This concept was applied in a recent in vitro study by Sharpe et al. The investigators demonstrated that applying oscillating magnetic fields with appropriate field strength, frequency, and on/off profiles could effectively arrest the cancer cells ETC, even in a nondividing status [178]. Considering the following information, the impact of external magnetic fields can be more potent in in vivo studies. Several studies have demonstrated that low-frequency external magnetic fields can modulate the tumor immune microenvironment and improve the antitumor immune response. Nie et al. demonstrated that the low-frequency magnetic field could enhance the survival of melanoma and hepatocellular murine models by reducing the number of regulatory T cells and increasing the number of CD8+ T cells and dendritic cells in TME [179,180]. It has been demonstrated that alternating magnetic fields can enhance ROS release from immune cells [181], representing mitochondrial metabolism [182]. These benefits were not reported for static magnetic fields [181]. As mentioned earlier, cancer cells’ functional mitochondria can make it more difficult for immune cells to penetrate the TME due to its harsh conditions, such as acidity, low glucose, and hypoxia. Therefore, cancer’s mitochondria-targeting magnetic field can remove the cancer cells’ support on TME modification and can increase the chance of cancer treatment. When cancer cells are exposed, their metabolism is disrupted and their support on the TME is reduced. Therefore, alternating magnetic fields can both weaken the tumor cells and activate the immune cells and facilitate their infiltration into TME to defeat the weakened cancer cells. These beneficial effects can potentially improve the response to different treatment modalities, given the importance of immune reactivation in radiotherapy [183], chemotherapy [121], targeted therapies [184], and immunotherapy [28]. This concept is in its preliminary phases and enfaces several shortcomings. For example, it is still unclear to what degree mitochondria contribute to the potential therapeutic effect of rotating external magnetic fields in cancer cells. In order to fully comprehend how magnetic fields, affect cancer cells, it is necessary to conduct multidisciplinary research that combines experimental studies and theoretical modeling (Figure 2).
9. Conclusions
This article illustrated how mitochondria is involved in the tumor response to different treatments as well as the normal tissue toxicity. Ever since, personalized treatment has been primarily based on genomic analyses. This paper put forward that considering the mitochondrial metabolism status of the cancer cells can provide additional information in selecting the appropriate treatment. With this concept in mind, future works can design more personalized treatments to improve the treatment results with fewer toxicities. Heteroplasmy analysis in personalized oncology provides insights into the genetic landscape of tumors, helps predict clinical outcomes, guides treatment decisions, and offers opportunities for the development of personalized therapeutic approaches. However, further research is needed to fully understand the impact of heteroplasmy and optimize its clinical utility in oncology.
Author Contributions
Conceptualization, B.B. and F.T.-H.; methodology, B.B. and F.T.-H.; software, F.T.-H.; validation, B.B. and F.T.-H.; investigation, B.B. and F.T.-H.; resources, B.B. and F.T.-H.; data curation, B.B. and F.T.-H.; writing—original draft preparation, F.T.-H.; writing—review and editing, B.B. and F.T.-H.; visualization, F.T.-H.; supervision, B.B.; project administration, B.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Trapani, D.; Franzoi, M.A.; Burstein, H.J.; Carey, L.A.; Delaloge, S.; Harbeck, N.; Hayes, D.F.; Kalinsky, K.; Pusztai, L.; Regan, M.M.; et al. Risk-adapted modulation through de-intensification of cancer treatments: An ESMO classification. Ann. Oncol. 2022, 33, 702–712. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022, 17, 98–106. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J.; Aznar, M.C.; Bacchus, C.; Coppes, R.P.; Deutsch, E.; Georg, D.; Haustermans, K.; Hoskin, P.; Krause, M.; Lartigau, E.F.; et al. Personalised radiation therapy taking both the tumour and patient into consideration. Radiother. Oncol. 2022, 166, A1–A5. [Google Scholar] [CrossRef]
- Abdelkarem, O.A.I.; Choudhury, A.; Burnet, N.G.; Summersgill, H.R.; West, C.M.L. Effect of Race and Ethnicity on Risk of Radiotherapy Toxicity and Implications for Radiogenomics. Clin. Oncol. 2022, 34, 653–669. [Google Scholar] [CrossRef]
- Ameri, A.; Heydarirad, G.; Choopani, R.; Poshtmahi, S.; Ameri, P.; Talebi, F.; Bagheri Pour, A.; Taghizadeh-Hesary, F. Sumac-rose water mouthwash versus benzydamine to prevent radiation-induced oral mucositis in head and neck cancers: A phase II randomized trial. J. Cancer Res. Clin. Oncol. 2023, 149, 7427–7439. [Google Scholar] [CrossRef]
- Ameri, A.; Rahnama, N.; Talebi, F.; Sourati, A.; Taghizadeh-Hesary, F. An evaluation of cancer aging research group (CARG) score to predict chemotherapy toxicity in older Iranian patients with cancer. Oncologie 2023, 25, 223–232. [Google Scholar] [CrossRef]
- Selvakumar, S.C.; Preethi, K.A.; Ross, K.; Tusubira, D.; Khan, M.W.A.; Mani, P.; Rao, T.N.; Sekar, D. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol. Cancer 2022, 21, 83. [Google Scholar] [CrossRef]
- Taghizadeh-Hesary, F.; Akbari, H.; Bahadori, M.; Behnam, B. Targeted Anti-Mitochondrial Therapy: The Future of Oncology. Genes 2022, 13, 1728. [Google Scholar] [CrossRef]
- van Gisbergen, M.W.; Offermans, K.; Voets, A.M.; Lieuwes, N.G.; Biemans, R.; Hoffmann, R.F.; Dubois, L.J.; Lambin, P. Mitochondrial Dysfunction Inhibits Hypoxia-Induced HIF-1α Stabilization and Expression of Its Downstream Targets. Front. Oncol. 2020, 10, 770. [Google Scholar] [CrossRef]
- Choudhury, F.K. Mitochondrial Redox Metabolism: The Epicenter of Metabolism during Cancer Progression. Antioxidants 2021, 10, 1838. [Google Scholar] [CrossRef]
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753. [Google Scholar] [CrossRef]
- Druker, J.; Wilson, J.W.; Child, F.; Shakir, D.; Fasanya, T.; Rocha, S. Role of Hypoxia in the Control of the Cell Cycle. Int. J. Mol. Sci. 2021, 22, 4874. [Google Scholar] [CrossRef]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef]
- Mandal, J.P.; Shiue, C.N.; Chen, Y.C.; Lee, M.C.; Yang, H.H.; Chang, H.H.; Hu, C.T.; Liao, P.C.; Hui, L.C.; You, R.I.; et al. PKCδ mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free Radic. Biol. Med. 2021, 163, 69–87. [Google Scholar] [CrossRef]
- Yu-Lee, L.-Y.; Lee, Y.-C.; Pan, J.; Lin, S.-C.; Pan, T.; Yu, G.; Hawke, D.H.; Pan, B.-F.; Lin, S.-H. Bone secreted factors induce cellular quiescence in prostate cancer cells. Sci. Rep. 2019, 9, 18635. [Google Scholar] [CrossRef]
- O’Reilly, E.; Zeinabad, H.A.; Szegezdi, E. Hematopoietic versus leukemic stem cell quiescence: Challenges and therapeutic opportunities. Blood Rev. 2021, 50, 100850. [Google Scholar] [CrossRef]
- Shiau, J.P.; Chuang, Y.T.; Cheng, Y.B.; Tang, J.Y.; Hou, M.F.; Yen, C.Y.; Chang, H.W. Impacts of Oxidative Stress and PI3K/AKT/mTOR on Metabolism and the Future Direction of Investigating Fucoidan-Modulated Metabolism. Antioxidants 2022, 11, 911. [Google Scholar] [CrossRef]
- Czowski, B.J.; Romero-Moreno, R.; Trull, K.J.; White, K.A. Cancer and pH Dynamics: Transcriptional Regulation, Proteostasis, and the Need for New Molecular Tools. Cancers 2020, 12, 2760. [Google Scholar] [CrossRef]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int. J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef] [PubMed]
- Towers, C.G.; Fitzwalter, B.E.; Regan, D.; Goodspeed, A.; Morgan, M.J.; Liu, C.W.; Gustafson, D.L.; Thorburn, A. Cancer Cells Upregulate NRF2 Signaling to Adapt to Autophagy Inhibition. Dev. Cell 2019, 50, 690–703.e696. [Google Scholar] [CrossRef]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Lambeth, D.O. What is the function of GTP produced in the Krebs citric acid cycle? IUBMB Life 2002, 54, 143–144. [Google Scholar] [CrossRef]
- Akbari, H.; Taghizadeh-Hesary, F.; Bahadori, M. Mitochondria determine response to anti-programmed cell death protein-1 (anti-PD-1) immunotherapy: An evidence-based hypothesis. Mitochondrion 2022, 62, 151–158. [Google Scholar] [CrossRef]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef]
- Kadonosono, T.; Miyamoto, K.; Sakai, S.; Matsuo, Y.; Kitajima, S.; Wang, Q.; Endo, M.; Niibori, M.; Kuchimaru, T.; Soga, T.; et al. AGE/RAGE axis regulates reversible transition to quiescent states of ALK-rearranged NSCLC and pancreatic cancer cells in monolayer cultures. Sci. Rep. 2022, 12, 9886. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Taghizadeh-Hesary, F.; Houshyari, M.; Farhadi, M. Mitochondrial metabolism: A predictive biomarker of radiotherapy efficacy and toxicity. J. Cancer Res. Clin. Oncol. 2023, 149, 6719–6741. [Google Scholar] [CrossRef]
- Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Brunsell, T.H.; Langerud, J.; Berg, K.C.G.; Røsok, B.I.; Bjørnbeth, B.A.; Nesbakken, A.; Lothe, R.A.; et al. Metastatic heterogeneity of the consensus molecular subtypes of colorectal cancer. NPJ Genom. Med. 2021, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Fazilaty, H.; Gardaneh, M.; Akbari, P.; Zekri, A.; Behnam, B. SLUG and SOX9 Cooperatively Regulate Tumor Initiating Niche Factors in Breast Cancer. Cancer Microenviron. 2016, 9, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Fazilaty, H.; Gardaneh, M.; Bahrami, T.; Salmaninejad, A.; Behnam, B. Crosstalk between breast cancer stem cells and metastatic niche: Emerging molecular metastasis pathway? Tumour Biol. 2013, 34, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Kim, M.; Jang, K.; Miller, P.; Picon-Ruiz, M.; Yeasky, T.M.; El-Ashry, D.; Slingerland, J.M. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene 2017, 36, 5199–5211. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers 2019, 11, 729. [Google Scholar] [CrossRef]
- Paupe, V.; Prudent, J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem. Biophys. Res. Commun. 2018, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- McCann, E.; O’Sullivan, J.; Marcone, S. Targeting cancer-cell mitochondria and metabolism to improve radiotherapy response. Transl. Oncol. 2021, 14, 100905. [Google Scholar] [CrossRef]
- Houshyari, M.; Taghizadeh-Hesary, F. Is Mitochondrial Metabolism a New Predictive Biomarker for Antiprogrammed Cell Death Protein-1 Immunotherapy? JCO Oncol. Pract. 2022, 19, 123–124. [Google Scholar] [CrossRef]
- Bentle, M.S.; Reinicke, K.E.; Bey, E.A.; Spitz, D.R.; Boothman, D.A. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J. Biol. Chem. 2006, 281, 33684–33696. [Google Scholar] [CrossRef]
- Lévy, N.; Martz, A.; Bresson, A.; Spenlehauer, C.; de Murcia, G.; Ménissier-de Murcia, J. XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res. 2006, 34, 32–41. [Google Scholar] [CrossRef]
- Kozlov, S.; Gueven, N.; Keating, K.; Ramsay, J.; Lavin, M.F. ATP activates ataxia-telangiectasia mutated (ATM) in vitro. Importance of autophosphorylation. J. Biol. Chem. 2003, 278, 9309–9317. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Rakotomalala, A.; Escande, A.; Furlan, A.; Meignan, S.; Lartigau, E. Hypoxia in Solid Tumors: How Low Oxygenation Impacts the “Six Rs” of Radiotherapy. Front. Endocrinol. 2021, 12, 742215. [Google Scholar] [CrossRef]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165715. [Google Scholar] [CrossRef]
- Syljuåsen, R.G. Cell cycle effects in radiation oncology. In Radiation Oncology; Wentz, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–8. [Google Scholar]
- Salazar-Roa, M.; Malumbres, M. Fueling the Cell Division Cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef]
- Suwa, T.; Kobayashi, M.; Nam, J.-M.; Harada, H. Tumor microenvironment and radioresistance. Exp. Mol. Med. 2021, 53, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef]
- Ericson, N.G.; Kulawiec, M.; Vermulst, M.; Sheahan, K.; O’Sullivan, J.; Salk, J.J.; Bielas, J.H. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Chen, L.; Yu, B.; Xue, Y.; Guo, R.; Su, J.; Liu, Y.; Sun, L. p53/PGC-1α-mediated mitochondrial dysfunction promotes PC3 prostate cancer cell apoptosis. Mol. Med. Rep. 2020, 22, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yu, D.; Wang, Z.; Li, S. Relationship between p53 status and the bioeffect of ionizing radiation. Oncol. Lett. 2021, 22, 661. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jarvis, I.W.H.; Bottai, M.; Dreij, K.; Stenius, U. TGF beta promotes repair of bulky DNA damage through increased ERCC1/XPF and ERCC1/XPA interaction. Carcinogenesis 2018, 40, 580–591. [Google Scholar] [CrossRef]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef]
- Hu, J.W.; Sun, P.; Zhang, D.X.; Xiong, W.J.; Mi, J. Hexokinase 2 regulates G1/S checkpoint through CDK2 in cancer-associated fibroblasts. Cell Signal 2014, 26, 2210–2216. [Google Scholar] [CrossRef]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 721–726. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Tran, A.N.; Lai, A.; Li, S.; Pope, W.B.; Teixeira, S.; Harris, R.J.; Woodworth, D.C.; Nghiemphu, P.L.; Cloughesy, T.F.; Ellingson, B.M. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol. 2014, 16, 414–420. [Google Scholar] [CrossRef]
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J. Exp. Med. 2021, 218, e20200924. [Google Scholar] [CrossRef]
- Li, S.; Sun, C.; Gu, Y.; Gao, X.; Zhao, Y.; Yuan, Y.; Zhang, F.; Hu, P.; Liang, W.; Cao, K.; et al. Mutation of IDH1 aggravates the fatty acid-induced oxidative stress in HCT116 cells by affecting the mitochondrial respiratory chain. Mol. Med. Rep. 2019, 19, 2509–2518. [Google Scholar] [CrossRef]
- Atlante, A.; Calissano, P.; Bobba, A.; Azzariti, A.; Marra, E.; Passarella, S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J. Biol. Chem. 2000, 275, 37159–37166. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Guillot, C.; Favaudon, V.; Herceg, Z.; Sagne, C.; Sauvaigo, S.; Merle, P.; Hall, J.; Chemin, I. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitrohepatocellular carcinoma models. BMC Cancer 2014, 14, 603. [Google Scholar] [CrossRef]
- Singh, D.D.; Parveen, A.; Yadav, D.K. Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines 2021, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, e1437. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yi, J.; Tao, L.; Huang, G.; Chu, X.; Song, H.; Chen, L. Wnt signaling induces radioresistance through upregulating HMGB1 in esophageal squamous cell carcinoma. Cell Death Dis. 2018, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; Van Houten, B.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011, 13, 701–711. [Google Scholar] [CrossRef]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-κB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef]
- Pour Khavari, A.; Liu, Y.; He, E.; Skog, S.; Haghdoost, S. Serum 8-Oxo-dG as a Predictor of Sensitivity and Outcome of Radiotherapy and Chemotherapy of Upper Gastrointestinal Tumours. Oxid. Med. Cell Longev. 2018, 2018, 4153574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sun, M.; Li, G.H.; Wu, Y.Z.; Wang, Y.; Jin, F.; Zhang, Y.Y.; Yang, L.; Wang, D.L. Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells. Oncol. Rep. 2013, 30, 1793–1801. [Google Scholar] [CrossRef]
- Eaton, J.S.; Lin, Z.P.; Sartorelli, A.C.; Bonawitz, N.D.; Shadel, G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Investig. 2007, 117, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Vaezi, A.; Feldman, C.H.; Niedernhofer, L.J. ERCC1 and XRCC1 as biomarkers for lung and head and neck cancer. Pharmgenomics Pers. Med. 2011, 4, 47–63. [Google Scholar] [CrossRef]
- Xu, C.; Xu, J.; Ji, G.; Liu, Q.; Shao, W.; Chen, Y.; Gu, J.; Weng, Z.; Zhang, X.; Wang, Y. Deficiency of X-ray repair cross-complementing group 1 in primordial germ cells contributes to male infertility. FASEB J. 2019, 33, 7427–7436. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, S.; Pang, J.; Yin, J.; Zhang, J.; Mu, D.; Tang, S.; Li, L.; Bao, H.; Wu, X. Genomic Profiling Reveals Novel Predictive Biomarkers for Chemo-Radiotherapy Toxicity and Efficacy in Non-Small-Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, e437. [Google Scholar] [CrossRef]
- Fan, F.; Zhuang, J.; Zhou, P.; Liu, X.; Luo, Y. MicroRNA-34a promotes mitochondrial dysfunction-induced apoptosis in human lens epithelial cells by targeting Notch2. Oncotarget 2017, 8, 110209–110220. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Choubey, V.; Gupta, R.; Kuum, M.; Safiulina, D.; Vaarmann, A.; Gogichaishvili, N.; Liiv, M.; Ilves, I.; Tämm, K.; et al. A novel role of KEAP1/PGAM5 complex: ROS sensor for inducing mitophagy. Redox Biol. 2021, 48, 102186. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Fan, J.; Chung, T.-W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine Phosphorylation of Mitochondrial Pyruvate Dehydrogenase Kinase 1 Is Important for Cancer Metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, B.; Xiao, H.; Dong, J.; Li, Y.; Zhu, C.; Jin, Y.; Li, H.; Cui, M.; Fan, S. LncRNA HOTAIR enhances breast cancer radioresistance through facilitating HSPA1A expression via sequestering miR-449b-5p. Thorac. Cancer 2020, 11, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xiong, Q.; Wu, Y.; Chen, Y.; Chen, Z.; Fleming, J.; Gao, D.; Bi, L.; Ge, F. Quantitative Proteomics Analysis Reveals Novel Insights into Mechanisms of Action of Long Noncoding RNA Hox Transcript Antisense Intergenic RNA (HOTAIR) in HeLa Cells. Mol. Cell Proteomics 2015, 14, 1447–1463. [Google Scholar] [CrossRef]
- Kong, L.; Zhou, X.; Wu, Y.; Wang, Y.; Chen, L.; Li, P.; Liu, S.; Sun, S.; Ren, Y.; Mei, M. Targeting HOTAIR induces mitochondria related apoptosis and inhibits tumor growth in head and neck squamous cell carcinoma in vitro and in vivo. Curr. Mol. Med. 2015, 15, 952–960. [Google Scholar] [CrossRef]
- Hashimoto, T.; Urushihara, Y.; Murata, Y.; Fujishima, Y.; Hosoi, Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem. Biophys. Res. Commun. 2022, 590, 82–88. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Pedersen, H.; Anne Adanma Obara, E.; Elbæk, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int. J. Mol. Sci. 2020, 21, 1588. [Google Scholar] [CrossRef]
- Liu, X.; Shan, G. Mitochondria Encoded Non-coding RNAs in Cell Physiology. Front. Cell Dev. Biol. 2021, 9, 713729. [Google Scholar] [CrossRef]
- Ma, J.; Lu, Y.; Zhang, S.; Li, Y.; Huang, J.; Yin, Z.; Ren, J.; Huang, K.; Liu, L.; Yang, K.; et al. β-Trcp ubiquitin ligase and RSK2 kinase-mediated degradation of FOXN2 promotes tumorigenesis and radioresistance in lung cancer. Cell Death Differ. 2018, 25, 1473–1485. [Google Scholar] [CrossRef]
- Mrozowski, R.M. Targeting the Ser/Thr Protein Kinase RSK to Reduce Breast Cancer Metastasis; Vanderbilt University: Nashville, TN, USA, 2015. [Google Scholar]
- Chu, C.; Niu, X.; Ou, X.; Hu, C. LAPTM4B knockdown increases the radiosensitivity of EGFR-overexpressing radioresistant nasopharyngeal cancer cells by inhibiting autophagy. Onco Targets Ther. 2019, 12, 5661–5677. [Google Scholar] [CrossRef]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Yadav, P.; Sainis, K.B.; Shankar, B.S. TNF-α and IGF-1 differentially modulate ionizing radiation responses of lung cancer cell lines. Cytokine 2018, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.; Jung, H.; Lee, H.; Singh, K.; Roy, M.; Gohel, D.; Kim, H.B.; Mane, M.; Vasiyani, H.; Currim, F.; et al. TNF-α differentially modulates subunit levels of respiratory electron transport complexes of ER/PR +ve/−ve breast cancer cells to regulate mitochondrial complex activity and tumorigenic potential. Cancer Metab. 2021, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondrial complex III: An essential component of universal oxygen sensing machinery? Respir. Physiol. Neurobiol. 2010, 174, 175–181. [Google Scholar] [CrossRef]
- Dörr, W. Radiobiology of tissue reactions. Ann. ICRP 2015, 44, 58–68. [Google Scholar] [CrossRef]
- Nicolatou-Galitis, O.; Bossi, P.; Orlandi, E.; René-Jean, B. The role of benzydamine in prevention and treatment of chemoradiotherapy-induced mucositis. Support. Care Cancer 2021, 29, 5701–5709. [Google Scholar] [CrossRef]
- Holley, A.K.; Miao, L.; St Clair, D.K.; St Clair, W.H. Redox-modulated phenomena and radiation therapy: The central role of superoxide dismutases. Antioxid. Redox Signal 2014, 20, 1567–1589. [Google Scholar] [CrossRef]
- Drobin, K.; Marczyk, M.; Halle, M.; Danielsson, D.; Papiez, A.; Sangsuwan, T.; Bendes, A.; Hong, M.G.; Qundos, U.; Harms-Ringdahl, M.; et al. Molecular Profiling for Predictors of Radiosensitivity in Patients with Breast or Head-and-Neck Cancer. Cancers 2020, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Groenendyk, J.; Michalak, M. Binding Proteins|Ca2+ Binding/Buffering Proteins: ER Luminal Proteins☆. In Encyclopedia of Biological Chemistry III, 3rd ed.; Jez, J., Ed.; Elsevier: Oxford, UK, 2021; pp. 534–546. [Google Scholar]
- Henke, N.; Albrecht, P.; Pfeiffer, A.; Toutzaris, D.; Zanger, K.; Methner, A. Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J. Biol. Chem. 2012, 287, 42042–42052. [Google Scholar] [CrossRef] [PubMed]
- Dewson, G. Bax to the wall: Bax-and Bak-induced mitochondrial dysfunction in apoptosis. Trends Biochem. Sci. 2001, 26, 353. [Google Scholar] [CrossRef]
- Bonanno, J.A.; Shyam, R.; Choi, M.; Ogando, D.G. The H(+) Transporter SLC4A11: Roles in Metabolism, Oxidative Stress and Mitochondrial Uncoupling. Cells 2022, 11, 197. [Google Scholar] [CrossRef] [PubMed]
- Sprung, C.N.; Forrester, H.B.; Siva, S.; Martin, O.A. Immunological markers that predict radiation toxicity. Cancer Lett. 2015, 368, 191–197. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef]
- Hernández, L.; Terradas, M.; Camps, J.; Martín, M.; Tusell, L.; Genescà, A. Aging and radiation: Bad companions. Aging Cell 2015, 14, 153–161. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Lawler, J.M.; Demaree, S.R. Relationship between NADP-specific isocitrate dehydrogenase and glutathione peroxidase in aging rat skeletal muscle. Mech. Ageing Dev. 2001, 122, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Tepper, J.E. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA Cancer J. Clin. 2021, 71, 437–454. [Google Scholar] [CrossRef]
- Madani, A.; Alack, K.; Richter, M.J.; Krüger, K. Immune-regulating effects of exercise on cigarette smoke-induced inflammation. J. Inflamm. Res. 2018, 11, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Willenborg, S.; Sanin, D.E.; Jais, A.; Ding, X.; Ulas, T.; Nüchel, J.; Popović, M.; MacVicar, T.; Langer, T.; Schultze, J.L.; et al. Mitochondrial metabolism coordinates stage-specific repair processes in macrophages during wound healing. Cell Metab. 2021, 33, 2398–2414.e2399. [Google Scholar] [CrossRef]
- Pratson, C.L.; Larkins, M.C.; Karimian, B.H.; Curtis, C.M.; Lepera, P.A.; Brodish, B.N.; Ju, A.W. The Impact of Smoking, Alcohol Use, Recurrent Disease, and Age on the Development of Neck Fibrosis in Head and Neck Cancer Patients Following Radiation Therapy. Front. Oncol. 2021, 11, 707418. [Google Scholar] [CrossRef]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J. Physiol. 2021, 599, 803–817. [Google Scholar] [CrossRef]
- Luoma, R.L.; Butler, M.W.; Stahlschmidt, Z.R. Plasticity of immunity in response to eating. J. Exp. Biol. 2016, 219, 1965–1968. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-Chain Amino Acid Supplementation Promotes Survival and Supports Cardiac and Skeletal Muscle Mitochondrial Biogenesis in Middle-Aged Mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Shanmugam, H.; Abdallah, H.; John Britto, J.S.; Galerati, I.; Gómez-Ambrosi, J.; Frühbeck, G.; Portincasa, P. The Potential of the Mediterranean Diet to Improve Mitochondrial Function in Experimental Models of Obesity and Metabolic Syndrome. Nutrients 2022, 14, 3112. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.R.; Macedo, G.E.; Martins, I.K.; Gomes, K.K.; de Carvalho, N.R.; Posser, T.; Franco, J.L. Short-term sleep deprivation with exposure to nocturnal light alters mitochondrial bioenergetics in Drosophila. Free Radic. Biol. Med. 2018, 120, 395–406. [Google Scholar] [CrossRef]
- De Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.A.; Singal, A.K. Mitochondrial dysfunction and alcohol-associated liver disease: A novel pathway and therapeutic target. Signal Transduct. Target. Ther. 2020, 5, 26. [Google Scholar] [CrossRef]
- Malińska, D.; Więckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymański, J.; Mathis, C.; Van der Toorn, M.; et al. Mitochondria as a possible target for nicotine action. J. Bioenerg. Biomembr. 2019, 51, 259–276. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef]
- Pizzorno, J. Mitochondria-Fundamental to Life and Health. Integr. Med. 2014, 13, 8–15. [Google Scholar]
- Burtscher, J.; Romani, M.; Bernardo, G.; Popa, T.; Ziviani, E.; Hummel, F.C.; Sorrentino, V.; Millet, G.P. Boosting mitochondrial health to counteract neurodegeneration. Prog. Neurobiol. 2022, 215, 102289. [Google Scholar] [CrossRef]
- Fendt, L.; Fazzini, F.; Weissensteiner, H.; Bruckmoser, E.; Schönherr, S.; Schäfer, G.; Losso, J.L.; Streiter, G.A.; Lamina, C.; Rasse, M.; et al. Profiling of Mitochondrial DNA Heteroplasmy in a Prospective Oral Squamous Cell Carcinoma Study. Cancers 2020, 12, 1933. [Google Scholar] [CrossRef]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Kloss-Brandstätter, A.; Schäfer, G.; Erhart, G.; Hüttenhofer, A.; Coassin, S.; Seifarth, C.; Summerer, M.; Bektic, J.; Klocker, H.; Kronenberg, F. Somatic mutations throughout the entire mitochondrial genome are associated with elevated PSA levels in prostate cancer patients. Am. J. Hum. Genet. 2010, 87, 802–812. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wei, Y.; Wang, Q.; Xu, H.; Wang, Y.; Yao, A.; Yang, H.; Gao, Y.; Zhou, F. Heteroplasmy of mutant mitochondrial DNA A10398G and analysis of its prognostic value in non-small cell lung cancer. Oncol. Lett. 2016, 12, 3081–3088. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.-S.; Wang, H.-S.; Mugaka, B.P.; Yang, G.-J.; Ding, Y. Mitochondria: Promising organelle targets for cancer diagnosis and treatment. Biomater. Sci. 2018, 6, 2786–2797. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef]
- Chen, Y.; Gong, L.; Liu, X.; Chen, X.; Yang, S.; Luo, Y. Mitochondrial DNA genomes revealed different patterns of high-altitude adaptation in high-altitude Tajiks compared with Tibetans and Sherpas. Sci. Rep. 2020, 10, 10592. [Google Scholar] [CrossRef]
- Dong, J.; Wong, L.J.; Mims, M.P. Mitochondrial inheritance and cancer. Transl. Res. 2018, 202, 24–34. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress. 2020, 4, 114–146. [Google Scholar] [CrossRef] [PubMed]
- Lajbner, Z.; Pnini, R.; Camus, M.F.; Miller, J.; Dowling, D.K. Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci. Rep. 2018, 8, 9500. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Fu, Q.; Xu, B.; Zhou, H.; Gao, J.; Shao, X.; Xiong, J.; Gu, Q.; Wen, S.; Li, F.; et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int. J. Cancer 2018, 142, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Motoi, M.; Nishimura, T.; Egashira, Y.; Kishida, F.; Watanuki, S. Relationship between mitochondrial haplogroup and physiological responses to hypobaric hypoxia. J. Physiol. Anthropol. 2016, 35, 12. [Google Scholar] [CrossRef]
- Toncheva, D.; Serbezov, D.; Karachanak-Yankova, S.; Nesheva, D. Ancient mitochondrial DNA pathogenic variants putatively associated with mitochondrial disease. PLoS ONE 2020, 15, e0233666. [Google Scholar] [CrossRef]
- Xiao, F.; Li, M.; Wang, J.; Liu, J.; Li, J.; Fang, H.; Lyu, J.; Shen, L. Association between mitochondrial DNA haplogroup variation and coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 960–966. [Google Scholar] [CrossRef]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef]
- Lechuga-Vieco, A.V.; Latorre-Pellicer, A.; Johnston, I.G.; Prota, G.; Gileadi, U.; Justo-Méndez, R.; Acín-Pérez, R.; Martínez-de-Mena, R.; Fernández-Toro, J.M.; Jimenez-Blasco, D.; et al. Cell identity and nucleo-mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Sci. Adv. 2020, 6, eaba5345. [Google Scholar] [CrossRef]
- Crooks, D.R.; Maio, N.; Lang, M.; Ricketts, C.J.; Vocke, C.D.; Gurram, S.; Turan, S.; Kim, Y.Y.; Cawthon, G.M.; Sohelian, F.; et al. Mitochondrial DNA alterations underlie an irreversible shift to aerobic glycolysis in fumarate hydratase-deficient renal cancer. Sci. Signal 2021, 14, eabc4436. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, J.H.; Kim, D.K.; Keum, D.Y. Nuclear and mitochondrial DNAs microsatellite instability and mitochondrial DNA copy number in adenocarcinoma and squamous cell carcinoma of lung: A pilot study. Apmis 2015, 123, 1048–1054. [Google Scholar] [CrossRef]
- Qiao, L.; Ru, G.; Mao, Z.; Wang, C.; Nie, Z.; Li, Q.; Huang-Yang, Y.; Zhu, L.; Liang, X.; Yu, J.; et al. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017, 8, 84373–84383. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wan, J.; Song, R.; Zhao, H. Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: A replication study. Carcinogenesis 2015, 36, 1307–1313. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Zorzano, A. Mitochondrial Dynamics and Liver Cancer. Cancers 2021, 13, 2571. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.S.; Mukherjee, S. Mitophagy in Carcinogenesis and Tumour Progression- A New Paradigm with Emerging Importance. Anti-Cancer Agents Med. Chem. 2021, 21, 2130–2141. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Pérez-Amado, C.J.; Bazan-Cordoba, A.; Hidalgo-Miranda, A.; Jiménez-Morales, S. Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. Int. J. Mol. Sci. 2021, 22, 7369. [Google Scholar] [CrossRef]
- Dickerson, T.; Jauregui, C.E.; Teng, Y. Friend or foe? Mitochondria as a pharmacological target in cancer treatment. Future Med. Chem. 2017, 9, 2197–2210. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 6014. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Eckl, E.M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Martin, T.D.; Cook, D.R.; Choi, M.Y.; Li, M.Z.; Haigis, K.M.; Elledge, S.J. A Role for Mitochondrial Translation in Promotion of Viability in K-Ras Mutant Cells. Cell Rep. 2017, 20, 427–438. [Google Scholar] [CrossRef]
- Gilmore, A.; King, L. Emerging approaches to target mitochondrial apoptosis in cancer cells. F1000Res 2019, 8, F1000. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef]
- Xu, J.; Shamul, J.G.; Kwizera, E.A.; He, X. Recent Advancements in Mitochondria-Targeted Nanoparticle Drug Delivery for Cancer Therapy. Nanomaterials 2022, 12, 743. [Google Scholar] [CrossRef]
- Xu, J.; Shamul, J.G.; Wang, H.; Lin, J.; Agarwal, P.; Sun, M.; Lu, X.; Tkaczuk, K.H.R.; He, X. Targeted Heating of Mitochondria Greatly Augments Nanoparticle-Mediated Cancer Chemotherapy. Adv. Healthc. Mater. 2020, 9, e2000181. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Q.; Xia, X.; Li, X.; Ruan, W.; Zheng, M.; Zou, Y.; Shi, B. Polymeric Nanoparticles for Mitochondria Targeting Mediated Robust Cancer Therapy. Front. Bioeng. Biotechnol. 2021, 9, 755727. [Google Scholar] [CrossRef]
- SAllemailem, K.; Almatroudi, A.; Alsahli, M.A.; Aljaghwani, A.; MEl-Kady, A.; Rahmani, A.H.; Khan, A.A. Novel Strategies for Disrupting Cancer-Cell Functions with Mitochondria-Targeted Antitumor Drug-Loaded Nanoformulations. Int. J. Nanomed. 2021, 16, 3907–3936. [Google Scholar] [CrossRef] [PubMed]
- David, H.; Staelin. Forces on Free Charges and Currents. Available online: https://phys.libretexts.org/Bookshelves/Electricity_and_Magnetism/Electromagnetics_and_Applications_(Staelin)/05%3A_Electromagnetic_Forces/5.01%3A_Forces_on_free_charges_and_currents (accessed on 28 June 2023).
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.A.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Helekar, S.A. Rotating Magnetic Fields Inhibit Mitochondrial Respiration, Promote Oxidative Stress and Produce Loss of Mitochondrial Integrity in Cancer Cells. Front. Oncol. 2021, 11, 768758. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Du, L.; Mou, Y.; Xu, Z.; Weng, L.; Du, Y.; Zhu, Y.; Hou, Y.; Wang, T. Effect of low frequency magnetic fields on melanoma: Tumor inhibition and immune modulation. BMC Cancer 2013, 13, 582. [Google Scholar] [CrossRef]
- Nie, Y.; Chen, Y.; Mou, Y.; Weng, L.; Xu, Z.; Du, Y.; Wang, W.; Hou, Y.; Wang, T. Low frequency magnetic fields enhance antitumor immune response against mouse H22 hepatocellular carcinoma. PLoS ONE 2013, 8, e72411. [Google Scholar] [CrossRef]
- Lei, H.; Pan, Y.; Wu, R.; Lv, Y. Innate Immune Regulation Under Magnetic Fields with Possible Mechanisms and Therapeutic Applications. Front. Immunol. 2020, 11, 582772. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef]
- Boustani, J.; Grapin, M.; Laurent, P.A.; Apetoh, L.; Mirjolet, C. The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers 2019, 11, 768758. [Google Scholar] [CrossRef]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).