
Genetics of ABCB1 in Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
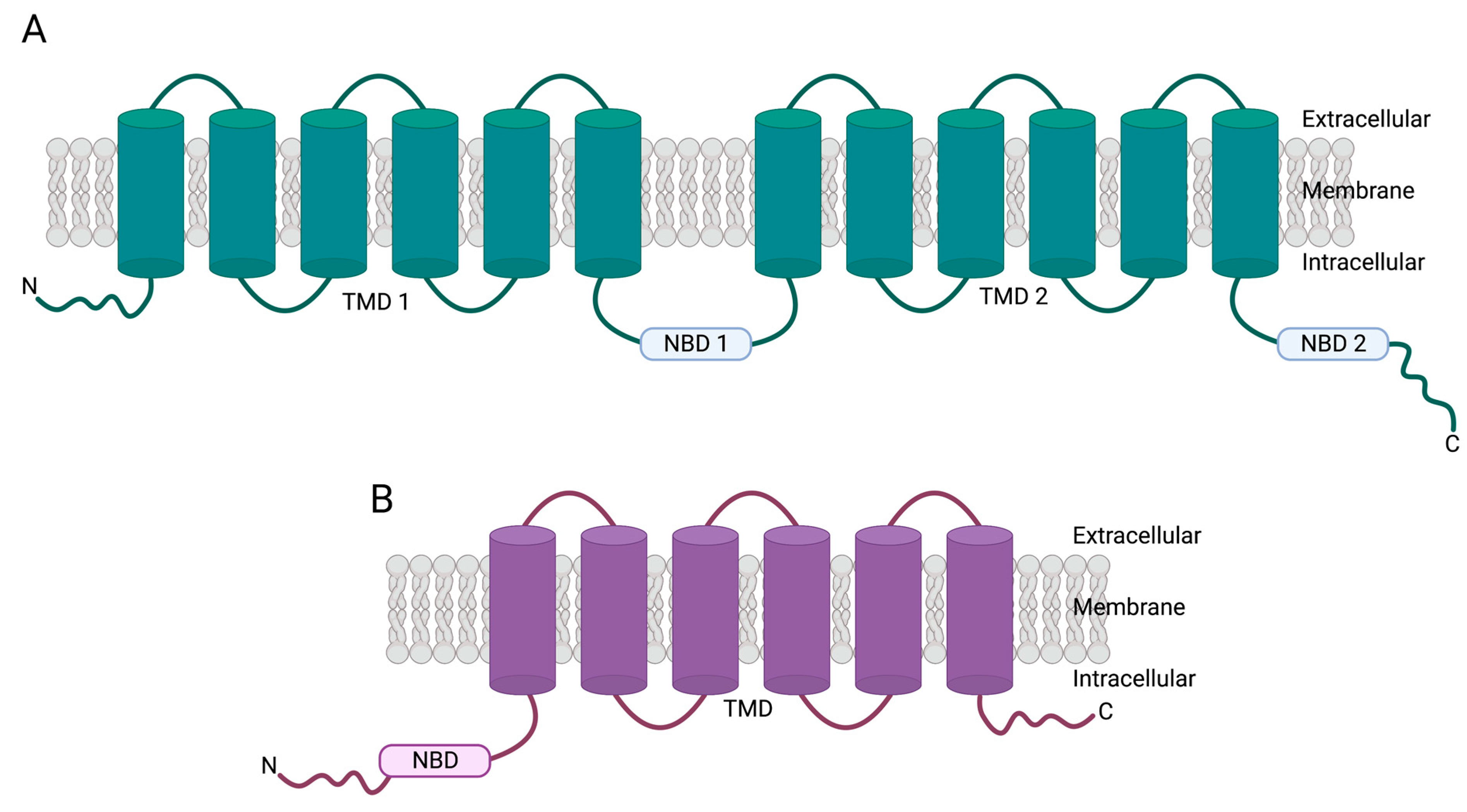
2. Overview of ABC Family of Transporters
3. ABCB1 Genetics
4. ABCB1 in Normal Tissues
4.1. Tissue Localization, Subcellular Localization, and Substrates
4.2. P-gp Mechanism of Action
5. ABCB1 in Cancers
5.1. P-gp Substrates: Chemotherapies and Targeted Therapies
5.2. Prevalence of ABCB1 Dysregulation in Cancer
5.3. Structural Variants Leading to ABCB1 Upregulation: Chromosome 7 Amplifications
5.4. Structural Variants Leading to ABCB1 Upregulation: Gene Fusions
5.5. SNPs Leading to Changes in ABCB1 Expression
5.6. SNPs Leading to Changes in ABCB1 ATPase Activity
5.7. Epigenetic Mechanisms of ABCB1 Upregulation
5.8. Transcriptional Regulation of ABCB1
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mao, Q.; Lai, Y.; Wang, J. Drug Transporters in Xenobiotic Disposition and Pharmacokinetic Prediction. Drug Metab. Dispos. 2018, 46, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Bosch, T.M.; Huitema, A.D.R.; Doodeman, V.D.; Jansen, R.; Witteveen, E.; Smit, W.M.; Jansen, R.L.; van Herpen, C.M.; Soesan, M.; Beijnen, J.H.; et al. Pharmacogenetic Screening of CYP3A and ABCB1 in Relation to Population Pharmacokinetics of Docetaxel. Clin. Cancer Res. 2006, 12, 5786–5793. [Google Scholar] [CrossRef] [PubMed]
- Wolking, S.; Schaeffeler, E.; Lerche, H.; Schwab, M.; Nies, A.T. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin. Pharmacokinet. 2015, 54, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. ABC Family Transporters. In Drug Transporters in Drug Disposition, Effects and Toxicity; Liu, X., Pan, G., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 13–100. ISBN 9789811376474. [Google Scholar]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ling, V. Reconstitution of Drug Transport by Purified P-glycoprotein. J. Biol. Chem. 1995, 270, 16167–16175. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J.; Yu, X.; Doige, C.A. Functional reconstitution of drug transport and ATPase activity in proteoliposomes containing partially purified P-glycoprotein. J. Biol. Chem. 1993, 268, 24197–24202. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Lelong, I.H.; Zhang, J.; Cardarelli, C. [36] Purification and reconstitution of human P-glycoprotein. Methods Enzymol. 1998, 292, 492–504. [Google Scholar] [CrossRef]
- Riordan, J.R.; Deuchars, K.; Kartner, N.; Alon, N.; Trent, J.; Ling, V. Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 1985, 316, 817–819. [Google Scholar] [CrossRef]
- Dean, M.; Allikmets, R. Complete Characterization of the Human ABC Gene Family. J. Bioenerg. Biomembr. 2001, 33, 475–479. [Google Scholar] [CrossRef]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Yabuuchi, H.; Takayanagi, S.-I.; Yoshinaga, K.; Taniguchi, N.; Aburatani, H.; Ishikawa, T. ABCC13, an unusual truncated ABC transporter, is highly expressed in fetal human liver. Biochem. Biophys. Res. Commun. 2002, 299, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Feng, J.; Yuan, D.; Zhou, J.; Miao, W. Tracing the structural evolution of eukaryotic ATP binding cassette transporter superfamily. Sci. Rep. 2015, 5, 16724. [Google Scholar] [CrossRef]
- Bhatia, A.; Schäfer, H.-J.; Hrycyna, C.A. Oligomerization of the Human ABC Transporter ABCG2: Evaluation of the Native Protein and Chimeric Dimers. Biochemistry 2005, 44, 10893–10904. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Jensen, U.; Hansen, A.; Covitz, K.-M.; Zhan, Z.; Fetsch, P.; Abati, A.; Hansen, P.R.; Horn, T.; Skovsgaard, T.; et al. Use of peptide antibodies to probe for the mitoxantrone resistance-associated protein MXR/BCRP/ABCP/ABCG2. Biochim. Biophys. Acta Biomembr. 2002, 1565, 6–16. [Google Scholar] [CrossRef] [PubMed]
- van Roermund, C.W.T.; Visser, W.F.; Ijlst, L.; van Cruchten, A.; Boek, M.; Kulik, W.; Waterham, H.R.; Wanders, R.J.A. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl–CoA esters. FASEB J. 2008, 22, 4201–4208. [Google Scholar] [CrossRef] [PubMed]
- Tordai, H.; Suhajda, E.; Sillitoe, I.; Nair, S.; Varadi, M.; Hegedus, T. Comprehensive Collection and Prediction of ABC Transmembrane Protein Structures in the AI Era of Structural Biology. Int. J. Mol. Sci. 2022, 23, 8877. [Google Scholar] [CrossRef]
- Chen, C.J.; Clark, D.; Ueda, K.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Genomic organization of the human multidrug resistance (MDR1) gene and origin of P-glycoproteins. J. Biol. Chem. 1990, 265, 506–514. [Google Scholar] [CrossRef]
- Raguz, S.; Randle, R.A.; Sharpe, E.R.; Foekens, J.A.; Sieuwerts, A.M.; Meijer-van Gelder, M.E.; Melo, J.V.; Higgins, C.F.; Yagüe, E. Production of P-glycoprotein from the MDR1 upstream promoter is insufficient to affect the response to first-line chemotherapy in advanced breast cancer. Int. J. Cancer 2008, 122, 1058–1067. [Google Scholar] [CrossRef]
- Raguz, S.; De Bella, M.T.; Tripuraneni, G.; Slade, M.J.; Higgins, C.F.; Coombes, R.C.; Yagüe, E. Activation of the MDR1 Upstream Promoter in Breast Carcinoma as a Surrogate for Metastatic Invasion. Clin. Cancer Res. 2004, 10, 2776–2783. [Google Scholar] [CrossRef]
- Reed, K.; Hembruff, S.L.; Laberge, M.L.; Villeneuve, D.J.; Côté, G.B.; Parissenti, A.M. Hypermethylation of the ABCB1 downstream gene promoter accompanies ABCB1 gene amplification and increased expression in docetaxel-resistant MCF-7 breast tumor cells. Epigenetics 2008, 3, 270–280. [Google Scholar] [CrossRef]
- Henrique, R.; Oliveira, A.I.; Costa, V.L.; Baptista, T.; Martins, A.T.; Morais, A.; Oliveira, J.; Jerónimo, C. Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer. BMC Genom. 2013, 14, 898. [Google Scholar] [CrossRef] [PubMed]
- Pappas, J.J.; Petropoulos, S.; Suderman, M.; Iqbal, M.; Moisiadis, V.; Turecki, G.; Matthews, S.G.; Szyf, M. The Multidrug Resistance 1 Gene Abcb1 in Brain and Placenta: Comparative Analysis in Human and Guinea Pig. PLoS ONE 2014, 9, e111135. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef]
- Role of P-Glycoprotein in Drug Disposition. Available online: https://oce-ovid-com.proxy.library.emory.edu/article/00007691-200002000-00029/HTML (accessed on 3 July 2023).
- Peeters, K.; Wilmer, M.J.; Schoeber, J.P.; Reijnders, D.; van den Heuvel, L.P.; Masereeuw, R.; Levtchenko, E. Role of P-Glycoprotein Expression and Function in Cystinotic Renal Proximal Tubular Cells. Pharmaceutics 2011, 3, 782–792. [Google Scholar] [CrossRef]
- Watanabe, T.; Suzuki, H.; Sawada, Y.; Naito, M.; Tsuruo, T.; Inaba, M.; Hanano, M.; Sugiyama, Y. Induction of hepatic P-glycoprotein enhances biliary excretion of vincristine in rats. J. Hepatol. 1995, 23, 440–448. [Google Scholar] [CrossRef]
- Leu, B.-L.; Huang, J.-D. Inhibition of intestinal P-glycoprotein and effects on etoposide absorption. Cancer Chemother. Pharmacol. 1995, 35, 432–436. [Google Scholar] [CrossRef]
- Mai, Y.; Dou, L.; Yao, Z.; Madla, C.M.; Gavins, F.K.H.; Taherali, F.; Yin, H.; Orlu, M.; Murdan, S.; Basit, A.W. Quantification of P-Glycoprotein in the Gastrointestinal Tract of Humans and Rodents: Methodology, Gut Region, Sex, and Species Matter. Mol. Pharm. 2021, 18, 1895–1904. [Google Scholar] [CrossRef]
- Cufer, T.; Pfeifer, M.; Vrhovec, I.; Frangez, R.; Kosec, M.; Mrhar, A.; Grabnar, I.; Golouh, R.; Vogric, S.; Sikic, B.I. Decreased cortisol secretion by adrenal glands perfused with the P-glycoprotein inhibitor valspodar and mitotane or doxorubicin. Anti-Cancer Drugs 2000, 11, 303–309. [Google Scholar] [CrossRef]
- Cufer, T.; Vrhovec, I.; Pfeifer, M.; Skrk, J.; Borstnar, S.; Sikic, B.I. Effect of the multidrug resistance modulator valspodar on serum cortisol levels in rabbits. Cancer Chemother. Pharmacol. 1998, 41, 517–521. [Google Scholar] [CrossRef]
- Bello-Reuss, E.; Ernest, S.; Holland, O.B.; Hellmich, M.R. Role of multidrug resistance P-glycoprotein in the secretion of aldosterone by human adrenal NCI-H295 cells. Am. J. Physiol. Physiol. 2000, 278, C1256–C1265. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kingdom, J.; Baczyk, D.; Lye, S.J.; Matthews, S.G.; Gibb, W. Expression of the Multidrug Resistance P-Glycoprotein, (ABCB1 glycoprotein) in the Human Placenta Decreases with Advancing Gestation. Placenta 2006, 27, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Kartner, N.; Riordan, J.R.; Ling, V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983, 221, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Kosztyu, P.; Dolezel, P.; Kimura, Y.; Cizkova, K.; Ruzickova, E. Estimation of ABCB1 concentration in plasma membrane. J. Cell. Biochem. 2019, 120, 18406–18414. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, F.; Beaune, P.; Tzourio, C.; Loriot, M.-A.; Elbaz, A. Interaction Between ABCB1 and Professional Exposure to Organochlorine Insecticides in Parkinson Disease. Arch. Neurol. 2010, 67, 739–745. [Google Scholar] [CrossRef]
- Narayan, S.; Sinsheimer, J.S.; Paul, K.C.; Liew, Z.; Cockburn, M.; Bronstein, J.M.; Ritz, B. Genetic variability in ABCB1, occupational pesticide exposure, and Parkinson’s disease. Environ. Res. 2015, 143, 98–106. [Google Scholar] [CrossRef]
- Theile, D.; Staffen, B.; Weiss, J. ATP-binding cassette transporters as pitfalls in selection of transgenic cells. Anal. Biochem. 2010, 399, 246–250. [Google Scholar] [CrossRef]
- Kino, K.; Taguchi, Y.; Yamada, K.; Komano, T.; Ueda, K. Aureobasidin A, an antifungal cyclic depsipeptide antibiotic, is a substrate for both human MDR1 and MDR2/P-glycoproteins. FEBS Lett. 1996, 399, 29–32. [Google Scholar] [CrossRef]
- Sugie, M.; Asakura, E.; Zhao, Y.L.; Torita, S.; Nadai, M.; Baba, K.; Kitaichi, K.; Takagi, K.; Takagi, K.; Hasegawa, T. Possible Involvement of the Drug Transporters P Glycoprotein and Multidrug Resistance-Associated Protein Mrp2 in Disposition of Azithromycin. Antimicrob. Agents Chemother. 2004, 48, 809–814. [Google Scholar] [CrossRef]
- Babić, Ž.; Kučišec-Tepeš, N.; Troskot, R.; Dorosulić, Z.; Svoboda-Beusan, I. The importance of P-glycoprotein multidrug transporter activity measurement in patients with Helicobacter pylori infection. Coll. Antropol. 2009, 33, 1145–1150. [Google Scholar] [PubMed]
- Mordi, I.R.; Chan, B.K.; Yanez, N.D.; Palmer, C.N.A.; Lang, C.C.; Chalmers, J.D. Genetic and pharmacological relationship between P-glycoprotein and increased cardiovascular risk associated with clarithromycin prescription: An epidemiological and genomic population-based cohort study in Scotland, UK. PLoS Med. 2020, 17, e1003372. [Google Scholar] [CrossRef] [PubMed]
- Römermann, K.; Wanek, T.; Bankstahl, M.; Bankstahl, J.P.; Fedrowitz, M.; Müller, M.; Löscher, W.; Kuntner, C.; Langer, O. (R)-[11C]verapamil is selectively transported by murine and human P-glycoprotein at the blood–brain barrier, and not by MRP1 and BCRP. Nucl. Med. Biol. 2013, 40, 873–878. [Google Scholar] [CrossRef]
- Ledwitch, K.V.; Gibbs, M.E.; Barnes, R.W.; Roberts, A.G. Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein. Biochem. Pharmacol. 2016, 118, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.L.; Gottesman, M.M.; Cardarelli, C.O.; Ramachandra, M.; Jeang, K.-T.; Ambudkar, S.V.; Pastan, I.; Dey, S. HIV-1 Protease Inhibitors Are Substrates for the MDR1 Multidrug Transporter. Biochemistry 1998, 37, 3594–3601. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Fromm, M.F.; Wandel, C.; Leake, B.; Wood, A.J.; Roden, D.M.; Wilkinson, G.R. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J. Clin. Investig. 1998, 101, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.J.; Tsai, T.F. ABCB1 in dermatology: Roles in skin diseases and their treatment. J. Mol. Med. 2021, 99, 1527–1538. [Google Scholar] [CrossRef]
- Ueda, K.; Okamura, N.; Hirai, M.; Tanigawara, Y.; Saeki, T.; Kioka, N.; Komano, T.; Hori, R. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J. Biol. Chem. 1992, 267, 24248–24252. [Google Scholar] [CrossRef]
- Ge, C.; Xu, D.; Yu, P.; Fang, M.; Guo, J.; Xu, D.; Qiao, Y.; Chen, S.; Zhang, Y.; Wang, H. P-gp expression inhibition mediates placental glucocorticoid barrier opening and fetal weight loss. BMC Med. 2021, 19, 311. [Google Scholar] [CrossRef]
- Kyle, C.J.; Nixon, M.; Homer, N.Z.M.; Morgan, R.A.; Andrew, R.; Stimson, R.H.; Walker, B.R. ABCC1 modulates negative feedback control of the hypothalamic-pituitary-adrenal axis in vivo in humans. Metabolism 2022, 128, 155118. [Google Scholar] [CrossRef]
- Parker, R.B.; Yates, C.R.; Laizure, S.C.; Weber, K.T. P-Glycoprotein Modulates Aldosterone Plasma Disposition and Tissue Uptake. J. Cardiovasc. Pharmacol. 2006, 47, 55–59. [Google Scholar] [CrossRef]
- Marques, P.; Courand, P.-Y.; Gouin-Thibault, I.; Zhygalina, V.; Bergerot, D.; Salem, J.-E.; Funck-Brentano, C.; Loriot, M.-A.; Azizi, M.; Blanchard, A. P-glycoprotein influences urinary excretion of aldosterone in healthy individuals. J. Hypertens. 2019, 37, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.J.; Waddell, B.J. P-Glycoprotein Restricts Access of Cortisol and Dexamethasone to the Glucocorticoid Receptor in Placental BeWo Cells. Endocrinology 2006, 147, 5147–5152. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.W.; Ammerman, L.; Chen, G.; Vogel, P.D.; Wise, J.G. Transport of Alzheimer’s associated amyloid-β catalyzed by P-glycoprotein. PLoS ONE 2021, 16, e0250371. [Google Scholar] [CrossRef] [PubMed]
- Solazzo, M.; Fantappiè, O.; Lasagna, N.; Sassoli, C.; Nosi, D.; Mazzanti, R. P-gp localization in mitochondria and its functional characterization in multiple drug-resistant cell lines. Exp. Cell Res. 2006, 312, 4070–4078. [Google Scholar] [CrossRef]
- Aryal, M.; Fischer, K.; Gentile, C.; Gitto, S.; Zhang, Y.-Z.; McDannold, N. Effects on P-Glycoprotein Expression after Blood-Brain Barrier Disruption Using Focused Ultrasound and Microbubbles. PLoS ONE 2017, 12, e0166061. [Google Scholar] [CrossRef]
- Ding, Y.; Zhong, Y.; Baldeshwiler, A.; Abner, E.L.; Bauer, B.; Hartz, A.M.S. Protecting P-glycoprotein at the blood–brain barrier from degradation in an Alzheimer’s disease mouse model. Fluids Barriers CNS 2021, 18, 10. [Google Scholar] [CrossRef]
- Brückmann, S.; Brenn, A.; Grube, M.; Niedrig, K.; Holtfreter, S.; Halbach, O.V.B.U.; Groschup, M.; Keller, M.; Vogelgesang, S. Lack of P-glycoprotein Results in Impairment of Removal of Beta-Amyloid and Increased Intraparenchymal Cerebral Amyloid Angiopathy after Active Immunization in a Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 656–667. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid- deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef]
- Anoshchenko, O.; Storelli, F.; Unadkat, J.D. Successful Prediction of Human Fetal Exposure to P-Glycoprotein Substrate Drugs Using the Proteomics-Informed Relative Expression Factor Approach and PBPK Modeling and Simulation. Drug Metab. Dispos. 2021, 49, 919–928. [Google Scholar] [CrossRef]
- Rubinchik-Stern, M.; Eyal, S. Drug Interactions at the Human Placenta: What Is the Evidence? Front. Pharm. 2012, 3, 126. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In Situ Localization of P-glycoprotein (ABCB1) in Human and Rat Brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrial Localization and Activity of P-Glycoprotein in Doxorubicin-Resistant K562 Cells–ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S0006295206000360?via%3Dihub#fig7 (accessed on 24 June 2023).
- Shen, Y.; Chu, Y.; Yang, Y.; Wang, Z. Mitochondrial localization of P-glycoprotein in the human breast cancer cell line MCF-7/ADM and its functional characterization. Oncol. Rep. 2012, 27, 1535–1540. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kim, I.-W.; Sauna, Z.E. The power of the pump: Mechanisms of action of P-glycoprotein (ABCB1). Eur. J. Pharm. Sci. 2006, 27, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Futamata, R.; Kimura, Y.; Kioka, N.; Nakatsu, T.; Kato, H.; Ueda, K. ABCB1/MDR1/P-gp employs an ATP-dependent twist-and-squeeze mechanism to export hydrophobic drugs. FEBS Lett. 2021, 595, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Pote, M.S.; Gacche, R.N. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov. Today 2023, 28, 103537. [Google Scholar] [CrossRef]
- Eytan, G.D.; Kuchel, P.W. Mechanism of Action of P-Glycoprotein in Relation to Passive Membrane Permeation. In International Review of Cytology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 190, pp. 175–250. [Google Scholar]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef]
- Eckford, P.D.W.; Sharom, F.J. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem. J. 2005, 389, 517–526. [Google Scholar] [CrossRef]
- Homolya, L.; Holló, Z.; Germann, U.; Pastan, I.; Gottesman, M.M.; Sarkadi, B. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J. Biol. Chem. 1993, 268, 21493–21496. [Google Scholar] [CrossRef]
- Raviv, Y.; Pollard, H.B.; Bruggemann, E.P.; Pastan, I.; Gottesman, M.M. Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells. J. Biol. Chem. 1990, 265, 3975–3980. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Gottesman, M.M. Is the multidrug transporter a flippase? Trends Biochem. Sci. 1992, 17, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Corder, A.B.; Ling, V. P-Glycoprotein-Mediated Hoechst 33342 Transport Out of the Lipid Bilayer. JBIC J. Biol. Inorg. Chem. 1997, 250, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. The Transmembrane Domains of the Human Multidrug Resistance P-glycoprotein Are Sufficient to Mediate Drug Binding and Trafficking to the Cell Surface. J. Biol. Chem. 1999, 274, 24759–24765. [Google Scholar] [CrossRef]
- Zoghbi, M.E.; Mok, L.; Swartz, D.J.; Singh, A.; Fendley, G.A.; Urbatsch, I.L.; Altenberg, G.A. Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer–associated P-glycoprotein during ATP hydrolysis. J. Biol. Chem. 2017, 292, 20412–20424. [Google Scholar] [CrossRef]
- Futamata, R.; Ogasawara, F.; Ichikawa, T.; Kodan, A.; Kimura, Y.; Kioka, N.; Ueda, K. In vivo FRET analyses reveal a role of ATP hydrolysis–associated conformational changes in human P-glycoprotein. J. Biol. Chem. 2020, 295, 5002–5011. [Google Scholar] [CrossRef]
- Safa, A.R.; Mehta, N.D.; Agresti, M. Photoaffinity labeling of P-glycoprotein in multidrug resistant cells with photoactive analogs of colchicine. Biochem. Biophys. Res. Commun. 1989, 162, 1402–1408. [Google Scholar] [CrossRef]
- Ishida, Y.; Ohtsu, T.; Hamada, H.; Sugimoto, Y.; Tobinai, K.; Minato, K.; Tsuruo, T.; Shimoyama, M. Multidrug Resistance in Cultured Human Leukemia and Lymphoma Cell Lines Detected by a Monoclonal Antibody, MRK16. Jpn. J. Cancer Res. 1989, 80, 1006–1013. [Google Scholar] [CrossRef]
- Dantzig, A.H.; Shepard, R.L.; Cao, J.; Law, K.L.; Ehlhardt, W.J.; Baughman, T.M.; Bumol, T.F.; Starling, J.J. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res. 1996, 56, 4171–4179. [Google Scholar]
- Veneroni, S.; Zaffaroni, N.; Daidone, M.G.; Benini, E.; Villa, R.; Silvestrini, R. Expression of P-glycoprotein and in vitro or in vivo resistance to doxorubicin and cisplatin in breast and ovarian cancers. Eur. J. Cancer 1994, 30, 1002–1007. [Google Scholar] [CrossRef]
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk. Lymphoma 2015, 56, 26–33. [Google Scholar] [CrossRef]
- Park, Y.B.; Kim, H.S.; Oh, J.H.; Lee, S.H. The co-expression of p53 protein and P-glycoprotein is correlated to a poor prognosis in osteosarcoma. Int. Orthop. 2001, 24, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Ohnishi, Y.; Yoshimura, M.; Ota, E.; Ozeki, Y.; Oshika, Y.; Tokunaga, T.; Yamazaki, H.; Ueyema, Y.; Ogata, T.; et al. P-glycoprotein-mediated acquired multidrug resistance of human lung cancer cells in vivo. Br. J. Cancer 1996, 74, 1929–1934. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.D.; Lee, O.W.; Brimacombe, K.R.; Chen, L.; Guha, R.; Lusvarghi, S.; Tebase, B.G.; Klumpp-Thomas, C.; Robey, R.W.; Ambudkar, S.V.; et al. A High-Throughput Screen of a Library of Therapeutics Identifies Cytotoxic Substrates of P-glycoprotein. Mol. Pharmacol. 2019, 96, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Wang, Y.C.; Juric, D.; Francisco, B.; Yu, R.X.; Duran, G.E.; Chen, K.G.; Chen, X.; Sikic, B.I. Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosom. Cancer 2006, 45, 365–374. [Google Scholar] [CrossRef]
- Ibrahim, S.M.; Karim, S.; Abusamra, H.; Pushparaj, P.N.; Khan, J.A.; Abuzenadah, A.M.; Gari, M.A.; Bakhashab, S.; Ahmed, F.; Al-Qahtan, M.H. Genomic amplification of chromosome 7 in the Doxorubicin resistant K562 cell line. Bioinformation 2018, 14, 587–593. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef]
- Priyadarshini, R.; Raj, G.M.; Kayal, S.; Ramesh, A.; Shewade, D.G. Influence of ABCB1 C3435T and C1236T gene polymorphisms on tumour response to docetaxel-based neo-adjuvant chemotherapy in locally advanced breast cancer patients of South India. J. Clin. Pharm. Ther. 2019, 44, 188–196. [Google Scholar] [CrossRef]
- Kim, H.J.; Keam, B.; Im, S.; Ham, H.S.; Oh, D.; Kim, J.; Han, W.S.; Kim, T.; Park, I.A.; Bang, Y.J. Use of MDR1/ABCB1 single nucleotide polymorphism (SNP) as a prognostic factor for breast cancer patients receiving docetaxel + doxorubicin neoadjuvant chemotherapy. J. Clin. Oncol. 2008, 26, 569. [Google Scholar] [CrossRef]
- Gréen, H.; Söderkvist, P.; Rosenberg, P.; Horvath, G.; Peterson, C. ABCB1 G1199A Polymorphism and Ovarian Cancer Response to Paclitaxel. J. Pharm. Sci. 2008, 97, 2045–2048. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, I.; Jeleń, A.; Pietrzak, J.; Żebrowska-Nawrocka, M.; Michalska, K.; Szmajda-Krygier, D.; Mirowski, M.; Łochowski, M.; Kozak, J.; Balcerczak, E. The impact of ABCB1 gene polymorphism and its expression on non-small-cell lung cancer development, progression and therapy–preliminary report. Sci. Rep. 2020, 10, 6188. [Google Scholar] [CrossRef] [PubMed]
- Xiaohui, S.; Aiguo, L.; Xiaolin, G.; Ying, L.; Hongxing, Z.; Yilei, Z. Effect of ABCB1 Polymorphism on the Clinical Outcome of Osteosarcoma Patients after Receiving Chemotherapy. Pak. J. Med. Sci. 2014, 30, 886–890. [Google Scholar]
- Mansoori, M.; Golalipour, M.; Alizadeh, S.; Jahangirerad, A.; Khandozi, S.R.; Fakharai, H.; Shahbazi, M. Genetic Variation in the ABCB1 Gene May Lead to mRNA Level Chabge: Application to Gastric Cancer Cases. Asian Pac. J. Cancer Prev. 2016, 16, 8467–8471. [Google Scholar] [CrossRef]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional Polymorphisms of the Human Multidrug-Resistance Gene: Multiple Sequence Variations and Correlation of One Allele with P-Glycoprotein Expression and Activity In Vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Johnson, A.D.; Papp, A.C.; Kroetz, D.L.; Sadée, W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet. Genom. 2005, 15, 693–704. [Google Scholar] [CrossRef]
- Siegsmund, M.; Brinkmann, U.; Scha[Combining Acute Accent]ffeler, E.; Weirich, G.; Schwab, M.; Eichelbaum, M.; Fritz, P.; Burk, O.; Decker, J.; Alken, P.; et al. Association of the P-Glycoprotein Transporter MDR1 C3435T Polymorphism with the Susceptibility to Renal Epithelial Tumors. J. Am. Soc. Nephrol. 2002, 13, 1847–1854. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakaeda, T.; Horinouchi, M.; Tamura, T.; Aoyama, N.; Shirakawa, T.; Matsuo, M.; Kasuga, M.; Okumura, K. Effect of the mutation (C3435T) at exon 26 of the MDR1 gene on expression level of MDR1 messenger ribonucleic acid in duodenal enterocytes of healthy Japanese subjects. Clin. Pharmacol. Ther. 2002, 71, 297–303. [Google Scholar] [CrossRef]
- Hitzl, M.; Drescher, S.; van der Kuip, H.; Schäffeler, E.; Fischer, J.; Schwab, M.; Eichelbaum, M.; Fromm, M.F. The C3435T mutation in the human MDR1 gene is associated with altered efflux of the P-glycoprotein substrate rhodamine 123 from CD56+ natural killer cells. Pharmacogenetics 2001, 11, 293–298. [Google Scholar] [CrossRef]
- Jiang, B.; Yan, L.-J.; Wu, Q. ABCB1(C1236T) Polymorphism Affects P-Glycoprotein-Mediated Transport of Methotrexate, Doxorubicin, Actinomycin D, and Etoposide. DNA Cell Biol. 2019, 38, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Leake, B.F.; Choo, E.F.; Dresser, G.K.; Kubba, S.V.; Schwarz, U.I.; Taylor, A.; Xie, H.-G.; McKinsey, J.; Zhou, S.; et al. Identification of Functionally Variant MDR1 Alleles among European Americans and African Americans. Clin. Pharmacol. Ther. 2001, 70, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Woodahl, E.L.; Yang, Z.; Bui, T.; Shen, D.D.; Ho, R.J.Y. Multidrug Resistance Gene G1199A Polymorphism Alters Efflux Transport Activity of P-Glycoprotein. Experiment 2004, 310, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Song, J.; Lai, Q.; Liu, B.; Wang, H.; Xu, Y.; Feng, X.; Sun, X.; Du, Z. Hypermethylation of ATP-binding cassette B1 (ABCB1) multidrug resistance 1 (MDR1) is associated with cisplatin resistance in the A549 lung adenocarcinoma cell line. Int. J. Exp. Pathol. 2016, 97, 412–421. [Google Scholar] [CrossRef]
- Sumarpo, A.; Ito, K.; Saiki, Y.; Ishizawa, K.; Wang, R.; Chen, N.; Sunamura, M.; Horii, A. Genetic and epigenetic aberrations of ABCB1 synergistically boost the acquisition of taxane resistance in esophageal squamous cancer cells. Biochem. Biophys. Res. Commun. 2020, 526, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.; Lebensohn, A.M.; Bahrami-Nejad, Z.; Marceau, C.; Champion, M.; Gevaert, O.; Sikic, B.I.; Carette, J.E.; Rohatgi, R. Chromatin-Remodeling Complex SWI/SNF Controls Multidrug Resistance by Transcriptionally Regulating the Drug Efflux Pump ABCB1. Cancer Res 2016, 76, 5810–5821. [Google Scholar] [CrossRef]
- Choi, E.J.; Seo, E.J.; Kim, D.K.; Lee, S.I.; Kwon, Y.W.; Jang, I.H.; Kim, K.-H.; Suh, D.-S.; Kim, J.H. FOXP1 functions as an oncogene in promoting cancer stem cell-like characteristics in ovarian cancer cells. Oncotarget 2015, 7, 3506–3519. [Google Scholar] [CrossRef]
- Chin, K.-V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of Activity of the Promoter of the Human MDR1 Gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef]
- De Angelis, P.; Stokke, T.; Smedshammer, L.; Lothe, R.A.; Lehne, G.; Chen, Y.; Clausen, O.P. P-glycoprotein is not expressed in a majority of colorectal carcinomas and is not regulated by mutant p53 in vivo. Br. J. Cancer 1995, 72, 307–311. [Google Scholar] [CrossRef]
- Corrêa, S.; Binato, R.; Du Rocher, B.; Castelo-Branco, M.T.; Pizzatti, L.; Abdelhay, E. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012, 12, 303. [Google Scholar] [CrossRef]
- Flahaut, M.; Muhlethaler, A.; Niggli, F.; Meier, R.; Coulon, A.; Bosman, F.; Joseph, J.-M.; Gross, N. The Wnt/Beta-Catenin Signalling Pathway Cooperates with MDR1 Gene-Encoded P-Glycoprotein in Multi-Drug Resistant Neuroblastoma Cells. Cancer Res. 2008, 68, 2455. [Google Scholar]
- Shen, D.-Y.; Zhang, W.; Zeng, X.; Liu, C.-Q. Inhibition of Wnt/β-catenin signaling downregulates P-glycoprotein and reverses multi-drug resistance of cholangiocarcinoma. Cancer Sci. 2013, 104, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, H.; Watanabe, M.; Sugita, K.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Sugano, S.; Tsujimoto, H. Regulations of ABCB1 and ABCG2 expression through MAPK pathways in acute lymphoblastic leukemia cell lines. Anticancer Res. 2013, 33, 5317–5323. [Google Scholar] [PubMed]
- Katayama, K.; Yoshioka, S.; Tsukahara, S.; Mitsuhashi, J.; Sugimoto, Y. Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol. Cancer Ther. 2007, 6, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Zhou, S.; Wang, Y.; Liu, X.; Zhou, L.; Yin, P.; Fan, Z.; Li, Q. COX-2 contributes to P-glycoprotein-mediated multidrug resistance via phosphorylation of c-Jun at Ser63/73 in colorectal cancer. Carcinog. 2011, 32, 667–675. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Li, Z.; You, J.; Wu, Q.-W.; Zhao, C.; Tzeng, C.-M.; Zhang, Z.-M. Interaction of WBP2 with ERα increases doxorubicin resistance of breast cancer cells by modulating MDR1 transcription. Br. J. Cancer 2018, 119, 182–192. [Google Scholar] [CrossRef]
- Mutoh, K.; Tsukahara, S.; Mitsuhashi, J.; Katayama, K.; Sugimoto, Y. Estrogen-mediated post transcriptional down-regulation of P-glycoprotein in MDR1-transduced human breast cancer cells. Cancer Sci. 2006, 97, 1198–1204. [Google Scholar] [CrossRef]
- Imai, Y.; Tsukahara, S.; Ishikawa, E.; Tsuruo, T.; Sugimoto, Y. Estrone and 17β-Estradiol Reverse Breast Cancer Resistance Protein-mediated Multidrug Resistance. Jpn. J. Cancer Res. 2002, 93, 231–235. [Google Scholar] [CrossRef]
- Brayboy, L.M.; Knapik, L.O.; Long, S.; Westrick, M.; Wessel, G.M. Ovarian hormones modulate multidrug resistance transporters in the ovary. Contracept. Reprod. Med. 2018, 3, 26. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Accession Number | Ensembl Accession Number | Number of Exons | mRNA Length (bp) | Protein Length (AA) |
|---|---|---|---|---|
| NM_001348945.2 | 32 | 5586 | 1350 | |
| NM_001348944.2 | 30 | 5387 | 1280 | |
| NM_000927.5 NM_001348946.2 | ENST00000622132.5 | 29 28 | 5534 5205 | 1280 1280 |
| ENST00000265724.8 | 29 | 4720 | 1280 | |
| ENST00000543898.5 | 29 | 4524 | 1216 | |
| ENST00000416177.1 | 6 | 461 | 48 |
| Tissue Localization | Subcellular Localization | Substrates |
|---|---|---|
| Adrenal gland [24,25] Kidneys [24,25] Colon [24,25] Rectum [24,25] Lungs [24,25] Liver [24,25] Blood–brain endothelial cells [34] Placenta [35] | On the cell surface/within the plasma membrane of: | Pesticides [38,39] Antibiotics:Calcium channel blockers [45,46] Protease inhibitors [47,48] Antihistamines [49] Hormones and steroids Cortisol [50,51,52] Aldosterone [50,53,54] Dexamethasone [50,55] Amyloid-β [56] Rhodamine 123 [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skinner, K.T.; Palkar, A.M.; Hong, A.L. Genetics of ABCB1 in Cancer. Cancers 2023, 15, 4236. https://doi.org/10.3390/cancers15174236
Skinner KT, Palkar AM, Hong AL. Genetics of ABCB1 in Cancer. Cancers. 2023; 15(17):4236. https://doi.org/10.3390/cancers15174236
Chicago/Turabian StyleSkinner, Katie T., Antara M. Palkar, and Andrew L. Hong. 2023. "Genetics of ABCB1 in Cancer" Cancers 15, no. 17: 4236. https://doi.org/10.3390/cancers15174236