

Regulation of IGF1R by MicroRNA-15b Contributes to the Anticancer Effects of Calorie Restriction in a Murine C3-TAg Model of Triple-Negative Breast Cancer
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Study Design
2.2. Assesment of Tumor Development Assessment
2.3. Tissue Processing and Histopathology
2.4. Serum Hormone Analysis
2.5. Cell Culture
2.6. Western Blot
2.7. RNA Extraction, miR and Gene Expression Analysis
2.8. Luciferase Assay and miR-15b Overexpression
2.9. Cellular Proliferation Assays
2.10. In Silico Analysis of miR-15b Target Prediction
2.11. Statistical Analyses
3. Results
3.1. Protective Effects of CR on Preventing Mammary Tumor Development
- CR promotes long-term weight maintenance and reduces levels of mitogenic hormones. Several studies have consistently shown that long-term CR without malnutrition is a robust intervention to inhibit cancer and increase life and health span in rodents and humans [9,10,32]. To investigate the impact of CR on mammary cancer we use the C3-TAg transgenic mice that develop spontaneous mammary tumors [22]. Our results show that the CR regimen resulted in an initial loss of body weight of ~2 g followed by long-term body weight maintenance (average 15.1 ± 0.64 g) over 20 weeks (survival and time course study, p < 0.0001), relative to mice on a control diet that continuously gained weight until the end of the study (25.1 ± 2.25 g) (Figure 1A).
- Consistently, CR reduced serum levels of metabolic hormones. A mixed-effect ANOVA revealed that there was a statistically significant interaction between the effects of the diet and the time course of the diet (presented as week of age) on the levels of insulin (F(3,17) = 3.871, p = 0.028), leptin (F(4,51) = 2.831, p = 0.034), and IGF1 (F(4,54) = 2.631, p = 0.044). In addition, simple main-effect analysis showed that CR and time had statistically significant effect on the levels of insulin (p = 0.034, and p = 0.024, respectively), leptin (p < 0.0001, and p = 0.020, respectively), and IGF1 (p < 0.0001, and p = 0.0089). Although the levels of insulin and leptin were reduced in the CR group, this reduction was not statistically significant over the time course, most likely due to the high variation in the measurements. However, the multi-comparison test showed statistically significant reduction in the levels of IGF1 relative to levels in mice under the control diet throughout the 12 weeks of the time-course study (from 8 to 20 weeks of age, Figure 1B). This result is consistent with previous studies in rodents showing a CR-mediated reduction in the levels of IGF1 [9].
- CR protects against tumor development and progression in a mouse model of TNBC. The CR regimen significantly delayed overall tumor development by 30% compared to the control diet, with a median time to palpable primary tumor of 23 weeks vs. 15 weeks, respectively (Figure 1C; p = 0.0002). Thus, the overall survival was increased by 50% in CR (median survival of 30 weeks) relative to the control diet (median survival of 20 weeks, p < 0.0001) (Figure 1D). Four mice in the CR group (30 and 34 weeks of age) and two mice on the control diet group (30 and 33 weeks of age) remained tumor free until the end of the survival study. Additionally, the CR regimen reduced the tumor burden by decreasing tumor multiplicity (p = 0.025) (Figure 1E) and secondary tumor weight (p = 0.050) (Figure 1F) compared with mice on the control diet. Also, in the CR group, relative to the control diet group, tumoral Igf1r expression was downregulated by 34% (p = 0.030) (Figure 1G) and a modest decrease in tumoral IGF1R protein levels was also observed (Figure 1H, PDF File S1). In our time course study, the axillary mammary glands from the control diet and CR mice were evaluated by histopathology every 3 weeks from 8 to 20 weeks of age. In the CR group, the majority of ductal structures remained normal across all time points; only a modest amount of MIN was observed in the glands, and low ductal density within the mammary gland was observed (Supplementary Table S2). In contrast, in mice on the control diet, the majority of mammary lesions progressed to MIN by 14 weeks and ductal development toward high density was observed (Supplementary Table S2). Together, these results demonstrate the protective effects of CR on preventing mammary tumor development by reducing the levels of mitogenic hormones, tumor burden, decreasing circulating levels of IGF1, and reducing the tumoral mRNA and protein levels of IGF1R; thus, increasing the life span of the C3-Tag mice.
3.2. miR-15b a Candidate Mediator of the Beneficial Effects of CR
- CR increases expression of several miRs known to regulate IGF signaling. We assessed the impact of CR on the tumoral expression of 10 miRs previously reported to target components of the IGF1 and mTOR pathways (Supplementary Table S3 [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51]). Six of the miRs were not differentially expressed (Supplementary Figure S1), while four were upregulated in secondary tumors from the CR group relative to the control diet: miR-15b (2.19-fold, p = 0.018); miR-199a-3p (2.07-fold, p = 0.021); miR-199a-5p (1.65-fold, p = 0.036); and miR-486 (6.26-fold, p = 0.046) (Figure 2A).
- Expression of miR-15b is affected by cell tumorigenicity. To investigate the impact of the four upregulated miRs on tumor progression, we evaluated their expression using the cell line series M28, M6, and M6C, derived from C3-TAg mice during different stages of tumor development [23]. In these cell lines, expression of miR-199a-3p and miR-199a-5p was not detected. The expression of miR-15b and miR-486 was reduced as the tumorigenicity of the cell-line progression increased (Figure 2B). In particular, miR-15b was significantly downregulated in metastatic M6C cells compared to M6 (invasive carcinoma) and M28 (weakly tumorigenic) cells. In contrast, miR-486 expression did not differ between M6 and M6C cells. These results suggest that miR-15b downregulation could be associated with increased tumor progression, and therefore we selected miR-15b as a candidate miR for further studies of the anticancer effects of CR in highly proliferative cells.
3.3. miR-15b Reduces Cell Proliferation and Targets Igf1r in Mammary Cancer Cell
- miR-15b overexpression inhibits mouse and human TNBC cell proliferation. To evaluate the impact of miR-15b on cellular proliferation, mouse (M6) and human (MDA-MB- 231) basal-like mammary cancer cell lines were dually transfected with a miR-15b mimic or control mimic in combination with a RenSP luciferase reporter plasmid carrying a synthetic miR-15b target site in the 3′ UTR of the luciferase gene. Seventy-two hours following transfection, increased activity of miR-15b was confirmed with a decreased luciferase signal (M6, p = 0.001; MDA-MB-231, p= 0.0074) (Figure 3A,B). Cellular proliferation, evaluated by BrdU incorporation, was reduced in both cell lines in response to miR-15b overexpression (M6, 36% reduction, p = 0.004; MDA-MB-231, 35% reduction, p = 0.002), relative to an miR-15b mimic control (Figure 3C,D). These results highlight the association between increased miR-15b expression and reduction in mammary-tumor-cell proliferation, suggesting a possible antiproliferative effect for miR-15b in mammary tumor cells.
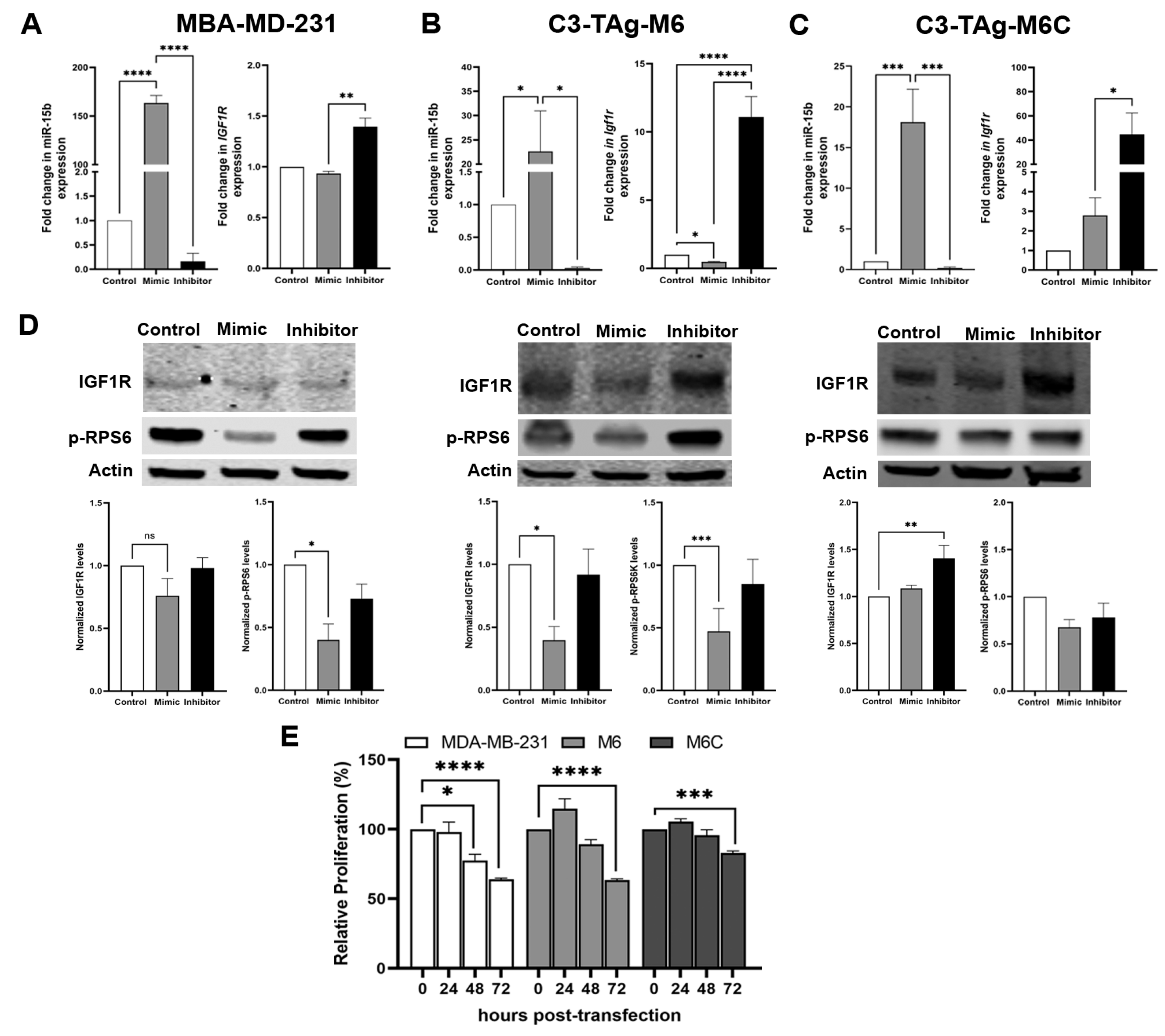
- miR-15b targets Igf1r to potentially inhibit mammary cancer cell proliferation. Studies using mouse models suggest that reduction of bioavailable IGF1 mediates many anticancer effects of CR [52,53,54,55]. Given that we measured increased tumoral expression of miR-15b in CR mice and the reduced tumor expression of Igf1r, we interrogated if upregulation of miR-15b affects the levels of IGF1R. To evaluate the impact of miR-15 on IGF1R expression, MDA-MB-231, M6 and M6C cells were transfected with an miR-15b mimic to enhance endogenous miR-15 activity, or with a miR-15b inhibitor to block miR-15b binding to its target genes. The transient expression of the miR-15b mimic resulted in the overexpression of miR-15b (Figure 4) in all cells lines; however, the effect on Igf1r expression was cell-line-dependent. In MDA-MB-231 (Figure 4A), the overexpression of miR-15b did not have significant effects on the levels on IGF1R. We detected a significant reduction of Igf1r in M6 cells (Figure 4B) and a slight increase in Igf1r in M6C cells (Figure 4C). In contrast, the treatment with an miR-15b hairpin inhibitor blocked miR-15b downregulation of Igf1r in all cell lines, causing a significant increase in Igf1r (Figure 4A). In addition, in MDA-MB-231 and M6 cells transfected with a miR-15b mimic, we observed a reduction in the protein levels of IGF1R and its downstream effector, the phospho-ribosomal protein S6 (p-RPS6), 48 h after overexpressing miR-15b (Figure 4D, PDF File S1). We did not observe significant changes in the levels of IGF1R and p-RPS6 in M6C cells or in the three cell lines transfected with the miR-15b hairpin inhibitor. Consistent with our results in Figure 3, we measured a mild effect on cellular proliferation at 48 h and a significant reduction at 72 h after miR-15B overexpression in MDA-MB-231 and M6 cells but not in M6C cells. These results suggest that the effects of miR-15b on IGF1R regulation in vitro are cell line dependent. However, the significant effect of the overexpression of miR-15b on cell proliferation suggest that miR-15b targets other genes related to cell cycle regulation.
3.4. Upregulation of miR-15b In Vitro Requires Low Serum and IGF1 Levels in M6 Cells
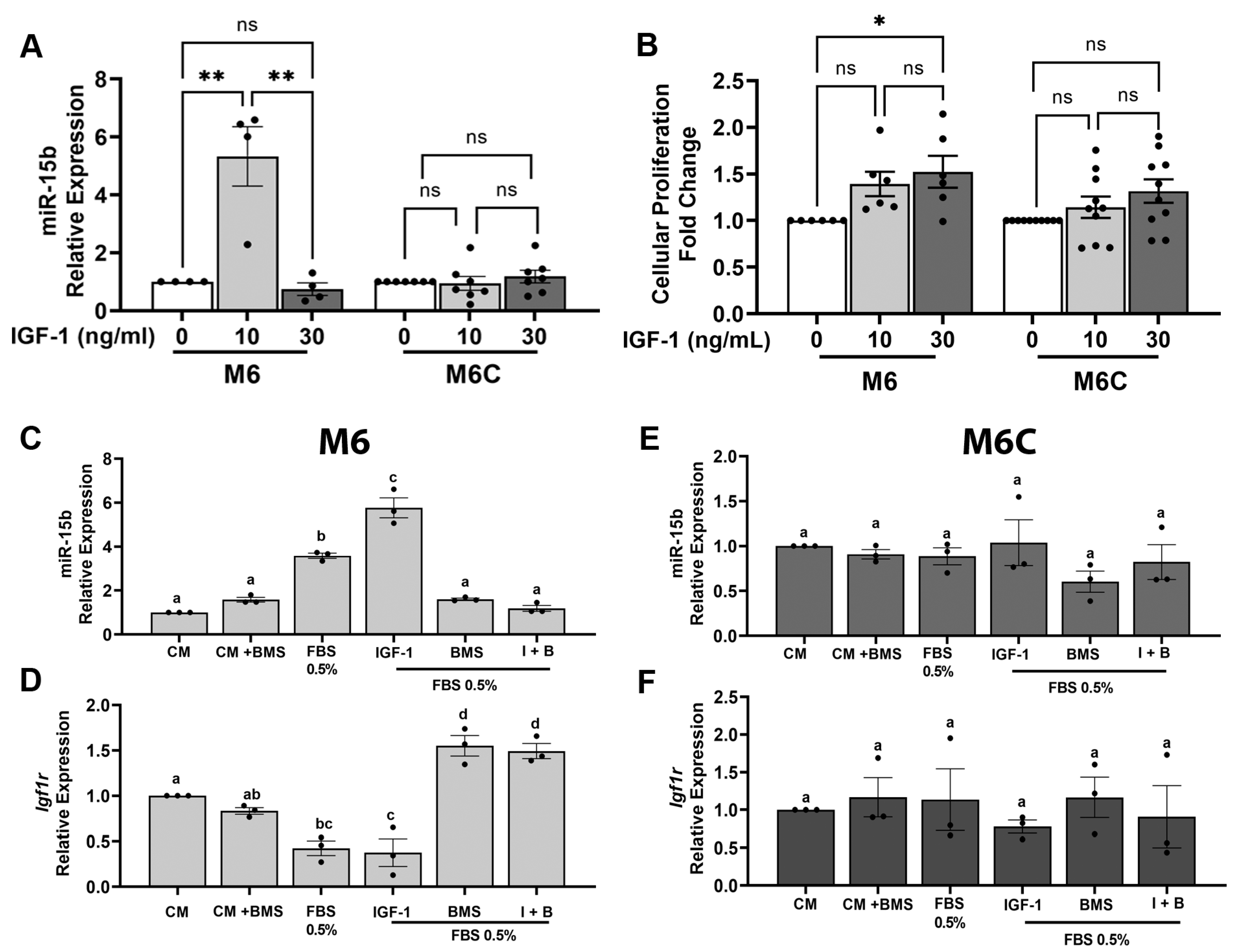
- miR-15b expression is regulated by low IGF1 levels. To further investigate the relationship between CR and miR-15b in vitro, we interrogated whether miR-15b is regulated by IGF1 and insulin levels. The M6 and M6C C3-TAg cell lines were treated with 0, 10 or 30 ng/mL of IGF1 (as described in methods). These IGF1 concentrations resemble the levels of IGF1 measured in the serum from CR and control mice, respectively (Figure 1B). In cultured cells, the treatment with 10 ng/mL of IGF1 induced the expression of miR-15b in the M6 cell lines but no in M6C cell lines; however, the addition of 30 ng/mL of IGF1 had not effects on miR-15b expression on both cell lines (Figure 5A). On the other hand, the treatment of M6 cells with different concentrations of insulin had no effects on miR-15b expression (Supplementary Figure S2). Additionally, the treatment of the M6 and M6C cells with 30 ng/mL of IGF1 increased cell proliferation compared to the cells treated with the lower dose of IGF1 (Figure 5B). These results indicate that IGF1 and not insulin regulate miR-15b expression and suggest a possible negative feedback loop between IGF1 levels and miR-15b expression, in which low IGF1 promote the expression of miR15 while high levels of IGF1 reduce miR-15b expression. Also, these results denote the differential sensitivity of the M6 and M6C cell lines to IGF1 in regulating miR-15b.
- Low serum and IGF1 levels regulate miR-15b expression. Next, we modified the cell culture media to mimic, in vitro, some of the CR-dependent systemic effects. For these experiments, we used M6 cells because of the high sensitivity to the overexpression of miR-15b and to the levels of IGF1, and we also use M6C, the less responsive cell line. To conduct these experiments, we considered that serum deprivation which reduces hormones, lipids, and growth factors, in combination with glucose reduction, could result in significant cellular stress. To partially mimic CR in vitro, we only reduced the concentration of FBS (see the Discussion, Section 4). First, we tested the expression of miR-15b under different concentrations of FBS (0.5, 1, 2% and 10%). The expression of miR-15b significantly increased only in the cells treated with 0.5% FBS (Supplementary Figure S3). Then, we cultured M6 and M6C for 18 h in complete media (CM, 10% FBS); CM + 100 nM BMS754807 (a potent and reversible inhibitor of the IGF1R/IR family kinases (Ki, <2 nmol/L)) [56]; 0.5% serum, 0.5% serum + 10 ng/mL IGF1; 0.5% serum + BMS754807; or 0.5% serum + 10 ng/mL IGF1 + 100 nM BMS754807. In both cell lines cultured with CM media (10% FBS) in the presence of IGF1R inhibitor (BMS), no significant changes in the expression of miR-15b or IGF1R were measured. In M6 cells (Figure 5C,D) but not in M6C cells (Figure 5E,F), serum restriction (0.5% FBS) alone elicited an increased expression of miR-15b (Figure 5C) and a decreased expression of Igf1r (Figure 5D). Treatment of M6 cells with serum restriction in the presence of 10 ng/mL IGF1 potentiated the expression of miR-15b but did not impact the expression of Igf1r relative to serum restriction alone. In media supplemented with 0.5% FBS, the inhibition of IGF1R with BMS754807 had no effect on the expression of miR-15b but it increased Igf1r expression, in contrast to what we observed in the cells cultured with complete media and BMS754807. However, the effects of 10 ng/mL IGF1 on miR-15b and Igf1r expression were abolished by the addition of BMS754807 to M6 cell cultured in media supplemented with 0.5% FBS and 10 ng/mL of IGF1. Together these results show that in M6 cells, partial serum starvation in combination with low levels of IGF1 can upregulate the levels of miR-15b via the activation of IGF1R. This effect is abolished by the presence of BMS754807, an IGF1R inhibitor, suggesting that in M6 cells, the expression of miR-15b and Igf1r requires an active IGF1/IGF1R interaction.
3.5. In Silico Analysis Corroborates miR-15b as a Regulator of IGF1/mTOR Signaling
- Identification of target of miR-15. To identify common target genes for the mature forms of miR-15b in humans and mice, we conducted in silico analysis using a series of webtools for predicting miR-15b targets. Combining the predicted miR-15b target from miRDB, miRabel, miRmap, and TargetScan databases, we identified 14,384 predicted human target genes (hsa-miR-15b-3p and hsa-miR-15b-5p, Supplementary Excel File S1). Moreover, combining predicted targets from miRDB, miRmap, and TargetScan, we predicted 8686 target genes for the mouse (mmu-miR-15b-5p and mmu-miR-15b-3p, Supplementary Excel File S2). Of these genes, 5924 were common between human and mouse (Figure 6A, Supplementary Data S3). The common list of miR-15b target genes was compared to a compiled list of 86 genes involved in the IGF1/mTOR pathway. This analysis showed that 52 out of 86 genes were predicted to be a target for miR-15b (Supplementary Excel File S3), suggesting that miR-15b may be involved in the regulation of the IGF1/mTOR pathway.
- Validation of miR-15 targets. Subsequently, the common list of 5924 target genes was subject to validation in miRTarBase, a database that compiles miR-gene interactions that have published experimental validation. This resulted in a list of 26 validated miR-15b target genes (Figure 6A). The expression of these genes was determined by qPCR in M6 and MDA-MB-231 cells transfected with an miR-15b mimic or with an miR-15b inhibitor. In M6 cells, Arl2, Axin2, Ccne1, and Igf1r were downregulated relative to control cells (Figure 6B, Supplementary Figure S4). In MDA-MB-231 cells expressing miR-15b mimic, most of the miR-15b target genes were downregulated, but only six were significant, including genes encoding suppressors of apoptosis, such as BCL2 and BCL2L2, and genes involved in cell cycle regulation, such as CCND1, CCNE1, CHEK1, and EIF4A1 (Supplementary Figure S5). These results suggest that miR-15b has a role in regulating cellular pathways related to cell metabolism and survival.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Agurs-Collins, T.; Ross, S.A.; Dunn, B.K. The Many Faces of Obesity and Its Influence on Breast Cancer Risk. Front. Oncol. 2019, 9, 765. [Google Scholar] [CrossRef] [PubMed]
- Lope, V.; Martín, M.; Castelló, A.; Ruiz, A.; Casas, A.M.; Baena-Cañada, J.M.; Antolín, S.; Ramos-Vázquez, M.; García-Sáenz, J.Á.; Muñoz, M.; et al. Overeating, caloric restriction and breast cancer risk by pathologic subtype: The EPIGEICAM study. Sci. Rep. 2019, 9, 3904. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Yang, W.; Chen, C.S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Bustamante-Marin, X.M.; Merlino, J.L.; Devericks, E.; Carson, M.S.; Hursting, S.D.; Stewart, D.A. Mechanistic Targets and Nutritionally Relevant Intervention Strategies to Break Obesity–Breast Cancer Links. Front. Endocrinol. 2021, 12, 632284. [Google Scholar] [CrossRef] [PubMed]
- Champ, C.E.; Baserga, R.; Mishra, M.V.; Jin, L.; Sotgia, F.; Lisanti, M.P.; Pestell, R.G.; Dicker, A.P.; Simone, N.L. Nutrient restriction and radiation therapy for cancer treatment: When less is more. Oncologist 2013, 18, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Simone, B.A.; Palagani, A.; Strickland, K.; Ko, K.; Jin, L.; Lim, M.K.; Dan, T.D.; Sarich, M.; Monti, D.A.; Cristofanilli, M.; et al. Caloric restriction counteracts chemotherapy-induced inflammation and increases response to therapy in a triple negative breast cancer model. Cell Cycle 2018, 17, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.M.; Dunlap, S.M.; Ford, N.A.; Hursting, S.D. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr. Relat. Cancer 2012, 19, 57–68. [Google Scholar] [CrossRef]
- Fontana, L.; Weiss, E.P.; Villareal, D.T.; Klein, S.; Holloszy, J.O. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell 2008, 7, 681–687. [Google Scholar] [CrossRef]
- Bowers, L.W.; Rossi, E.L.; O’Flanagan, C.H.; de Graffenried, L.A.; Hursting, S.D. The Role of the Insulin/IGF System in Cancer: Lessons Learned from Clinical Trials and the Energy Balance-Cancer Link. Front. Endocrinol. 2015, 6, 77. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.-I. 40 YEARS OF IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yin, Z.; Tao, K.; Wang, G.; Gao, J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy (Review). Oncol. Lett. 2018, 15, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.W. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: Pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010, 11, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, A.H.; Björner, S.; Ygland Rödström, M.; Jirström, K.; Borgquist, S.; Ingvar, C.; Pollak, M.N.; Jernström, H. Pre- and Postoperative Circulating IGF-I, IGFBP-3, and IGFBP-7 Levels in Relation to Endocrine Treatment and Breast Cancer Recurrence: A Nested Case-Control Study. Front. Oncol. 2021, 11, 626058. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Casa, A.; Lazard, Z.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Lee, A.V. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J. Clin. Oncol. 2008, 26, 4078–4085. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Suh, Y. Regulation of IGF -1 signaling by microRNAs. Front. Genet. 2015, 5, 472. [Google Scholar] [CrossRef]
- Alqurashi, N.; Hashimi, S.M.; Wei, M.Q. Chemical Inhibitors and microRNAs (miRNA) Targeting the Mammalian Target of Rapamycin (mTOR) Pathway: Potential for Novel Anticancer Therapeutics. Int. J. Mol. Sci. 2013, 14, 3874–3900. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Qu, J.H.; Liu, B.; Zhang, P.; Zhang, T.; Fan, P.C.; Wang, X.M.; Xiao, G.Y.; Su, Y.; et al. Caloric Restriction Induces MicroRNAs to Improve Mitochondrial Proteostasis. iScience 2019, 17, 155–166. [Google Scholar] [CrossRef]
- Olivo-Marston, S.E.; Hursting, S.D.; Perkins, S.N.; Schetter, A.; Khan, M.; Croce, C.; Harris, C.C.; Lavigne, J. Effects of calorie restriction and diet-induced obesity on murine colon carcinogenesis, growth and inflammatory factors, and microRNA expression. PLoS ONE 2014, 9, e94765. [Google Scholar] [CrossRef]
- Shastri, A.A.; Saleh, A.; Savage, J.E.; DeAngelis, T.; Camphausen, K.; Simone, N.L. Dietary alterations modulate the microRNA 29/30 and IGF-1/AKT signaling axis in breast Cancer liver metastasis. Nutr. Metab. 2020, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Green, J.E.; Shibata, M.A.; Yoshidome, K.; Liu, M.L.; Jorcyk, C.; Anver, M.R.; Wigginton, J.; Wiltrout, R.; Shibata, E.; Kaczmarczyk, S.; et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: Ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 2000, 19, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Holzer, R.G.; MacDougall, C.; Cortright, G.; Atwood, K.; Green, J.E.; Jorcyk, C.L. Development and characterization of a progressive series of mammary adenocarcinoma cell lines derived from the C3(1)/SV40 Large T-antigen transgenic mouse model. Breast Cancer Res. Treat. 2003, 77, 65–76. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.M.; Kerin, M.J.; Miller, N. Identification and Validation of miRNAs as Endogenous Controls for RQ-PCR in Blood Specimens for Breast Cancer Studies. PLoS ONE 2014, 8, e83718. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, G.; Hackl, H.; Gampenrieder, S.P.; Hamacher, F.; Hufnagl, C.; Hauser-Kronberger, C.; Zehentmayr, F.; Fastner, G.; Sedlmayer, F.; Mlineritsch, B.; et al. miR-16-5p Is a Stably-Expressed Housekeeping MicroRNA in Breast Cancer Tissues from Primary Tumors and from Metastatic Sites. Int. J. Mol. Sci. 2016, 17, 156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. The Guideline of the Design and Validation of MiRNA Mimics. In MicroRNA and Cancer: Methods and Protocols; Wu, W., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 211–223. [Google Scholar]
- Bowers, L.W.; Cavazos, D.A.; Maximo, I.X.; Brenner, A.J.; Hursting, S.D.; de Graffenried, L.A. Obesity enhances nongenomic estrogen receptor crosstalk with the PI3K/Akt and MAPK pathways to promote in vitro measures of breast cancer progression. Breast Cancer Res. BCR 2013, 15, R59. [Google Scholar] [CrossRef] [PubMed]
- Vejnar, C.E.; Blum, M.; Zdobnov, E.M. miRmap web: Comprehensive microRNA target prediction online. Nucleic Acids Res. 2013, 41, W165–W168. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2019, 48, D127–D131. [Google Scholar] [CrossRef]
- Quillet, A.; Saad, C.; Ferry, G.; Anouar, Y.; Vergne, N.; Lecroq, T.; Dubessy, C. Improving Bioinformatics Prediction of microRNA Targets by Ranks Aggregation. Front. Genet. 2020, 10, 1330. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef]
- Fontana, L.; Klein, S.; Holloszy, J.O.; Premachandra, B.N. Effect of Long-Term Calorie Restriction with Adequate Protein and Micronutrients on Thyroid Hormones. J. Clin. Endocrinol. Metab. 2006, 91, 3232–3235. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Feng, Y.; Deng, M.; Li, J.; Cai, H.; Meng, Q.; Fang, W.; Li, Y.; Ke, J.; Long, X. MiR-15b is a key regulator of proliferation and apoptosis of chondrocytes from patients with condylar hyperplasia by targeting IGF1, IGF1R and BCL2. Osteoarthr. Cartil. 2019, 27, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, H.; Tian, L.; Wang, F.; Han, L.; Zhang, W.; Bai, Y.-A. miR-15b Inhibits the Progression of Glioblastoma Cells Through Targeting Insulin-like Growth Factor Receptor 1. Horm. Cancer 2016, 8, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Li, F.; Li, Y.; Qin, S.; Wen, C.; Fu, Y.; Xiao, Q. Inhibition of microRNA-19b promotes ovarian granulosa cell proliferation by targeting IGF-1 in polycystic ovary syndrome. Mol. Med. Rep. 2018, 17, 4889–4898. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Jang, J.-Y.; Kim, P.-J.; Kim, Y.-G.; Nam, S.J.; Paik, J.H.; Kim, T.M.; Heo, D.S.; Kim, C.-W.; Jeon, Y.K. MicroRNA-21 plays an oncogenic role by targeting FOXO1 and activating the PI3K/AKT pathway in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 15035–15049. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Liang, R.; Yang, Y.; Hou, X.; Wang, Z.; Zhu, S.; Wang, C.; Tang, Z.; Li, K. MicroRNA-21 Regulates PI3K/Akt/mTOR Signaling by Targeting TGFβI during Skeletal Muscle Development in Pigs. PLoS ONE 2015, 10, e0119396. [Google Scholar] [CrossRef] [PubMed]
- Sahay, S.; Tiwari, P.; Pandey, M.; Gupta, K.P. PI3K/Akt Pathway and miR-21 are Involved in N-Ethyl-N-Nitrosourea-Induced F1 Mouse Lung Tumorigenesis: Effect of Inositol Hexaphosphate. J. Environ. Pathol. Toxicol. Oncol. 2019, 38, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Lerman, G.; Avivi, C.; Mardoukh, C.; Barzilai, A.; Tessone, A.; Gradus, B.; Pavlotsky, F.; Barshack, I.; Polak-Charcon, S.; Orenstein, A.; et al. MiRNA Expression in Psoriatic Skin: Reciprocal Regulation of hsa-miR-99a and IGF-1R. PLoS ONE 2011, 6, e20916. [Google Scholar] [CrossRef]
- Geng, Y.; Sui, C.; Xun, Y.; Lai, Q.; Jin, L. MiRNA-99a can regulate proliferation and apoptosis of human granulosa cells via targeting IGF-1R in polycystic ovary syndrome. J. Assist. Reprod. Genet. 2018, 36, 211–221. [Google Scholar] [CrossRef]
- Cheng, H.; Xue, J.; Yang, S.; Chen, Y.; Wang, Y.; Zhu, Y.; Wang, X.; Kuang, D.; Ruan, Q.; Duan, Y.; et al. Co-targeting of IGF1R/mTOR pathway by miR-497 and miR-99a impairs hepatocellular carcinoma development. Oncotarget 2017, 8, 47984–47997. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, J.; Kang, Z.; Liu, F.; Lin, Z. miR-101 down-regulates mTOR expression and attenuates neuropathic pain in chronic constriction injury rat models. Neurosci. Res. 2019, 158, 30–36. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, M.; Li, Q.; Zhu, P. MiR-101 reduces cell proliferation and invasion and enhances apoptosis in endometrial cancer via regulating PI3K/Akt/mTOR. Cancer Biomarkers 2021, 21, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.-S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, C.; Zhang, A.; Wang, K.; Jia, Z.; Wang, G.; Han, L.; Kang, C.; Pu, P. miR-221/222 is the regulator of Cx43 expression in human glioblastoma cells. Oncol. rep. 2012, 27, 1504–1510. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Peng, R.; Liu, H.; He, W.; Zhang, L.; Peng, H.; Zhang, Z. The Long Noncoding RNA 150Rik Promotes Mesangial Cell Proliferation via miR-451/IGF1R/p38 MAPK Signaling in Diabetic Nephropathy. Cell. Physiol. Biochem. 2018, 51, 1410–1428. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Shen, F.; Lechauve, C.; Xu, P.; Zhao, G.; Itkow, J.; Wu, F.; Hou, Y.; Wu, X.; Yu, L.; et al. miR-144/451 represses the LKB1/AMPK/mTOR pathway to promote red cell precursor survival during recovery from acute anemia. Haematologica 2018, 103, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, J.; Wang, X.; Yang, B.; Cui, L. MicroRNA-199a-5p Induced Autophagy and Inhibits the Pathogenesis of Ankylosing Spondylitis by Modulating the mTOR Signaling via Directly Targeting Ras Homolog Enriched in Brain (Rheb). Cell. Physiol. Biochem. 2017, 42, 2481–2491. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, C.; Fan, L.; Wang, J.; Li, T.; Liu, Z.; Sheng, J.; Qian, R.; Duan, A.; Lu, D. MiR-199a-5p Targets ZEB1 to Inhibit the Epithelial-Mesenchymal Transition of Ovarian Ectopic Endometrial Stromal Cells Via PI3K/Akt/mTOR Signal Pathway In Vitro and In Vivo. Reprod. Sci. 2020, 27, 110–118. [Google Scholar] [CrossRef]
- Peng, Y.; Dai, Y.; Hitchcock, C.; Yang, X.; Kassis, E.S.; Liu, L.; Luo, Z.; Sun, H.-L.; Cui, R.; Wei, H.; et al. Insulin growth factor signaling is regulated by microRNA-486, an underexpressed microRNA in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 15043–15048. [Google Scholar] [CrossRef]
- Zhu, M.; Yi, M.; Kim, C.H.; Deng, C.; Li, Y.; Medina, D.; Stephens, R.M.; E Green, J. Integrated miRNA and mRNA expression profiling of mouse mammary tumor models identifies miRNA signatures associated with mammary tumor lineage. Genome Biol. 2011, 12, R77. [Google Scholar] [CrossRef]
- Dunn, S.E.; Kari, F.W.; French, J.; Leininger, J.R.; Travlos, G.; Wilson, R.; Barrett, J.C. Dietary Restriction Reduces Insulin-like Growth Factor I Levels, Which Modulates Apoptosis, Cell Proliferation, and Tumor Progression in p53-deficient Mice. Cancer Res. 1997, 57, 4667. [Google Scholar] [PubMed]
- Harvey, A.E.; Lashinger, L.M.; Hays, D.; Harrison, L.M.; Lewis, K.; Fischer, S.M.; Hursting, S.D. Calorie restriction decreases murine and human pancreatic tumor cell growth, nuclear factor-κB activation, and inflammation-related gene expression in an insulin-like growth factor-1-dependent manner. PLoS ONE 2014, 9, e94151. [Google Scholar] [CrossRef] [PubMed]
- Lashinger, L.M.; Harrison, L.M.; Rasmussen, A.J.; Logsdon, C.D.; Fischer, S.M.; McArthur, M.J.; Hursting, S.D. Dietary energy balance modulation of Kras- and Ink4a/Arf+/--driven pancreatic cancer: The role of insulin-like growth factor-I. Cancer Prev. Res. 2013, 6, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.M.; Lavigne, J.A.; Chandramouli, G.V.; Lui, H.; Barrett, J.C.; Hursting, S.D. Dose-dependent effects of calorie restriction on gene expression, metabolism, and tumor progression are partially mediated by insulin-like growth factor-1. Cancer Med. 2012, 1, 275–288. [Google Scholar] [CrossRef]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef]
- Rickman, A.D.; Williamson, D.A.; Martin, C.K.; Gilhooly, C.H.; Stein, R.I.; Bales, C.W.; Roberts, S.; Das, S.K. The CALERIE Study: Design and methods of an innovative 25% caloric restriction intervention. Contemp. Clin. Trials 2011, 32, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Ingram, D.K.; de Cabo, R. Calorie restriction in rodents: Caveats to consider. Ageing Res. Rev. 2017, 39, 15–28. [Google Scholar] [CrossRef]
- Ibrahim, E.M.; Al-Foheidi, M.H.; Al-Mansour, M.M. Energy and caloric restriction, and fasting and cancer: A narrative review. Support. Care Cancer 2021, 29, 2299–2304. [Google Scholar] [CrossRef]
- Hofer, S.J.; Carmona-Gutierrez, D.; Mueller, M.I.; Madeo, F. The ups and downs of caloric restriction and fasting: From molecular effects to clinical application. EMBO Mol. Med. 2022, 14, e14418. [Google Scholar] [CrossRef]
- Pomatto-Watson, L.C.D.; Bodogai, M.; Bosompra, O.; Kato, J.; Wong, S.; Carpenter, M.; Duregon, E.; Chowdhury, D.; Krishna, P.; Ng, S.; et al. Daily caloric restriction limits tumor growth more effectively than caloric cycling regardless of dietary composition. Nat. Commun. 2021, 12, 6201. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Sun, H.; Zou, J.; Chen, L.; Zu, X.; Wen, G.; Zhong, J. Triple-negative breast cancer and its association with obesity. Mol. Clin. Oncol. 2017, 7, 935–942. [Google Scholar] [CrossRef]
- Berger, E.R.; Iyengar, N.M. Obesity and Energy Balance Considerations in Triple-Negative Breast Cancer. Cancer J. 2021, 27, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Ferraresi, A.; Esposito, A.; Maheshwari, C.; Dhanasekaran, D.N.; Mollace, V.; Isidoro, C. Calorie Restriction for Cancer Prevention and Therapy: Mechanisms, Expectations, and Efficacy. J. Cancer Prev. 2021, 26, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Galet, C.; Gray, A.; Said, J.W.; Castor, B.; Wan, J.; Beltran, P.J.; Calzone, F.J.; Elashoff, D.; Cohen, P.; Aronson, W.J. Effects of calorie restriction and IGF-1 receptor blockade on the progression of 22Rv1 prostate cancer xenografts. Int. J. Mol. Sci. 2013, 14, 13782–13795. [Google Scholar] [CrossRef] [PubMed]
- Usary, J.; Zhao, W.; Darr, D.; Roberts, P.J.; Liu, M.; Balletta, L.; Karginova, O.; Jordan, J.; Combest, A.; Bridges, A.; et al. Predicting drug responsiveness in human cancers using genetically engineered mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4889–4899. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Darr, D.B.; Santos, C.M.; Ross, M.; Valdivia, A.; Jordan, J.L.; Midkiff, B.R.; Cohen, S.; Nikolaishvili-Feinberg, N.; Miller, C.R.; et al. Effects of tumor microenvironment heterogeneity on nanoparticle disposition and efficacy in breast cancer tumor models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6083–6095. [Google Scholar] [CrossRef]
- Roberts, P.J.; Usary, J.E.; Darr, D.B.; Dillon, P.M.; Pfefferle, A.D.; Whittle, M.C.; Duncan, J.S.; Johnson, S.M.; Combest, A.J.; Jin, J.; et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5290–5303. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Rossi, E.L.; McDonell, S.B.; Chen, X.; Tsai, Y.H.; Parker, J.S.; Usary, J.; Perou, C.M.; Hursting, S.D. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer 2017, 3, 26. [Google Scholar] [CrossRef]
- Smith, L.A.; O’Flanagan, C.H.; Bowers, L.W.; Allott, E.H.; Hursting, S.D. Translating Mechanism-Based Strategies to Break the Obesity-Cancer Link: A Narrative Review. J. Acad. Nutr. Diet. 2018, 118, 652–667. [Google Scholar] [CrossRef]
- Vella, V.; Lappano, R.; Bonavita, E.; Maggiolini, M.; Clarke, R.B.; Belfiore, A.; De Francesco, E.M. Insulin/IGF Axis and the Receptor for Advanced Glycation End Products: Role in Meta-inflammation and Potential in Cancer Therapy. Endocr. Rev. 2023, 44, 693–723. [Google Scholar] [CrossRef] [PubMed]
- Blando, J.; Moore, T.; Hursting, S.; Jiang, G.; Saha, A.; Beltran, L.; Shen, J.; Repass, J.; Strom, S.; DiGiovanni, J. Dietary energy balance modulates prostate cancer progression in Hi-Myc mice. Cancer Prev. Res. 2011, 4, 2002–2014. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, A.; Speakman, J.R.; Soltani, S.; Djafarian, K. Effect of calorie restriction or protein intake on circulating levels of insulin like growth factor I in humans: A systematic review and meta-analysis. Clin. Nutr. 2020, 39, 1705–1716. [Google Scholar] [CrossRef]
- Redman, L.M.; Veldhuis, J.D.; Rood, J.; Smith, S.R.; Williamson, D.; Ravussin, E.; Pennington CALERIE Team. The effect of caloric restriction interventions on growth hormone secretion in nonobese men and women. Aging Cell 2010, 9, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Kaiser, D.L. Effects of calorie restriction and weight loss on glucose and insulin levels in obese humans. J. Am. Coll. Nutr. 1985, 4, 411–419. [Google Scholar] [CrossRef]
- Shah, M. Calorie Restriction and Insulin Sensitivity in Obesity. In Handbook of Famine, Starvation, and Nutrient Deprivation: From Biology to Policy; Preedy, V.R., Patel, V.B., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1127–1138. [Google Scholar]
- Kökten, T.; Hansmannel, F.; Ndiaye, N.C.; Heba, A.C.; Quilliot, D.; Dreumont, N.; Arnone, D.; Peyrin-Biroulet, L. Calorie Restriction as a New Treatment of Inflammatory Diseases. Adv. Nutr. 2021, 12, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Calin, G.A.; Grazi, G.L.; Pollutri, D.; Croce, C.M.; Bolondi, L.; Gramantieri, L. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010, 70, 5184–5193. [Google Scholar] [CrossRef]
- Li, S.Q.; Wang, Z.H.; Mi, X.G.; Liu, L.; Tan, Y. MiR-199a/b-3p suppresses migration and invasion of breast cancer cells by downregulating PAK4/MEK/ERK signaling pathway. IUBMB Life 2015, 67, 768–777. [Google Scholar] [CrossRef]
- Kim, S.; Lee, U.J.; Kim, M.N.; Lee, E.J.; Kim, J.Y.; Lee, M.Y.; Choung, S.; Kim, Y.J.; Choi, Y.C. MicroRNA miR-199a* regulates the MET proto-oncogene and the downstream extracellular signal-regulated kinase 2 (ERK2). J. Biol. Chem. 2008, 283, 18158–18166. [Google Scholar] [CrossRef]
- Leroy, C.; Ramos, P.; Cornille, K.; Bonenfant, D.; Fritsch, C.; Voshol, H.; Bentires-Alj, M. Activation of IGF1R/p110β/AKT/mTOR confers resistance to α-specific PI3K inhibition. Breast Cancer Res. 2016, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; You, K.S.; Park, J.-S.; Lee, S.-G.; Seong, Y.-S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2021, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- John, S.A.; Ottolia, M.; Weiss, J.N.; Ribalet, B. Dynamic modulation of intracellular glucose imaged in single cells using a FRET-based glucose nanosensor. Pflugers Arch. Eur. J. Physiol. 2008, 456, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Wollman, A.J.M.; Kioumourtzoglou, D.; Ward, R.; Gould, G.W.; Bryant, N.J. Large scale, single-cell FRET-based glucose uptake measurements within heterogeneous populations. iScience 2022, 25, 104023. [Google Scholar] [CrossRef] [PubMed]
- MacLean, J.A., 2nd; King, M.L.; Okuda, H.; Hayashi, K. WNT7A Regulation by miR-15b in Ovarian Cancer. PLoS ONE 2016, 11, e0156109. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Iliopoulos, D.; Struhl, K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc. Natl. Acad. Sci. USA 2012, 109, 14470–14475. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gu, X.; Sun, L.; Flowers, A.B.; Rademaker, A.W.; Zhou, Y.; Kiyokawa, H. Downregulation of Smurf2, a tumor-suppressive ubiquitin ligase, in triple-negative breast cancers: Involvement of the RB-microRNA axis. BMC Cancer 2014, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Lovat, F.; Fassan, M.; Gasparini, P.; Rizzotto, L.; Cascione, L.; Pizzi, M.; Vicentini, C.; Balatti, V.; Palmieri, D.; Costinean, S.; et al. miR-15b/16-2 deletion promotes B-cell malignancies. Proc. Natl. Acad. Sci. USA 2015, 112, 11636–11641. [Google Scholar] [CrossRef]
- Wu, B.; Liu, G.; Jin, Y.; Yang, T.; Zhang, D.; Ding, L.; Zhou, F.; Pan, Y.; Wei, Y. miR-15b-5p Promotes Growth and Metastasis in Breast Cancer by Targeting HPSE2. Front. Oncol. 2020, 10, 108. [Google Scholar] [CrossRef]
- Rizzo, S.; Cangemi, A.; Galvano, A.; Fanale, D.; Buscemi, S.; Ciaccio, M.; Russo, A.; Castorina, S.; Bazan, V. Analysis of miRNA expression profile induced by short term starvation in breast cancer cells treated with doxorubicin. Oncotarget 2017, 8, 71924–71932. [Google Scholar] [CrossRef]
- Arun, R.P.; Cahill, H.F.; Marcato, P. Breast Cancer Subtype-Specific miRNAs: Networks, Impacts, and the Potential for Intervention. Biomedicines 2022, 10, 651. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef]
- Solomon-Zemler, R.; Sarfstein, R.; Werner, H. Nuclear insulin-like growth factor-1 receptor (IGF1R) displays proliferative and regulatory activities in non-malignant cells. PLoS ONE 2017, 12, e0185164. [Google Scholar] [CrossRef] [PubMed]
- Packham, S.; Warsito, D.; Lin, Y.; Sadi, S.; Karlsson, R.; Sehat, B.; Larsson, O. Nuclear translocation of IGF-1R via p150Glued and an importin-β/RanBP2-dependent pathway in cancer cells. Oncogene 2015, 34, 2227–2238. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Fei, K.; Feldman, S.M.; Port, E.; Friedman, N.B.; Boolbol, S.K.; Killelea, B.; Pilewskie, M.; Choi, L.; King, T.; et al. Insulin resistance contributes to racial disparities in breast cancer prognosis in US women. Breast Cancer Res. 2020, 22, 40. [Google Scholar] [CrossRef]
- Popp, C.J.; Zhou, B.; Manigrasso, M.B.; Li, H.; Curran, M.; Hu, L.; St-Jules, D.E.; Alemán, J.O.; Vanegas, S.M.; Jay, M.; et al. Soluble Receptor for Advanced Glycation End Products (sRAGE) Isoforms Predict Changes in Resting Energy Expenditure in Adults with Obesity during Weight Loss. Curr. Dev. Nutr. 2022, 6, nzac046. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A.; Kotani, K.; Taing, J.; Matsuoka, Y.; Sano, Y.; Yoshimura, M.; Egawa, K.; Horikawa, C.; Kitagawa, Y.; Kiso, Y.; et al. Short-Term Low Calorie Diet Intervention Reduces Serum Advanced Glycation End Products in Healthy Overweight or Obese Adults. Ann. Nutr. Metab. 2009, 54, 197–201. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustamante-Marin, X.; Devlin, K.L.; McDonell, S.B.; Dave, O.; Merlino, J.L.; Grindstaff, E.J.; Ho, A.N.; Rezeli, E.T.; Coleman, M.F.; Hursting, S.D. Regulation of IGF1R by MicroRNA-15b Contributes to the Anticancer Effects of Calorie Restriction in a Murine C3-TAg Model of Triple-Negative Breast Cancer. Cancers 2023, 15, 4320. https://doi.org/10.3390/cancers15174320
Bustamante-Marin X, Devlin KL, McDonell SB, Dave O, Merlino JL, Grindstaff EJ, Ho AN, Rezeli ET, Coleman MF, Hursting SD. Regulation of IGF1R by MicroRNA-15b Contributes to the Anticancer Effects of Calorie Restriction in a Murine C3-TAg Model of Triple-Negative Breast Cancer. Cancers. 2023; 15(17):4320. https://doi.org/10.3390/cancers15174320
Chicago/Turabian StyleBustamante-Marin, Ximena, Kaylyn L. Devlin, Shannon B. McDonell, Om Dave, Jenna L. Merlino, Emma J. Grindstaff, Alyssa N. Ho, Erika T. Rezeli, Michael F. Coleman, and Stephen D. Hursting. 2023. "Regulation of IGF1R by MicroRNA-15b Contributes to the Anticancer Effects of Calorie Restriction in a Murine C3-TAg Model of Triple-Negative Breast Cancer" Cancers 15, no. 17: 4320. https://doi.org/10.3390/cancers15174320