Simple Summary
In blood cancers and solid tumors, genetic changes serve to initiate and promote cancer. These genetic changes can include rearrangements to genes which can alter gene function. Identification of gene rearrangements through molecular laboratory tests may help guide clinical care in patients with cancer. Different types of gene rearrangements can occur, including those within a gene and those between genes. Rearrangements occurring within a gene can be difficult to identify using current computational approaches. Our clinical laboratory designed sequencing panels for blood cancers and solid tumors to detect rearrangements within and between genes. In this study, we discuss our three-year experience using a laboratory method of targeted sequencing to detect gene rearrangements. We highlight our approach and the clinical utility for the reporting of rearrangements both within and between genes.
Abstract
Gene fusions are a form of structural rearrangement well established as driver events in pediatric and adult cancers. The identification of such events holds clinical significance in the refinement, prognostication, and provision of treatment in cancer. Structural rearrangements also extend beyond fusions to include intragenic rearrangements, such as internal tandem duplications (ITDs) or exon-level deletions. These intragenic events have been increasingly implicated as cancer-promoting events. However, the detection of intragenic rearrangements may be challenging to resolve bioinformatically with short-read sequencing technologies and therefore may not be routinely assessed in panel-based testing. Within an academic clinical laboratory, over three years, a total of 608 disease-involved samples (522 hematologic malignancy, 86 solid tumors) underwent clinical testing using Anchored Multiplex PCR (AMP)-based RNA sequencing. Hematologic malignancies were evaluated using a custom Pan-Heme 154 gene panel, while solid tumors were assessed using a custom Pan-Solid 115 gene panel. Gene fusions, ITDs, and intragenic deletions were assessed for diagnostic, prognostic, or therapeutic significance. When considering gene fusions alone, we report an overall diagnostic yield of 36% (37% hematologic malignancy, 41% solid tumors). When including intragenic structural rearrangements, the overall diagnostic yield increased to 48% (48% hematologic malignancy, 45% solid tumor). We demonstrate the clinical utility of reporting structural rearrangements, including gene fusions and intragenic structural rearrangements, using an AMP-based RNA sequencing panel.
1. Introduction
Hematologic and solid malignancies develop as the result of acquired somatic alterations or chromosomal rearrangements that promote aberrant activation of oncogenic signaling pathways or inactivation of tumor suppressor genes. Structural rearrangements, including gene fusions and intragenic deletions or duplications, are becoming increasingly recognized as drivers of pediatric and adult tumors (benign and malignant). It is estimated that >20% of human cancers are associated with a gene fusion event [1]. In addition, complex structural rearrangements are becoming increasingly recognized as driving events in cancer [2,3,4,5,6,7,8,9,10,11,12,13,14,15]. While fusions result from balanced or unbalanced structural rearrangements [16], the biological mechanisms underlying intragenic duplications or deletions are varied and gene dependent. An internal tandem duplication (ITD) results from an in-frame duplication of variable length that alters gene function.
ITDs have clinical significance in numerous cancer types. For example, an ITD occurring predominantly in exon 14 of the receptor tyrosine kinase, FLT3, is a well-established prognostic marker for acute myeloid leukemia [12,17]. The FLT3 in-frame tandem duplication encodes for a fragment of the juxtamembrane domain, a regulatory domain involved in maintaining an inactive conformation in the absence of ligand [18] resulting in ligand-independent dimerization and phosphorylation to promote constitutive signaling. Furthermore, ITDs in the growth factor receptors EGFR and FGFR1 are characteristic driver events described in pediatric and adult solid and central nervous system (CNS) tumors. The EGFR and FGFR1 intragenic structural rearrangements result in the tandem duplication of the entire tyrosine kinase domain, promoting protein autophosphorylation and constitutive receptor activation [2,5,6,8,10,13,14,15,19,20]. Notably, in the setting of both FLT3 and EGFR ITDs, targeted therapies have demonstrated clinical utility [4,6,12,13]. Intragenic deletions occur when a portion of genomic material within a gene is lost. Dependent on the gene and the deletion event, mechanistically these can result in the inactivation or altered function of the translated protein product. A classic example in pediatric acute lymphoblastic leukemia involves the IKZF1 gene, whereby variably deleted exons generate multiple alternatively spliced transcript isoforms [11]. The presence of this event helps guide patient risk stratification [21,22].
Identification of structural rearrangements may occur by both cytogenetic and molecular methodologies, including conventional karyotype, fluorescence in situ hybridization (FISH), reverse transcription PCR (RT-PCR), microarray analysis, and targeted next generation sequencing (NGS)-based assays [16,23,24,25]. Each methodology has inherent advantages and disadvantages that need to be taken into consideration during clinical interpretation (Table 1). For example, conventional karyotype analysis relies on the ability to culture disease-involved cells, and resolution is limited to what is detectable by microscopic analysis of chromosome morphology and banding patterns. FISH may provide a rapid turnaround time and information about numerical and structural abnormalities; however, data are limited to that observable in interphase or metaphase studies. Atypical and novel signal patterns, in particular, can be challenging to interpret relative to the predicted downstream impact on the gene products. Targeted RT-PCR is rapid and cost-effective for interrogating a limited number of well-characterized structural rearrangements at the level of the transcript; however, atypical breakpoints or poor-quality tissue (e.g., highly fragmented or cross-linked nucleic acid derived from some formalin-fixed paraffin-embedded (FFPE) samples) may hinder assay performance and result interpretation. NGS methods have high throughput capability to detect structural rearrangements by either a targeted or unbiased approach at the transcript level. Although the cost of sequencing has decreased over time, this approach may still be cost-prohibitive in some clinical settings and requires a strong bioinformatics infrastructure. In addition, identification of certain alterations, such as ITDs from paired-end short-read sequencing data, can still prove to be bioinformatically challenging. Bioinformatic tools such as CICERO [26] and arriba [27] may aid in an unbiased detection of ITDs and gene fusions in RNA-sequencing data, while tools such as Pindel [28] (small to moderate size insertions/deletions) may be useful for DNA-based NGS technologies.

Table 1.
Comparison of methodology.
Herein, we describe a three-year experience with an anchored multiplex PCR (AMP)-based assay to generate targeted RNA sequencing libraries for the Illumina platform. Additionally, we highlight the utility of AMP-based targeted RNA-sequencing to identify complex intragenic structural rearrangements, including internal tandem duplications (ITDs) and intragenic deletions in tumors from individuals with solid cancers and hematologic malignancies.
2. Materials and Methods
2.1. Study Participants
This retrospective study evaluated tumors and disease-involved samples (n = 608) from individuals undergoing diagnostic testing for targeted gene fusion analysis on the basis of external and internal clinical orders received by The Steve and Cindy Rasmussen Institute for Genomic Medicine Clinical Laboratory at Nationwide Children’s Hospital from 1 January 2019 through 31 December 2021. Testing was performed using a gene panel specific for either hematologic malignancy (FusionPlex Pan-Heme (Part #DB0247; FusionPlex Pan-Heme EPOR add v1.2; ArcherDx, Boulder, CO, USA) or solid tumors (FusionPlex Pan-Solid (Part #DB0177; FusionPlex NWC Pan Solid Tumor v1.0; ArcherDx, Boulder, CO, USA), depending on the indication for study. The Pan-Heme panel was offered throughout the entirety of the three-year timeframe, while the Pan-Solid panel was clinically offered starting 1 April 2019. Demographic information, including sex, age, specimen source, and indication for testing, was collected from the requisition form. This study was approved by the Institutional Review Board at Nationwide Children’s Hospital.
2.2. Library Preparation and Sequencing
This assay evaluates 154 genes for Pan-Heme and 115 genes for Pan-Solid, in addition to four control genes (Table S1). Disease-involved specimens, including peripheral blood, bone marrow, pleural fluid, and lymph node tissue for hematologic analysis, and snap frozen or FFPE for solid tumor analysis, underwent internal pathology review to assess for adequate disease involvement (>10% disease content for disease-involved peripheral blood/bone marrow/pleural fluid or snap frozen tissue, and >25% for FFPE tissue). Custom AMP-based targeted RNA sequencing assays comprising separate panels for hematologic and solid tumors were designed, with the methodology utilizing a unidirectional gene-specific primer (GSP2) which targets specific exons as part of a designed panel to enable identification of both known (recurrently described) and novel gene fusions and isoforms (see Table S1 for gene content and GSP2 primer coordinates). GSP2s were designed to cover the common structural rearrangement breakpoints and may not target all exons in the gene. Due to the unidirectional nature of the GSP2s, PCR products can amplify across unknown breakpoints, allowing for the identification of novel gene fusions and intragenic structural rearrangements. Single nucleotide variation, small intraexon insertions/deletions (with the exception of BCOR ITD and FLT3 ITD, both of which have been curated within the Archer database), and whole gene duplications/deletions cannot be detected by this assay.
2.3. Sequencing
Libraries were generated following the manufacturer’s recommendations, in addition to performing quantitative PCR (qPCR) amplification for sample quality. If the qPCR value was below 30 CT, the sample quality was deemed sufficient to continue to library generation. This assay utilizes unidirectional GSP2 to amplify molecular barcodes ligated onto cDNA fragment ends. Amplified products were sequenced using the MiSeq (Illumina, San Diego, CA, USA) sequencer. Each FusionPlex library preparation batch included a control sample comprising a 10% dilution of the SeraSeq Fusion RNA mix v3 control (Item No. 0710-0431, SeraCare Life Sciences Inc., Milford, MA, USA), from which a library was generated to assess for batch and run-level quality and accuracy in sensitively calling fusions and structural rearrangements.
2.4. Sequence Data Analysis
BCL conversion was performed in an Amazon Web Services (AWS) cloud process using bcl2fastq v2.20.0.422 (Illumina, San Diego, CA, USA). The resulting FASTQ file was transferred to the Archer Analysis Software v6.2.7 virtual instance hosted internally. Samples originally processed using Archer Analysis Software v5.1.3 were reprocessed through v6.2.7. The data analysis was configured with default settings using a gene transfer file (GTF) specific to the utilized gene panel (Pan-Solid (dSA09260 Fusion Plex NWC Pan Solid Tumor v1.0, Table S1) or Pan-Heme (FusionPlex pan Heme EPOR add v1.2, Table S1). We required a minimum Average Unique RNA Start Sites per GSP2 Controls ≥ 10 and Read Depth Normalization ≥ 3,500,000 (for downsampling). Minimum passing quality control metrics per sample include: Q30 ≥70%, FASTQ Read Count ≥ 4,500,000 (if snap frozen tissue, peripheral blood, bone marrow, pleural fluid) or 8,000,000 (if FFPE tissue). The following metrics were only evaluated for snap frozen tissue, peripheral blood, bone marrow, and pleural fluid: On-Target Reads ≥ 80%, Average RNA Reads per GSP2 ≥ 90, Average Unique Start Sites per GSP2 ≥ 30, and GSP2s with greater than 20 Unique Start Sites ≥ 40%. FFPE tissues are only required to pass minimum thresholds for Q30 reads, FASTQ reads, Percent On-Target Reads, and Average Unique RNA Start Sites per GSP2 Controls. Reads were aligned to the human reference genome (GRCh37/hg19). Gene fusions and additional complex structural rearrangements (also referred to as isoforms), including ITDs and intragenic exon deletions, were ordered in a systematized schema using a laboratory-developed analysis workflow. Fusions and isoforms were prioritized for further interpretation if they were (1) gene-to-gene events and associated with strong evidence as described by the manufacturer; (2) gene-to-gene events, associated with weak evidence as described by the manufacturer, and deemed of high quality (i.e., is not classified as a mispriming event, known Ensembl paralog, transcript readthrough, containing intronic sequence, and out of reading frame); (3) intergenic event associated with genes described with immunoglobulin gene fusions and associated with strong evidence; or (4) intergenic event associated with immunoglobulin genes (IGH, IGK, IGL) and associated with weak evidence and deemed of high quality. Events previously reported were also prioritized for additional review. Additionally, we manually reviewed genes known to be associated with intragenic rearrangement (solid tumors: BCOR, EGFR, FGFR1, MET; hematological malignancy: ERG, FLT3, IKZF1, PAX5) within the software user interface for complex structural rearrangements, filtering for rarely observed novel and oncogenic isoforms noting exons skipped or exons out of order. Structural rearrangements were interpreted using the standard guidelines set forth by the Association for Molecular Pathology (AMP), American Society of Clinical Oncology (ASCO), and College of American Pathologists (CAP) [29].
2.5. Orthogonal Confirmation
Orthogonal confirmation of structural events was performed by multiple methodologies. Most frequently, structural rearrangements were confirmed by RT-PCR followed by bi-directional Sanger sequencing. For RT-PCR, 500 ng of RNA with MultiScribe reverse transcriptase (ThermoFisher, Waltham, MA, USA) and random hexamers (Applied Biosystems, Foster City, CA, USA) was used. PCR of the cDNA product was performed with primers designed to amplify across the breakpoint junction (primer sequences are available upon request). The PCR products were purified using the QIAquick purification kit (Qiagen, Germantown, MD, USA). Forward and reverse Sanger sequencing reactions were performed using the Big Dye v3.1 terminator mix (ThermoFisher, Waltham, MA, USA). Sequencing was performed on an Applied Biosystems 3730 or 3730XL instrument. For internal tandem duplications and selected gene fusions, FASTQ files from RNA sequencing generated from the Pan-Heme or Pan-Solid assays were run through the CICERO fusion calling software to confirm the reported event [26]. Additionally, in some cases, FISH was used for orthogonal confirmation.
2.6. Statistical Analysis
A chi-squared test or unpaired t-test was used to determine the significance between groups. p < 0.05 was considered significant.
3. Results
3.1. Overall Clinical Cohort Characteristics
Among the analyzed cohort, the average age at the time of testing was 25 years old (range: 0–82 years). Males underwent testing at a slightly higher frequency (57%) compared to females (43%). The Pan-Heme panel (n = 522, 86%) was ordered at a higher frequency compared to the Pan-Solid panel (n = 86, 14%).
3.2. Three-Year Diagnostic Yield for Hematologic Malignancies
We evaluated samples from 522 individuals with hematologic malignancies using an AMP-based targeted RNA sequencing hematologic panel (Table 2 and Table S2). The mean disease involvement (as measured in percentage of blasts) for the evaluated sample was 69% (median: 80%, range: 0–95%). A single patient with hypereosinophilic syndrome had an estimated disease involvement of 0%, as this diagnosis does not typically present with increased blasts. The mean age of testing was 28 years old (median: 20 years, range: 0–82 years). Overall, 382 (73%) and 137 (26%) samples were derived from disease-involved bone marrow and peripheral blood, respectively. Other tissue types analyzed included pleural fluid (n = 1), snap frozen lymph node tissue (n = 1), and FFPE lymph node tissue (n = 1). The diagnostic yield did not differ with regard to tissue type, with 51% of peripheral blood and 46% of bone marrow samples returning clinically actionable findings (p = 0.33, Figure 1). The indications for study were captured from the provided order requisition and predominantly included individuals with acute lymphoid leukemia, not otherwise specified (n = 222, 43%), and B-cell acute lymphoid leukemia (n = 226, 43%) (Table S2). Of the 522 total cases evaluated, 261 had a finding reported, with 251 of those classified as Tier I or Tier II for strong or potential clinical significance, respectively, according to the AMP/ASCO/CAP standard guidelines [29]. Findings reported as Tier III (variants of uncertain significance) were not considered positive for diagnostic yield calculations. Among gene fusions alone, we reported a diagnostic yield of 37% (183/522 cases). With the inclusion of intragenic structural rearrangements, we observed an increase in diagnostic yield to 48% (251/522 cases, Table S2). To further evaluate diagnostic yield across age groups, individuals were classified as ≤18 years old (pediatric adolescent/young adult (AYA)) and >18 years old (adults). No differences were appreciated in diagnostic yield between the pediatric/AYA (49%, 123/249 cases) and adult groups (47%, 128/273 cases) (p = 0.57) (Figure 1).
Table 2.
Demographics for individuals undergoing testing on the Pan-Heme assay.
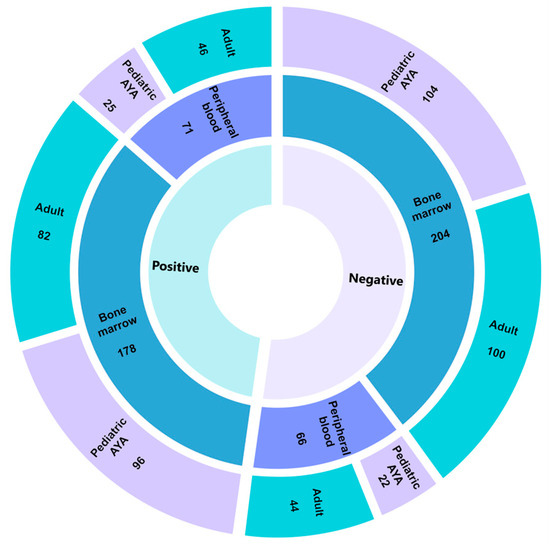
Figure 1.
Pan-Heme diagnostic yield. Sunburst plot highlighting diagnostic yield across 522 individuals who underwent testing on the Pan-Heme panel. The innermost circle denotes positive (defined as either a Tier I or II event according to AMP/ASCO/CAP guidelines) or negative findings with tissue type and age group reported in the middle and outermost circles, respectively. AYA, adolescent/young adults.
Among clinically significant fusions in the adult cohort, ABL1 and ABL2 fusions were the most frequent, with BCR::ABL1 fusion identified in 39/51 (76%) individuals (Figure 2). Of those 39 individuals with BCR::ABL1 gene fusion, 37 (95%) presented with acute lymphoid leukemia. Among the pediatric/AYA cohort, the ETV6::RUNX1 fusion typically associated with a favorable outcome in acute lymphoblastic leukemia was most frequent (Figure 2). Among both pediatric and adult cohorts, IKZF1 deletion was the most common intragenic structural rearrangement, frequently involving deletion of exons 2-3, exons 2-6, exons 2-7, and exons 4-7 (Figure 2 and Figure S1). By expanding our analysis to include intragenic structural rearrangements, we identified that 10% (51/522) of individuals harbored an intragenic rearrangement concurrently with a gene fusion, with a statistically significant preference towards adults (12%, 34/273) relative to the pediatric/AYA (7%, 17/249) group (p = 0.03, Figure S1, Table S2). Additionally, we identified a single individual with B-cell precursor acute lymphoid leukemia with two intragenic deletions involving IKZF1 and PAX5, in the absence of an ERG deletion, suggestive of the IKZF1plus phenotype [30].
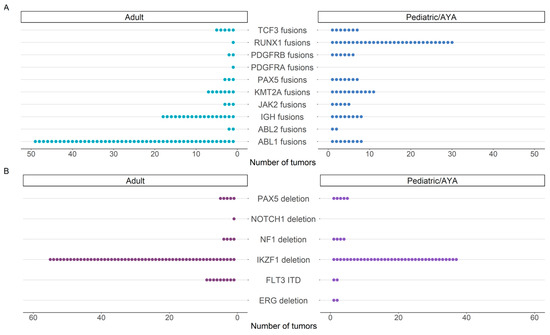
Figure 2.
Frequency of events identified by the Pan-Heme assay. The number of (A) gene fusions and (B) intragenic structural rearrangements identified in individuals with hematologic malignancy according to age group.
3.3. Three-Year Diagnostic Yield for CNS and Non-CNS Solid Tumors
We evaluated 86 CNS and non-CNS solid tumors using an AMP-based targeted RNA sequencing solid tumor panel (Table 3 and Table S3). The mean disease involvement for the evaluated tumor sections was 78% (median: 85%, range: 10–100%). The mean age at testing was 9 years old (median: 8 years, range: 0–32 years), with a strong bias towards the pediatric population due to a predominant institutional patient population undergoing study. Only five individuals >18 years of age were evaluated by the Pan-Solid panel in this cohort. Fifty-seven (66%) samples were from snap frozen tissue, while the remaining 29 (34%) were derived from FFPE sections. This cohort included 38 (44%) individuals with non-CNS solid tumors and 48 (56%) individuals with CNS tumors. Of the 40 cases with a reportable finding, 39 (98%) were classified with strong or potential clinical significance (Tier I or Tier II) according to AMP/ASCO/CAP standard guidelines (Supplementary Table S3). Among reportable gene fusions alone, a diagnostic yield of 41% (35/86 cases) was observed. With the inclusion of intragenic structural rearrangements, the diagnostic yield increased to 45% (39/86 cases). CNS tumors (23/48 (48%)) and non-CNS tumors (16/38 (42%)) demonstrated a similar proportion of cases returning a clinically significant finding (p = 0.59, Figure 3A). Despite the differences in sequencing metrics between snap frozen and FFPE tissues, the diagnostic yield of these samples was comparable, with 38% of FFPE and 49% of snap frozen tumors returning a clinically significant finding (p = 0.32, Figure 3A).
Table 3.
Demographics for individuals undergoing testing on the Pan-Solid assay.
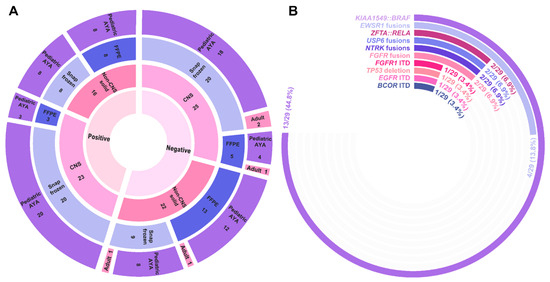
Figure 3.
Pan-Solid diagnostic yield. (A) Sunburst plot highlighting diagnostic yield across 37 individuals who underwent testing on the Pan-Solid panel. The innermost circle denotes positive (defined as either a Tier I or II event according to AMP/ASCO/CAP guidelines) or negative findings with disease type, tissue type, and age group represented by the outer circles. CNS, central nervous system; FFPE, formalin-fixed paraffin embedded; AYA, adolescent/young adult. (B) Radial bar chart demonstrating the frequency of fusions and structural rearrangements identified by the Pan-Solid panel.
Among detected fusions and isoforms, the majority (88%) represented events previously described in the literature and/or databases on the basis of gene partners (Table S3). The well-described KIAA1549::BRAF fusion was most frequently observed, being identified in 13/23 (57%) CNS tumors with a clinically significant finding (Figure 3B). While the majority of clinically significant events were gene fusions (n = 33), three cases harbored a complex structural rearrangement resulting in tandem duplication, including EGFR ITD (Solid_19), FGFR1 ITD (Solid_86), and BCOR ITD (Solid_68). In total, 36/40 (90%) cases harbored a single reportable finding. Four cases harbored multiple isoforms of the reportable gene fusion event: MYH9::USP6 (Solid_35), SOX10::NTRK3 (Solid_69), SQSTM1::NTRK2 (Solid_64), and CHCHD7::PLAG1 (Solid_21) (Table S3). Additionally, Solid_21 also harbored a second, independent PLAG1 fusion (ZFHX4::PLAG1).
3.4. Clinical Utility of Identified Events
We evaluated the clinical utility of the identified genomic events as related to the tumor type provided for testing. Events reported as Tier I or Tier II according to the AMP/ASCO/CAP guidelines were evaluated for clinical utility. In total, 217 of 608 (36%) patient samples harbored a gene fusion event classified as Tier I or Tier II providing meaningful information to inform diagnosis. In relation to the observed genomic event, our testing provided prognostic information for 90% (n = 196) of cases and demonstrated potential for change to therapeutic management in 64% (n = 138) of cases (Table S4). In total, 127 cases were identified with an intragenic deletion or ITD. Of those, there were nine unique, non-redundant events identified, including ITDs of the BCOR (n = 1), EGFR (n = 1), FGFR1 (n = 1), and FLT3 (n = 11) genes, and intragenic deletions of ERG (n = 2), IKZF1 (n = 92), NF1 (n = 8), NOTCH1 (n = 1), and PAX5 (n = 10) genes. These intragenic events provided information relating to diagnostic refinement (7/9 (78%) events), prognostication (3/9 (33%) events), and/or identification of potential targeted therapy (3/9 (33%) events) (Table S5).
The clinical utility and implications of this testing have been reported on five individuals within this cohort. We reported on an individual with an epithelioid tumor of the omentum which was found to harbor a PRRC2B::ALK fusion (Solid_13). Based on the presence of an ALK rearrangement (initially identified by FISH and refined by AMP-based RNA sequencing), the individual was offered targeted therapy with the ALK inhibitor, crizotinib [31]. We also presented a 2-year-old with a congenital inguinal mass [32]. Through AMP-based RNA sequencing, we identified two different isoforms of a CHCHD7::PLAG1 fusion and a ZFHX4::PLAG1 fusion (Solid_21). These fusion events are predicted to result in promoter swapping, whereby the 5′ gene partners CHCHD7 or ZFHX4 may drive expression of full-length PLAG1. PLAG1-rearranged tumors may present with multiple 5′ fusion partners, and emerging data suggest that the histologic spectrum of this entity is expanding [33,34,35]. Furthermore, we also previously reported on an infant with a congenital midline lesion associated with a closed spinal dysraphism [36]. In this atypical lesion, we identified an FGFR1 ITD (Solid_86) which is exceedingly rare outside of CNS tumors, highlighting the expanding phenotypic spectrum to include benign lesions. Additionally, we reported on an infant who presented with a renal mass for which an EGFR ITD (Solid_19) was identified [37]. The presence of the EGFR ITD confirmed a diagnosis of congenital mesoblastic nephroma. Lastly, we described a 2-year-old female with a CIC::NUTM1 sarcoma (Solid_47) and highlighted the histological overlap with primitive myxoid mesenchymal tumor of infancy [38].
4. Discussion
Clinical testing to identify a wide breadth of gene fusion or complex structural rearrangement events across many genes and breakpoints is becoming increasingly critical for guiding diagnostic, prognostic, and therapeutic information. The capabilities of NGS-based assays to simultaneously evaluate gene fusions and complex structural rearrangements has the potential to improve patient care by reducing time to molecular diagnosis and improving diagnostic yield. Notably, the 2021 World Health Organization (WHO) Classification of Tumours of the Central Nervous System transformed the reporting schema for CNS tumor diagnostics, promoting the integration of molecular and histopathological data to delineate a final integrated diagnosis [39]. Herein, we describe our three-year experience using AMP-based targeted RNA sequencing among pediatric and adult populations and the expansion of our reporting paradigm to include both gene fusions and complex structural rearrangements. This study highlights the capability and utility of AMP-based targeted RNA fusion panels as a diagnostic tool for reporting clinically significant complex structural rearrangements, in addition to gene fusions.
Multiple groups have demonstrated the feasibility and utility of AMP-based targeted RNA sequencing panels to identify gene fusions across a breadth of hematologic, CNS, and non-CNS solid tumors in the diagnostic setting [40,41,42,43]. Panel composition across studies varied from 53 to 112 genes, with enrichment for targets in both solid tumors (n = 4 studies) and hematologic malignancies (n = 1 study). Across these studies, the reported diagnostic yield was broad (3.8–61%), owing to differences in patient populations and tumor types under evaluation [40,41,42,43]. Notably, complex structural rearrangements were not well represented among these prior studies, with only a single group reporting the well-described MET and EGFR exon skipping events [42]. Expanding on these prior studies, we evaluated diagnostic yield across a wide breadth of hematologic malignancies and solid tumors, reporting clinically significant complex structural rearrangements in addition to gene fusions. Similar to prior studies, the diagnostic yield of our assay was 36% (37% hematologic malignancies, 41% solid tumors) when considering only gene fusions. By including a report of complex structural rearrangements, we increased our combined diagnostic yield to 48% (48% hematologic malignancies, 45% solid tumors). Thus, we provide evidence demonstrating the clinical utility of AMP-based targeted RNA sequencing methodologies for the identification of complex structural rearrangements.
The identification of complex structural rearrangement may have clinical significance for individuals with hematologic and solid malignancies. For example, IKZF1 encodes for IKAROS, a regulator of lymphoid differentiation. The IKZF1 deletion was the most common event observed amongst both pediatric/AYA and adult Pan-Heme cohorts (Figure 2). Deletion of IKZF1, most frequently whole gene deletion, is found in 25–35% of adults and 12–17% of children with acute lymphoid leukemia and confers a poorer prognosis [11,21,44]. A high correlation between the BCR::ABL1 fusion and IKZF1 deletion has been reported in individuals with B-cell acute lymphoid leukemia, with both events serving as independent risk factors associated with poor prognosis [45]. PAX5 intragenic deletions were also frequently identified in our hematologic cohort (Figure 2). PAX5 encodes paired box 5, a transcription factor essential for regulating the B cell lineage differentiation [46]. Previously described intragenic deletions have been shown to abolish the homeodomain-like domain and part of the transactivation domain, resulting in a loss of function [47,48,49]. PAX5 loss arrests B cell differentiation at immature precursor stages to promote leukemogenesis [46]. FLT3 was the most frequently observed ITD among the Pan-Heme cohort. FLT3 encodes a receptor tyrosine kinase that is widely expressed among hematopoietic progenitor cells [50]. FLT3 ITD is observed in about 30% of acute myeloid leukemia and is associated with poor risk [12,50,51]. FLT3 inhibitors have demonstrated clinical efficacy among this patient population, although resistance remains a challenge [4]. While intragenic structural rearrangements were not as frequent among our Pan-Solid cohort, we identified three cases harboring an ITD in our solid tumor cohort. In the setting of a CNS tumor, the presence of a BCOR ITD may provide diagnostic information, defining a molecular entity in the WHO Classification of CNS Tumours known as CNS Tumour with BCOR ITD [39,52,53,54,55,56,57]. In the kidney, the identification of a BCOR ITD is diagnostic for clear cell sarcoma of the kidney [57,58,59]. In the soft tissue, BCOR genetic alterations, including BCOR ITD, are diagnostically defining for a sarcoma that includes undifferentiated round cell sarcoma and primitive myxoid mesenchymal tumor of infancy morphologic types [60]. Similarly, EGFR ITDs are described in the setting of infantile fibrosarcoma and congenital mesoblastic nephroma, particularly in classic or mixed subtype, and aids in diagnostics if the tumor is negative for the more commonly identified ETV6::NTRK3 fusion. This scenario further highlights the utility of broader panel testing and analysis to encompass recurrent events to enable alignment for an integrated diagnosis along with histopathologic characteristics [19,20,37,61]. FGFR1 ITDs are commonly reported in the setting of low-grade glial tumors for which the therapeutic significance is still under study [5,8,15].
There are technical aspects of this multiplexed targeted RNA panel that offer improvements over the current methodologies. For example, FISH utilizes fluorescent probes targeted to regions of interest for a particular gene or locus. FISH is a rapid and cost-effective method to identify regions of genomic loss or gain or structural rearrangement; however, it is highly targeted and cannot accurately determine the downstream impact on transcripts or proteins. If a tumor harbors an atypical fusion partner or breakpoint, a very focal event, or a complex event, FISH may be unable to detect it. Multiplexed NGS assays have demonstrated cost-benefit and reduced turnaround time compared to single gene testing in a variety of cancer types [62,63,64]. AMP-based chemistry allows for the identification of both recurrent fusions and structural rearrangements and importantly those that are novel or may harbor atypical breakpoints [65]. The ability to fully characterize a structural rearrangement can have clinical impact. For example, the EWSR1 gene generates fusion transcripts with multiple gene partners. While break-apart EWSR1 FISH probes are frequently used for diagnostic testing to determine if an EWSR1 rearrangement is present, this assay is not able to identify the fusion partner. The 3′ gene partner is important for diagnostic classification as various partners (e.g., FLI1 in Ewing sarcoma, WT1 in desmoplastic small round cell tumor) are characteristic of distinct tumor entities [66]. While some ITDs have recurrent breakpoints, the majority are not recurrent, may be different sizes, and can include unaligned or intronic linker sequence. Notably, we have demonstrated that AMP chemistry can call intragenic structural rearrangements across a wide breadth of sizes, with an average size for FLT3 ITD (n = 11) of 64 bp (range: 21–198 bp) (Table S2). Intragenic deletions for hematologic malignancies (n = 113) typically spanned multiple exons, with an average size of 108,545 bp (range: 41,536–139,851 bp). While ITDs were less commonly seen amongst solid tumors (n = 3), the size tended to be larger than what was observed for FLT3 ITD (Table S3), with a BCOR ITD of 90 bp and an EGFR ITD of 27,434 bp representing the smallest and largest events in this cohort, respectively.
This assay also demonstrates technical limitations that should be considered when evaluating and clinically interpreting intragenic, complex structural rearrangements. In tumors for which a FLT3 ITD is identified, the frequency or allelic ratio is necessary to fully delineate the prognostic implications of this event [51,67]. Our implementation of AMP-based targeted RNA sequencing does not enable a determination of the allelic ratio. Additionally, the ability of this assay to detect very small ITD events is expected to be limited. Furthermore, our assay was not designed to detect whole gene (or larger) gains or losses in the form of copy number variation. Gene-level and larger IKZF1 deletions associated with haploinsufficiency would not be reportable through this assay [11,68]. Moreover, the bioinformatic identification of complex structural rearrangements using the manufacturer’s analysis software user interface may pose challenges if the event is not curated within the Quiver database (http://quiver.archerdx.com/ accessed on 31 July 2023). Curated events, such as the EGFR ITD and IKZF1 deletion, are classified as oncogenic isoforms within the manufacturer’s analysis software and thus prioritized for analysis. Many of the newly described clinically relevant intragenic deletions and ITDs (e.g., FGFR1 ITDs) are not curated. Therefore, identifying these events typically requires manual review amongst many false positive fusions and isoforms and readthrough events. Additionally, the reporting of small intraexon insertions/deletions (termed “intraexon gap” in the Archer UI) which have not been curated within the Quiver database (i.e., FLT3 ITD and BCOR ITD) remains a challenge as fragmented nucleic acids, such as those from FFPE samples, tend to have an elevated level of false positive intraexon gap events based on our experience. While calling these events is technically possible with this methodology, it will require significant validation to ensure accurate reporting. The incorporation of other bioinformatic tools, such as CICERO [26], may assist in identifying complex structural rearrangements, particularly ITDs, and improve the ability to distinguish artifacts from true, medically meaningful events. While our laboratory testing scheme currently incorporates orthogonal confirmation for many reportable events, the use of orthogonal confirmation may vary between laboratories.
There are limitations to consider for this retrospective study. While our institution serves a pediatric population, we also serve as a reference lab for the testing of both pediatric and adult samples, as evident in our Pan-Heme cohort, where 52% of cases were >18 years old. However, our Pan-Solid cohort consisted predominantly of a pediatric population (94% of individuals ≤18 years old at the time of testing) derived predominantly from in-house testing. The inherent differences in the etiology of pediatric and adult tumors should be considered when interpreting these data as pediatric tumors are more likely to have a low somatic mutation burden and be driven by gene fusions or structural rearrangements [69,70]. The genetic landscape for pediatric and adult tumors is constantly evolving and ensuring gene content for panel-based testing relevant to the patient population under study is critical. Both Pan-Heme and Pan-Solid panels used in our laboratory were customized to include additional gene specific primers targeting genes associated with pediatric cancers.
5. Conclusions
In summary, we describe the three-year diagnostic yield of clinical targeted RNA sequencing using AMP-based library preparation. The cohort tested at our institution represents a wide spectrum of demographics, including pediatric/AYA and adult cohorts evaluated for hematologic and solid cancers, using diverse tissue types, including FFPE, snap frozen, peripheral blood, and bone marrow. We demonstrate an increase in diagnostic yield when complex structural rearrangements, including intragenic deletions and ITDs, are reported by this methodology, expanding on the provision of clinically useful information as related to diagnostic classification, prognostication, and therapeutic determination in this cohort.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers15174394/s1, Figure S1: Co-occurrence of gene fusions and intragenic structural rearrangements, Table S1: Pan-Heme and Pan-Solid gene lists, Table S2: Pan-Heme Gene Fusions and Structural Rearrangements, Table S3: Pan-Solid Gene Fusions and Structural Rearrangements, Table S4: Gene Fusion Clinical Outcomes, Table S5: Intragenic Structural Rearrangement Clinical Outcomes.
Author Contributions
Conceptualization, K.M.S., K.L. and C.E.C.; methodology, K.M.S., K.L. and C.E.C.; formal analysis, K.M.S., A.M. (Amanda Moccia), B.A.B., E.S., V.J., H.J., A.M. (Aimee McKinney), S.C.K., M.T.M., K.L., S.C.R. and C.E.C.; investigation, K.M.S., A.M. (Amanda Moccia), E.S., V.J., H.J., S.C.K., M.T.M., K.L., S.C.R. and C.E.C.; resources, E.R.M.; data curation, K.M.S., A.M. (Amanda Moccia), B.A.B., E.S., V.J., H.J. and A.M. (Aimee McKinney); writing—original draft preparation, K.M.S. and B.A.B.; writing—review and editing, K.M.S., A.M. (Amanda Moccia), B.A.B., E.S., V.J, H.J., A.M. (Aimee McKinney), S.C.K., M.T.M., E.R.M., K.L., S.C.R. and C.E.C.; visualization, K.M.S. and B.A.B.; supervision, K.L. and C.E.C.; project administration, K.M.S., K.L., C.E.C. and E.R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of Nationwide Children’s Hospital (IRB17-00206, approved 9 May 2017). For patients with solely clinical testing, ethical review and approval were waived for this study, as the study was deemed to be no greater than minimal risk to patients (STUDY00001767, granted 14 June 2021).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Acknowledgments
We thank the Nationwide Foundation Pediatric Innovation Fund for generously supporting sequencing, data production, and analysis. Additionally, we would like to acknowledge Ruthann Pfau for her commitment and guidance in the realm of gene fusion clinical reporting and counsel in study design.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Baik, C.S.; Wu, D.; Smith, C.; Martins, R.G.; Pritchard, C.C. Durable response to tyrosine kinase inhibitor therapy in a lung cancer patient harboring epidermal growth factor receptor tandem kinase domain duplication. J. Thorac. Oncol. 2015, 10, e97–e99. [Google Scholar] [CrossRef]
- Chiang, S.; Lee, C.H.; Stewart, C.J.R.; Oliva, E.; Hoang, L.N.; Ali, R.H.; Hensley, M.L.; Arias-Stella, J.A.; Frosina, D.; Jungbluth, A.A.; et al. BCOR is a robust diagnostic immunohistochemical marker of genetically diverse high-grade endometrial stromal sarcoma, including tumors exhibiting variant morphology. Mod. Pathol. 2017, 30, 1251–1261. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Fina, F.; Barets, D.; Colin, C.; Bouvier, C.; Padovani, L.; Nanni-Metellus, I.; Ouafik, L.H.; Scavarda, D.; Korshunov, A.; Jones, D.T.W.; et al. Droplet digital PCR is a powerful technique to demonstrate frequent FGFR1 duplication in dysembryoplastic neuroepithelial tumors. Oncotarget 2017, 8, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.N.; Sheehan, J.H.; Shaver, T.M.; Bailey, M.; Lipson, D.; Chandramohan, R.; Brewer, M.R.; York, S.J.; Kris, M.G.; Pietenpol, J.A.; et al. EGFR kinase domain duplication (EGFR -KDD) is a novel oncogenic driver in lung cancer that is clinically responsive to afatinib. Cancer Discov. 2015, 5, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Butler, E.; Wilson, R.; Roy, A.; Zheng, Y.; Liem, P.; Rakheja, D.; Pavlick, D.; Young, L.L.; Rosenzweig, M. Novel PDGFRB rearrangement in multifocal infantile myofibromatosis is tumorigenic and sensitive to imatinib. Mol. Case Stud. 2019, 5, a004440. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Hutter, B.; Jäger, N.; Korshunov, A.; Kool, M.; Warnatz, H.J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.K.; et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef]
- Ryall, S.; Zapotocky, M.; Fukuoka, K.; Nobre, L.; Stucklin, A.G.; Bennett, J.; Siddaway, R.; Li, C.; Pajovic, S.; Arnoldo, A. Integrated molecular and clinical analysis of 1000 pediatric low-grade gliomas. Cancer Cell 2020, 37, 569–583.e5. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Cavé, H.; Moorman, A.V. IKZF1 deletions in pediatric acute lymphoblastic leukemia: Still a poor prognostic marker? Blood 2020, 135, 252–260. [Google Scholar] [CrossRef]
- Tallman, M.S.; Wang, E.S.; Altman, J.K.; Appelbaum, F.R.; Bhatt, V.R.; Bixby, D.; Coutre, S.E.; De Lima, M.; Fathi, A.T.; Fiorella, M.; et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 721–749. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.; Xue, X.; Ou, Q.; Wu, X.; Liang, Y.; Wang, X.; You, M.; Shao, Y.W.; Zhang, Z. Clinical outcomes of EGFR kinase domain duplication to targeted therapies in NSCLC. Int. J. Cancer 2019, 144, 2677–2682. [Google Scholar] [CrossRef] [PubMed]
- Wegert, J.; Vokuhl, C.; Collord, G.; Del Castillo Velasco-Herrera, M.; Farndon, S.J.; Guzzo, C.; Jorgensen, M.; Anderson, J.; Slater, O.; Duncan, C.; et al. Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nat. Commun. 2018, 9, 2378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The Structural Basis for Autoinhibition of FLT3 by the Juxtamembrane Domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Lei, L.; Stohr, B.A.; Berry, S.; Lockwood, C.M.; Davis, J.L.; Rudzinski, E.R.; Kunder, C.A. Recurrent EGFR alterations in NTRK3 fusion negative congenital mesoblastic nephroma. Pract. Lab. Med. 2020, 21, e00164. [Google Scholar] [CrossRef]
- Zhao, M.; Yin, M.; Kuick, C.H.; Chen, H.; Aw, S.J.; Merchant, K.; Ng, E.H.Q.; Gunaratne, S.; Loh, A.H.P.; Gu, W.; et al. Congenital mesoblastic nephroma is characterised by kinase mutations including EGFR internal tandem duplications, the ETV6–NTRK3 fusion, and the rare KLHL7–BRAF fusion. Histopathology 2020, 77, 611–621. [Google Scholar] [CrossRef]
- Brown, P.; Inaba, H.; Annesley, C.; Beck, J.; Colace, S.; Dallas, M.; DeSantes, K.; Kelly, K.; Kitko, C.; Lacayo, N.; et al. Pediatric acute lymphoblastic leukemia, version 2.2020. JNCCN J. Natl. Compr. Cancer Netw. 2020, 18, 81–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.M.; Harvey, R.C.; Mullighan, C.G.; Gastier-Foster, J.; Wharton, W.; Kang, H.; Borowitz, M.J.; Camitta, B.M.; Carroll, A.J.; Devidas, M.; et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood 2012, 119, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A. The role of cytogenetics and molecular diagnostics in the diagnosis of soft-tissue tumors. Mod. Pathol. 2014, 27, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Paratala, B.S.; Dolfi, S.C.; Khiabanian, H.; Rodriguez-Rodriguez, L.; Ganesan, S.; Hirshfield, K.M. Emerging Role of Genomic Rearrangements in Breast Cancer: Applying Knowledge from Other Cancers. Biomark. Cancer 2016, 8s1, BIC.S34417. [Google Scholar] [CrossRef]
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M.; Dal Cin, P.; Diez Campelo, M.; Gallego, M.S.; Granada Font, I.; Haase, D.T.; et al. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2022, 139, 2273–2284. [Google Scholar] [CrossRef]
- Tian, L.; Li, Y.; Edmonson, M.N.; Zhou, X.; Newman, S.; McLeod, C.; Thrasher, A.; Liu, Y.; Tang, B.; Rusch, M.C.; et al. CICERO: A versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol. 2020, 21, 126. [Google Scholar] [CrossRef]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Bennett, J.T.; Tan, T.Y.; Alcantara, D.; Tétrault, M.; Timms, A.E.; Jensen, D.; Collins, S.; Nowaczyk, M.J.; Lindhurst, M.J.; Christensen, K.M. Mosaic activating mutations in FGFR1 cause encephalocraniocutaneous lipomatosis. Am. J. Hum. Genet. 2016, 98, 579–587. [Google Scholar] [CrossRef]
- Gupta, A.; Liu, H.; Schieffer, K.M.; Koo, S.C.; Cottrell, C.E.; Mardis, E.R.; Roberts, R.D.; Yeager, N.D. Targeted Therapy in a Young Adult with a Novel Epithelioid Tumor Driven by a PRRC2B-ALK Fusion. J. Natl. Compr. Cancer Netw. 2021, 19, 1116–1121. [Google Scholar] [CrossRef]
- Logan, S.J.; Schieffer, K.M.; Conces, M.R.; Stonerock, E.; Miller, A.R.; Fitch, J.; LaHaye, S.; Voytovich, K.; McGrath, S.; Magrini, V. Novel morphologic findings in PLAG1-rearranged soft tissue tumors. Genes Chromosom. Cancer 2021, 60, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Brčić, I.; Igrec, J.; Halbwedl, I.; Viertler, C.; Liegl-Atzwanger, B. Expanding the spectrum of PLAG1-rearranged lipoblastomas arising in patients over 45, with identification of novel fusion partners. Mod. Pathol. 2022, 35, 283–285. [Google Scholar] [CrossRef]
- Chung, C.T.; Antonescu, C.R.; Dickson, B.C.; Chami, R.; Marrano, P.; Fan, R.; Shago, M.; Hameed, M.; Thorner, P.S. Pediatric fibromyxoid soft tissue tumor with PLAG1 fusion: A novel entity? Genes Chromosomes Cancer 2021, 60, 263–271. [Google Scholar] [CrossRef]
- Fritchie, K.; Wang, L.; Yin, Z.; Nakitandwe, J.; Hedges, D.; Horvai, A.; Mora, J.T.; Folpe, A.L.; Bahrami, A. Lipoblastomas presenting in older children and adults: Analysis of 22 cases with identification of novel PLAG1 fusion partners. Mod. Pathol. 2021, 34, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Kautto, E.A.; Schieffer, K.M.; McGrath, S.; Miller, A.R.; Hernandez-Gonzalez, M.E.; Choi, S.; Conces, M.R.; Fernandez-Faith, E.; Ho, M.L.; Lee, K.; et al. Expanding the clinical phenotype of FGFR1 internal tandem duplication. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006174. [Google Scholar] [CrossRef]
- Koo, S.C.; Schieffer, K.M.; Lee, K.; Gupta, A.; Pfau, R.B.; Avenarius, M.R.; Stonerock, E.; LaHaye, S.; Fitch, J.; Setty, B.A.; et al. EGFR internal tandem duplications in fusion-negative congenital and neonatal spindle cell tumors. Genes Chromosomes Cancer 2023, 62, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Biederman, L.E.; Lee, K.; Yeager, N.D.; Sribnick, E.A.; Shenoy, A. CIC::NUTM1 sarcoma mimicking primitive myxoid mesenchymal tumour of infancy: Report of a case. Histopathology 2022, 81, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Avenarius, M.R.; Miller, C.R.; Arnold, M.A.; Koo, S.; Roberts, R.; Hobby, M.; Grossman, T.; Moyer, Y.; Wilson, R.K.; Mardis, E.R.; et al. Genetic Characterization of Pediatric Sarcomas by Targeted RNA Sequencing. J. Mol. Diagn. 2020, 22, 1238–1245. [Google Scholar] [CrossRef]
- Chang, F.; Lin, F.; Cao, K.; Surrey, L.F.; Aplenc, R.; Bagatell, R.; Resnick, A.C.; Santi, M.; Storm, P.B.; Tasian, S.K.; et al. Development and Clinical Validation of a Large Fusion Gene Panel for Pediatric Cancers. J. Mol. Diagn. 2019, 21, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Hindi, I.; Shen, G.; Tan, Q.; Cotzia, P.; Snuderl, M.; Feng, X.; Jour, G. Feasibility and clinical utility of a pan-solid tumor targeted RNA fusion panel: A single center experience. Exp. Mol. Pathol. 2020, 114, 104403. [Google Scholar] [CrossRef]
- Selvam, P.; Kelly, K.; Hesse, A.N.; Spitzer, D.; Reddi, H.V. Evaluating gene fusions in solid tumors—Clinical experience using an RNA based 53 gene next-generation sequencing panel. Cancer Genet. 2019, 233, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Marke, R.; Leeuwen, F.N.v.; Scheijen, B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 103, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Medvedovic, J.; Ebert, A.; Tagoh, H.; Busslinger, M. Pax5: A master regulator of B cell development and leukemogenesis. Adv. Immunol. 2011, 111, 179–206. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5 -driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Schwab, C.; Nebral, K.; Chilton, L.; Leschi, C.; Waanders, E.; Boer, J.M.; Žaliová, M.; Sutton, R.; Öfverholm, I.I.; Ohki, K.; et al. Intragenic amplification of PAX5: A novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv. 2017, 1, 1473. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Appay, R.; Macagno, N.; Padovani, L.; Korshunov, A.; Kool, M.; André, N.; Scavarda, D.; Pietsch, T.; Figarella-Branger, D. HGNET-BCOR Tumors of the Cerebellum: Clinicopathologic and Molecular Characterization of 3 Cases. Am. J. Surg. Pathol. 2017, 41, 1254–1260. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nobusawa, S.; Nakata, S.; Nakada, M.; Arakawa, Y.; Mineharu, Y.; Sugita, Y.; Yoshioka, T.; Araki, A.; Sato, Y.; et al. CNS high-grade neuroepithelial tumor with BCOR internal tandem duplication: A comparison with its counterparts in the kidney and soft tissue. Brain Pathol. 2018, 28, 710–720. [Google Scholar] [CrossRef]
- Ferris, S.P.; Velazquez Vega, J.; Aboian, M.; Lee, J.C.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Saunders, T.; Chen, Y.Y.; Banerjee, A.; et al. High-grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication—A comprehensive clinical, radiographic, pathologic, and genomic analysis. Brain Pathol. 2020, 30, 46–62. [Google Scholar] [CrossRef]
- De Lima, L.; Sürme, M.B.; Gessi, M.; Mastronuzzi, A.; Miele, E.; Tamburrini, G.; Massimi, L. Central nervous system high-grade neuroepithelial tumor with BCOR alteration (CNS HGNET-BCOR)-case-based reviews. Childs Nerv. Syst. 2020, 36, 1589–1599. [Google Scholar] [CrossRef]
- Al-Ibraheemi, A.; Putra, J.; Tsai, H.K.; Cano, S.; Lip, V.; Pinches, R.S.; Restrepo, T.; Alexandrescu, S.; Janeway, K.A.; Duraisamy, S.; et al. Assessment of BCOR Internal Tandem Duplications in Pediatric Cancers by Targeted RNA Sequencing. J. Mol. Diagn. JMD 2021, 23, 1269–1278. [Google Scholar] [CrossRef]
- Ueno-Yokohata, H.; Okita, H.; Nakasato, K.; Akimoto, S.; Hata, J.; Koshinaga, T.; Fukuzawa, M.; Kiyokawa, N. Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat. Genet. 2015, 47, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kumar, V.; Zorman, B.; Fang, E.; Haines, K.M.; Doddapaneni, H.; Hampton, O.A.; White, S.; Bavle, A.A.; Patel, N.R.; et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat. Commun. 2015, 6, 8891. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef]
- Kao, E.; Pinto, N.; Trobaugh-Lotrario, A.; Deutsch, G.H.; Wu, Y.; Wang, W.; Rudzinski, E.R.; Liu, Y.J. Tyrosine kinase-altered spindle cell neoplasms with EGFR internal tandem duplications. Genes Chromosom. Cancer 2022, 61, 616–621. [Google Scholar] [CrossRef]
- De Unamuno Bustos, B.; Estal, R.M.; Simó, G.P.; De Juan Jimenez, I.; Muñoz, B.E.; Serna, M.R.; De Miquel, V.A.; Ros, M.L.; Sánchez, R.B.; Enguídanos, E.N.; et al. Towards Personalized Medicine in Melanoma: Implementation of a Clinical Next-Generation Sequencing Panel. Sci. Rep. 2017, 7, 495. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, A.; Wordsworth, S.; Fermont, J.M.; Page, S.; Kaur, K.; Camps, C.; Kaisaki, P.; Gupta, A.; Talbot, D.; Middleton, M.; et al. Clinical applicability and cost of a 46-gene panel for genomic analysis of solid tumours: Retrospective validation and prospective audit in the UK National Health Service. PLoS Med. 2017, 14, e1002230. [Google Scholar] [CrossRef] [PubMed]
- Pennell, N.A.; Mutebi, A.; Zhou, Z.-Y.; Ricculli, M.L.; Tang, W.; Wang, H.; Guerin, A.; Arnhart, T.; Dalal, A.; Sasane, M.; et al. Economic Impact of Next-Generation Sequencing Versus Single-Gene Testing to Detect Genomic Alterations in Metastatic Non–Small-Cell Lung Cancer Using a Decision Analytic Model. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Krystel-Whittemore, M.; Taylor, M.S.; Rivera, M.; Lennerz, J.K.; Le, L.P.; Dias-Santagata, D.; Iafrate, A.J.; Deshpande, V.; Chebib, I.; Nielsen, G.P.; et al. Novel and established EWSR1 gene fusions and associations identified by next-generation sequencing and fluorescence in-situ hybridization. Hum. Pathol. 2019, 93, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Straube, J.; Ling, V.Y.; Hill, G.R.; Lane, S.W. The impact of age, NPM1(mut), and FLT3(ITD) allelic ratio in patients with acute myeloid leukemia. Blood 2018, 131, 1148–1153. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.H.; et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef]
- Dupain, C.; Harttrampf, A.C.; Urbinati, G.; Geoerger, B.; Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Mol. Ther. Nucleic Acids 2017, 6, 315–326. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).