
Evaluation of Temozolomide and Fingolimod Treatments in Glioblastoma Preclinical Models

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Cells and Cell Culture
2.2. In Vitro Treatments
2.3. 2D Cell Growth Assay
2.4. 3D Tumor Spheroid Assay
2.5. Animals
2.6. In Vivo Treatments
2.7. Subcutaneous Graft Animal Model
2.8. Brain Orthotopic Graft Animal Model
2.9. Animal Ethical Consideration and Limit Points
2.10. Patient Data Analysis
2.11. Statistics
3. Results
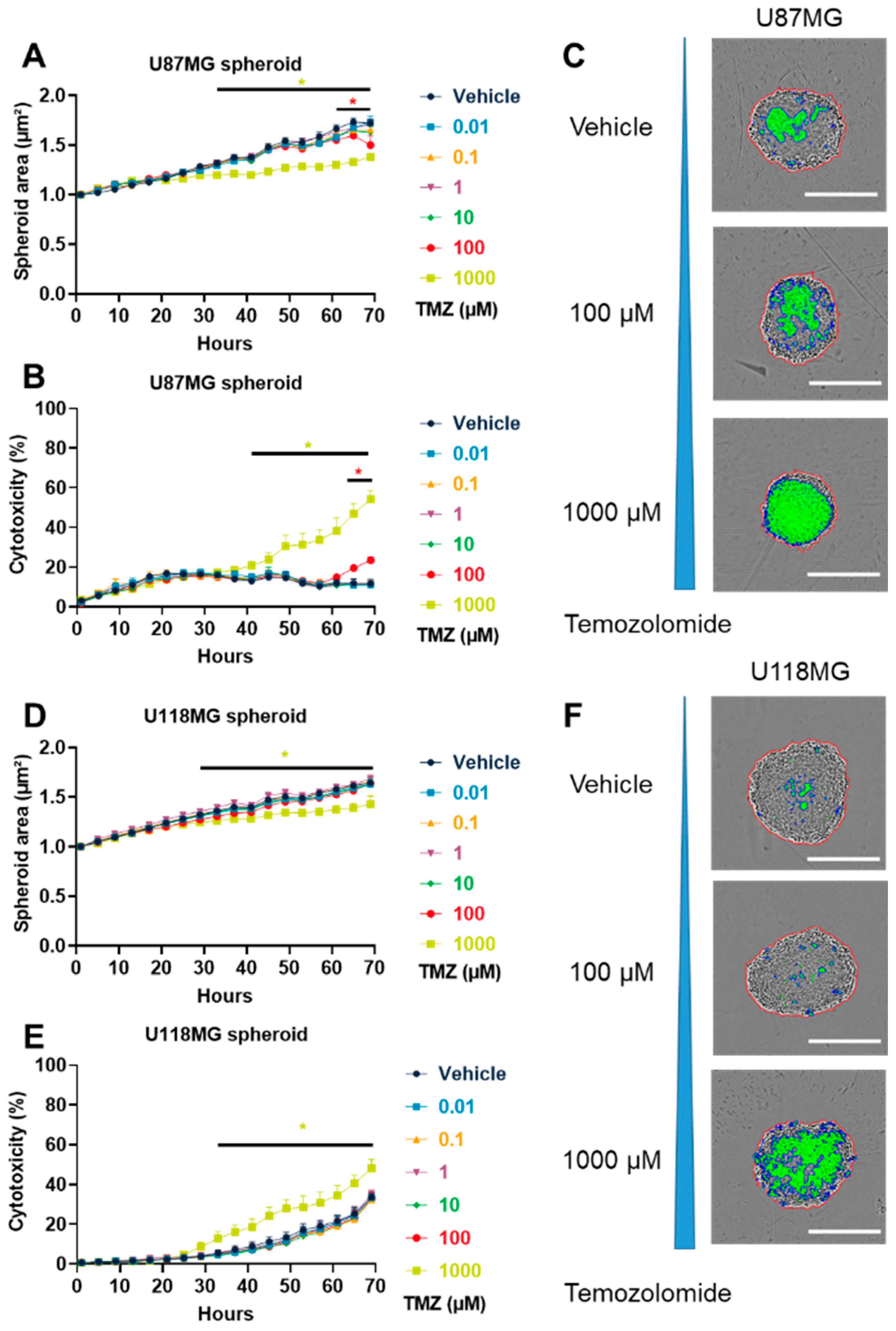
3.1. TMZ Induced GBM Cytotoxicity in Both 2D and 3D In Vitro Models
3.2. TMZ Affected GBM In Vivo Tumor Growth Differently in Subcutaneous Model
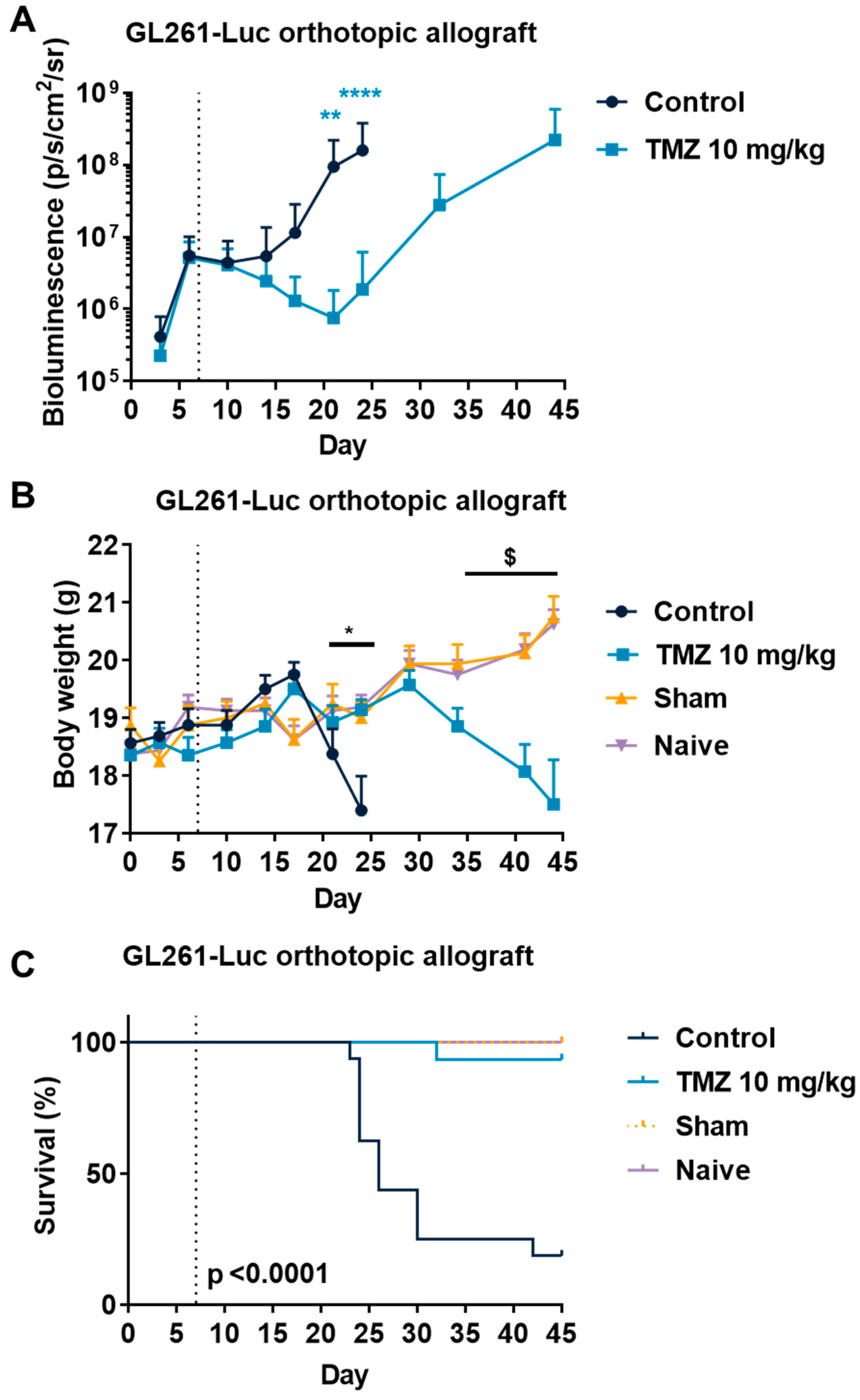
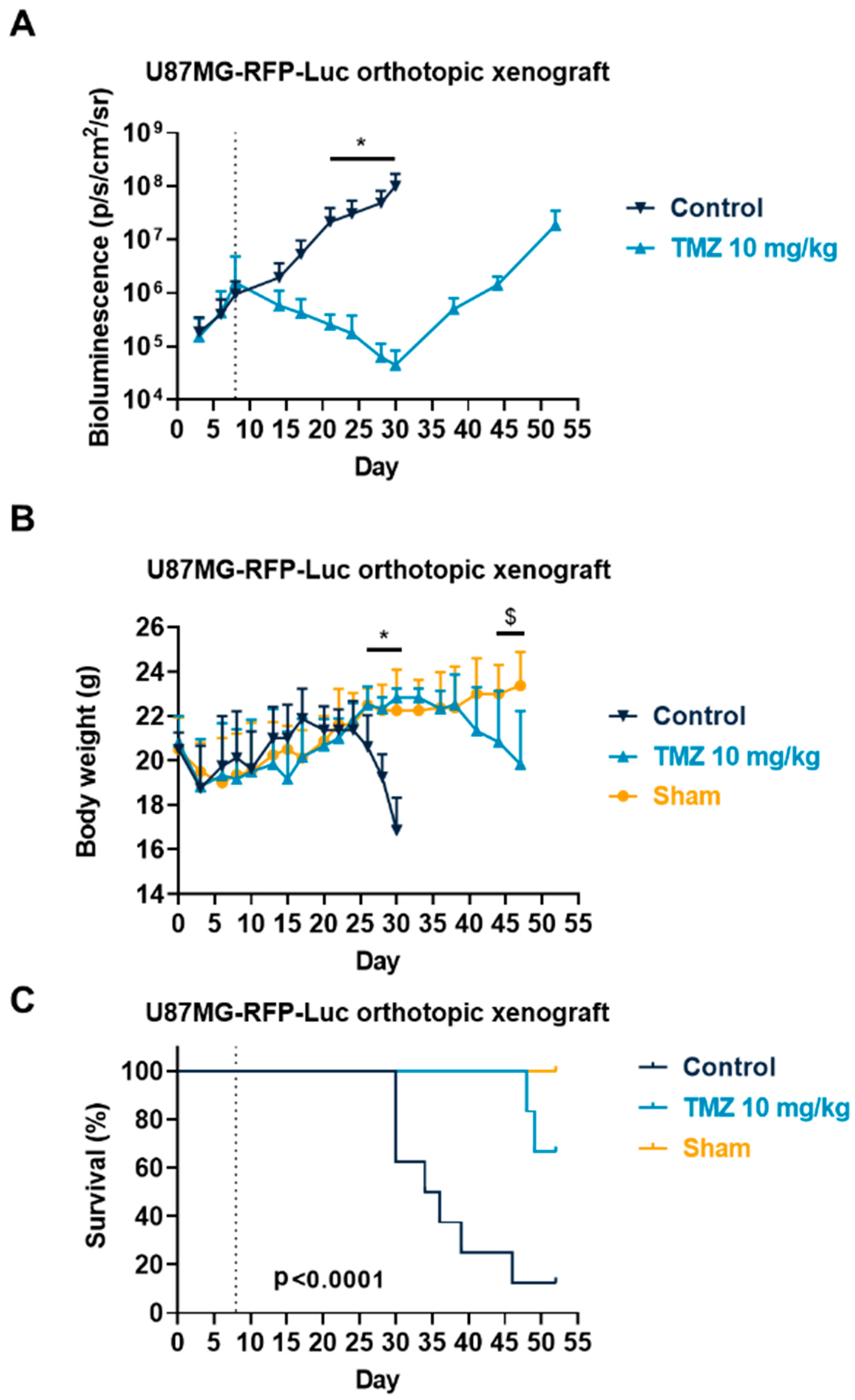
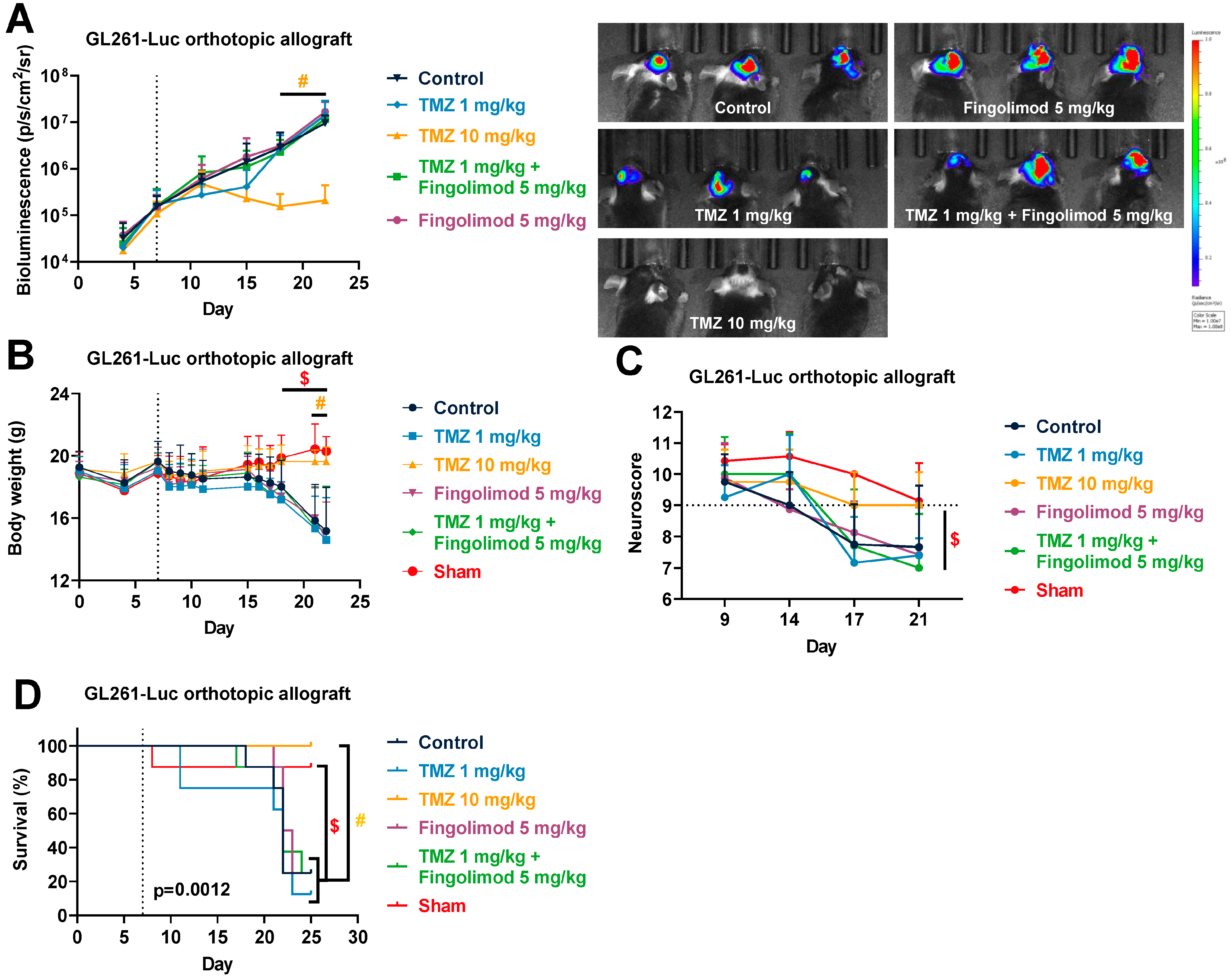
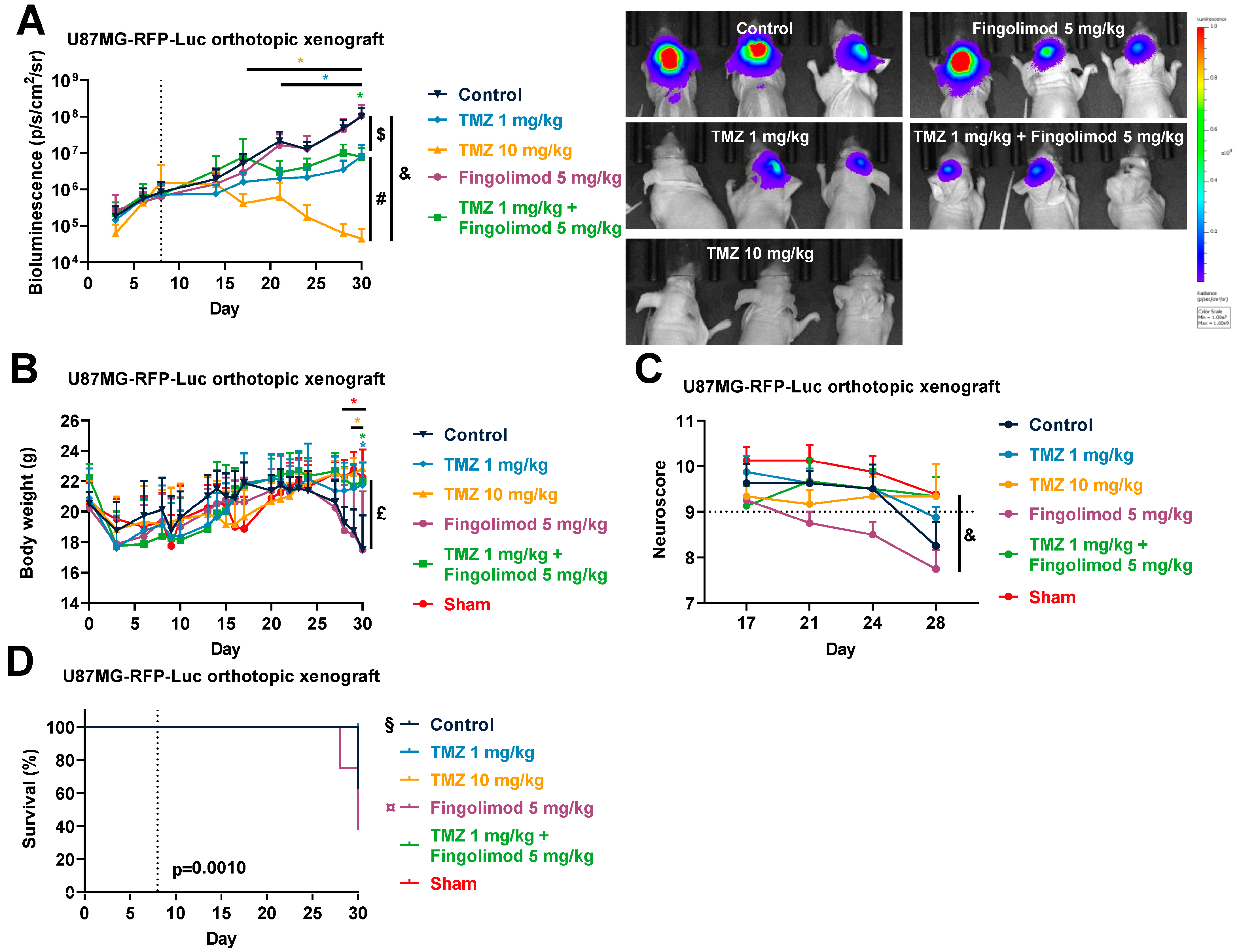
3.3. Temozolomide Reduced GBM Tumor Growth in Syngenic and Xenograft Orthopic Models Associated with Acquired Resistance
3.4. Analysis of Public Clinical Data: Expression of Downstream Target of Fingolimod Demonstrating That S1PR4 Is Correlated with Worse Prognostic Value in TCGA GBM Patient Cohort
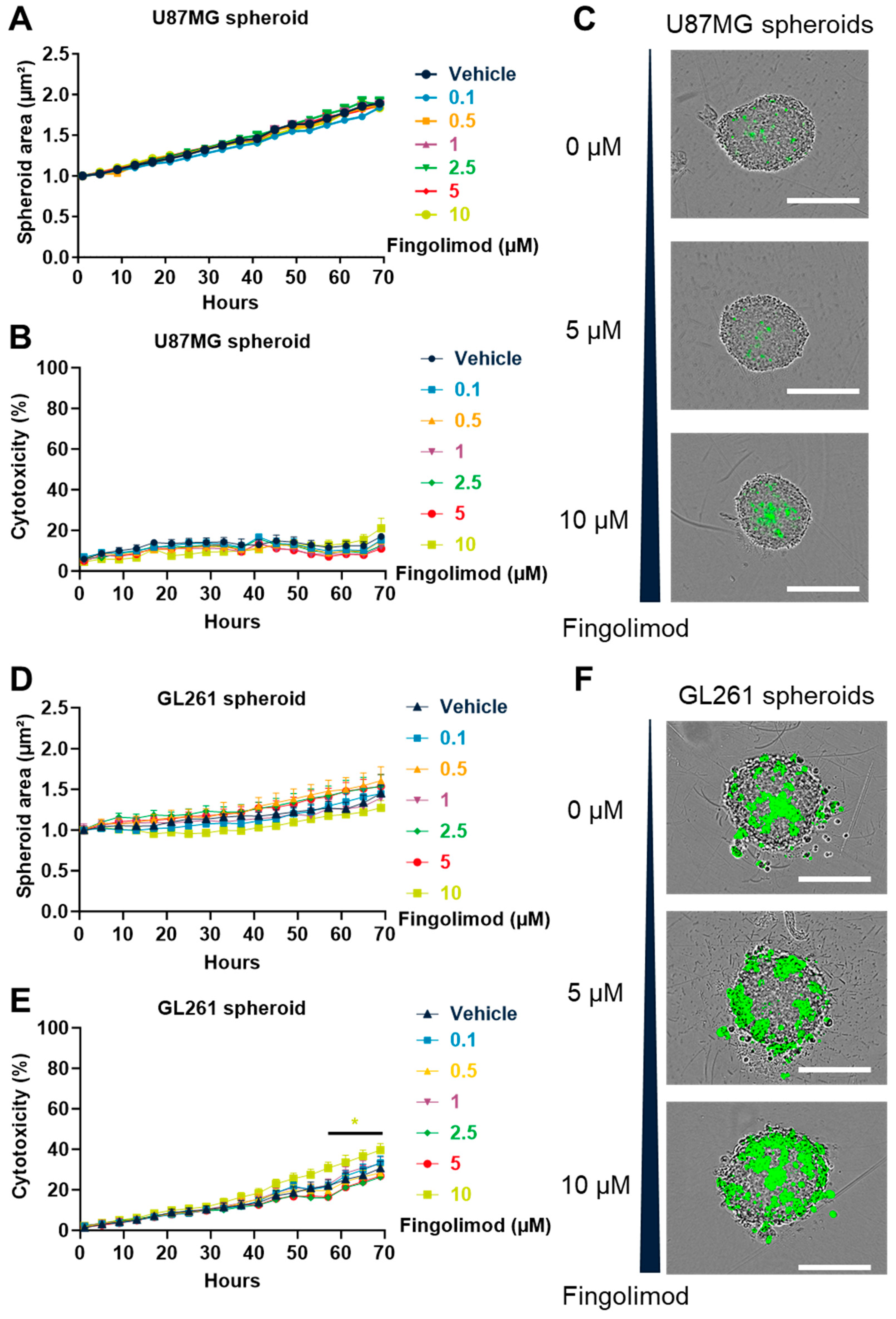
3.5. Fingolimod Induced Cytotoxicty in 2D Culture but Weakly Affected 3D Spheroid Culture
3.6. Fingolimod Did Not Affect GBM Progression In Vivo in Immunocompetent and Immunodeficient Orthotopic Mouse Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMSO | dimethyl sulfoxide |
| GBM | glioblastoma |
| i.p. | intraperitoneal |
| LGG | low-grade glioma |
| OS | overall survival |
| PBS | phosphate-buffered saline |
| PEG | polyethylene glycol |
| p.o. | per os |
| S1P | sphingosine-1-phosphate |
| S1PR | sphingosine-1-phosphate receptor |
| SD | standard deviation |
| SEM | standard error to the mean |
| TMZ | Temozolomide |
References
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2015, 17, iv1–iv62. [Google Scholar] [CrossRef]
- Fabbro-Peray, P.; Zouaoui, S.; Darlix, A.; Fabbro, M.; Pallud, J.; Rigau, V.; Mathieu-Daude, H.; Bessaoud, F.; Bauchet, F.; Riondel, A.; et al. Association of Patterns of Care, Prognostic Factors, and Use of Radiotherapy–Temozolomide Therapy with Survival in Patients with Newly Diagnosed Glioblastoma: A French National Population-Based Study. J. Neuro-Oncol. 2019, 142, 91–101. [Google Scholar] [CrossRef]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of Glioblastoma: A Systematic Review. ESMO Open 2017, 1, e000144. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Lemée, J.-M.; Clavreul, A.; Menei, P. Intratumoral Heterogeneity in Glioblastoma: Don’t Forget the Peritumoral Brain Zone. Neuro-Oncology 2015, 17, 1322–1332. [Google Scholar] [CrossRef]
- Audureau, E.; Chivet, A.; Ursu, R.; Corns, R.; Metellus, P.; Noel, G.; Zouaoui, S.; Guyotat, J.; Le Reste, P.-J.; Faillot, T.; et al. Prognostic Factors for Survival in Adult Patients with Recurrent Glioblastoma: A Decision-Tree-Based Model. J. Neuro-Oncol. 2018, 136, 565–576. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune Microenvironment of Gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Erlotinib (Tarceva) Tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef]
- Quillien, V.; Lavenu, A.; Ducray, F.; Joly, M.-O.; Chinot, O.; Fina, F.; Sanson, M.; Carpentier, C.; Karayan-Tapon, L.; Rivet, P.; et al. Validation of the High-Performance of Pyrosequencing for Clinical MGMT Testing on a Cohort of Glioblastoma Patients from a Prospective Dedicated Multicentric Trial. Oncotarget 2016, 7, 61916–61929. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning from 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Szatmári, T.; Lumniczky, K.; Désaknai, S.; Trajcevski, S.; Hídvégi, E.J.; Hamada, H.; Sáfrány, G. Detailed Characterization of the Mouse Glioma 261 Tumor Model for Experimental Glioblastoma Therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef]
- Bae, S.H.; Park, M.-J.; Lee, M.M.; Kim, T.M.; Lee, S.-H.; Cho, S.Y.; Kim, Y.-H.; Kim, Y.J.; Park, C.-K.; Kim, C.-Y. Toxicity Profile of Temozolomide in the Treatment of 300 Malignant Glioma Patients in Korea. J. Korean Med. Sci. 2014, 29, 980–984. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, S.; Yang, Z.; Li, Z.-A.; Hu, W.; Dai, L.; Liang, W.; Wang, X. Cholesterol Metabolism and Its Implication in Glioblastoma Therapy. J. Cancer 2022, 13, 1745–1757. [Google Scholar] [CrossRef]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The Role of Sphingosine-1-phosphate in Inflammation and Cancer Progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef]
- Mahajan-Thakur, S.; Bien-Möller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme—A Systematic Review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate and Cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Geffken, K.; Spiegel, S. Sphingosine kinase 1 in breast cancer. Adv. Biol. Regul. 2018, 67, 59–65. [Google Scholar] [CrossRef]
- Patmanathan, S.N.; Yap, L.F.; Murray, P.G.; Paterson, I.C. The Antineoplastic Properties of FTY720: Evidence for the Repurposing of Fingolimod. J. Cell. Mol. Med. 2015, 19, 2329–2340. [Google Scholar] [CrossRef]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of Multiple Sclerosis—Success from Bench to Bedside. Nat. Rev. Neurol. 2019, 15, 53–58. [Google Scholar] [CrossRef]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The Emerging Role of FTY720 (Fingolimod) in Cancer Treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef]
- Rupp, T.; Pelouin, O.; Genest, L.; Legrand, C.; Froget, G.; Castagné, V. Therapeutic Potential of Fingolimod in Triple Negative Breast Cancer Preclinical Models. Transl. Oncol. 2021, 14, 100926. [Google Scholar] [CrossRef]
- Rupp, T.; Debasly, S.; Genest, L.; Froget, G.; Castagné, V. Therapeutic Potential of Fingolimod and Dimethyl Fumarate in Non-Small Cell Lung Cancer Preclinical Models. Int. J. Mol. Sci. 2022, 23, 8192. [Google Scholar] [CrossRef]
- Loewe, R.; Valero, T.; Kremling, S.; Pratscher, B.; Kunstfeld, R.; Pehamberger, H.; Petzelbauer, P. Dimethylfumarate Impairs Melanoma Growth and Metastasis. Cancer Res. 2006, 66, 11888–11896. [Google Scholar] [CrossRef]
- Foster, C.A.; Howard, L.M.; Schweitzer, A.; Persohn, E.; Hiestand, P.C.; Balatoni, B.; Reuschel, R.; Beerli, C.; Schwartz, M.; Billich, A. Brain Penetration of the Oral Immunomodulatory Drug FTY720 and Its Phosphorylation in the Central Nervous System during Experimental Autoimmune Encephalomyelitis: Consequences for Mode of Action in Multiple Sclerosis. J. Pharmacol. Exp. Ther. 2007, 323, 469–475. [Google Scholar] [CrossRef]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2016, 30, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, M.A.; Al Barim, B.; Nagl, J.; Weigand, M.A.; Uhl, E.; Uhle, F.; Di Fazio, P.; Schwarm, F.P.; Stein, M. Sphingosine-1-phosphate Analogue FTY720 Exhibits a Potent Anti-Proliferative Effect on Glioblastoma Cells. Int. J. Oncol. 2020, 57, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H. FTY720 Inhibits the Nrf2/ARE Pathway in Human Glioblastoma Cell Lines and Sensitizes Glioblastoma Cells to Temozolomide. Pharmacol. Rep. 2017, 69, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Ding, K.; Xu, J. FTY720 Induces Autophagy-Related Apoptosis and Necroptosis in Human Glioblastoma Cells. Toxicol. Lett. 2015, 236, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-D.; Ji, J.; Xue, T.-F.; Sun, Y.-Q.; Guo, R.-B.; Cheng, H.; Sun, X.-L. FTY720 Exerts Anti-Glioma Effects by Regulating the Glioma Microenvironment Through Increased CXCR4 Internalization by Glioma-Associated Microglia. Front. Immunol. 2020, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; Zhu, J.; Ding, K.; Xu, J. FTY720 Reduces Migration and Invasion of Human Glioblastoma Cell Lines via Inhibiting the PI3K/AKT/MTOR/P70S6K Signaling Pathway. Tumor Biol. 2014, 35, 10707–10714. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Bernal, A.; Palanichamy, K.; Ray Chaudhury, A.; Van Brocklyn, J.R. Induction of Brain Tumor Stem Cell Apoptosis by FTY720: A Potential Therapeutic Agent for Glioblastoma. Neuro-Oncology 2012, 14, 405–415. [Google Scholar] [CrossRef]
- Dent, P.; Booth, L.; Roberts, J.L.; Poklepovic, A.; Hancock, J.F. Fingolimod Augments Monomethylfumarate Killing of GBM Cells. Front. Oncol. 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH Mutation and MGMT Promoter Methylation in Glioblastoma: Results of a Prospective Registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef]
- Oh, S.-J.; Yang, J.-I.; Kim, O.; Ahn, E.-J.; Kang, W.D.; Lee, J.-H.; Moon, K.-S.; Lee, K.-H.; Cho, D. Human U87 Glioblastoma Cells with Stemness Features Display Enhanced Sensitivity to Natural Killer Cell Cytotoxicity through Altered Expression of NKG2D Ligand. Cancer Cell Int. 2017, 17, 22. [Google Scholar] [CrossRef]
- Li, F.; Deng, Z.; Zhang, L.; Wu, C.; Jin, Y.; Hwang, I.; Vladimirova, O.; Xu, L.; Yang, L.; Lu, B.; et al. ATRX Loss Induces Telomere Dysfunction and Necessitates Induction of Alternative Lengthening of Telomeres during Human Cell Immortalization. EMBO J. 2019, 38, e96659. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Marshall, L.; Perryman, L.; Bax, D.A.; Little, S.E.; Viana-Pereira, M.; Sharp, S.Y.; Vassal, G.; Pearson, A.D.J.; Reis, R.M.; et al. MGMT-independent temozolomide resistance in paediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010, 70, 9243–9252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gilbert, M.R.; Kyprianou, N.; Rangnekar, V.M.; Horbinski, C. The Tumor Suppressor Prostate Apoptosis Response-4 (Par-4) is Regulated by Mutant IDH1 and Kills Glioma Stem Cells. Acta Neuropathol. 2014, 128, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Moen, E.L.; Stark, A.L.; Zhang, W.; Dolan, M.E.; Godley, L.A. The Role of Gene Body Cytosine Modifications in MGMT Expression and Sensitivity to Temozolomide. Mol. Cancer Ther. 2014, 13, 1334–1344. [Google Scholar] [CrossRef]
- Su, F.; Tang, X.; Li, G.; Koeberle, A.; Liu, B. SIRT7–SREBP1 Restrains Cancer Cell Metabolic Reprogramming by Upregulating IDH1. Genome Instab. Dis. 2021, 2, 126–137. [Google Scholar] [CrossRef]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patanè, M.; Zucca, I.; Riccardi Sirtori, F.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective Immuno-Targeting of the IDH1 Mutation R132H in a Murine Model of Intracranial Glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef]
- Fan, D.; Yue, Q.; Chen, J.; Wang, C.; Yu, R.; Jin, Z.; Yin, S.; Wang, Q.; Chen, L.; Liao, X.; et al. Reprogramming the Immunosuppressive Microenvironment of IDH1 Wild-Type Glioblastoma by Blocking Wnt Signaling between Microglia and Cancer Cells. OncoImmunology 2021, 10, 1932061. [Google Scholar] [CrossRef]
- Swenson, S.; Minea, R.O.; Tuan, C.D.; Thein, T.-Z.; Chen, T.C.; Markland, F.S. A Novel Venom-Derived Peptide for Brachytherapy of Glioblastoma: Preclinical Studies in Mice. Molecules 2018, 23, 2918. [Google Scholar] [CrossRef]
- Minea, R.O.; Duc, T.C.; Swenson, S.D.; Cho, H.-Y.; Huang, M.; Hartman, H.; Hofman, F.M.; Schönthal, A.H.; Chen, T.C. Developing a Clinically Relevant Radiosensitizer for Temozolomide-Resistant Gliomas. PLoS ONE 2020, 15, e0238238. [Google Scholar] [CrossRef]
- Rupp, T.; Legrand, C.; Hunault, M.; Genest, L.; Babin, D.; Froget, G.; Castagné, V. A Face-To-Face Comparison of Tumor Chicken Chorioallantoic Membrane (TCAM) In Ovo with Murine Models for Early Evaluation of Cancer Therapy and Early Drug Toxicity. Cancers 2022, 14, 3548. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; De Schlichting, E.; Dupic, G.; Pereira, B.; Fogli, A.; Müller-Barthélémy, M.; Dalloz, P.; Khalil, T.; Dillies, A.F.; et al. Radiotherapy plus Temozolomide in Elderly Patients with Glioblastoma: A “Real-Life” Report. Radiat. Oncol. 2017, 12, 197. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Takahara, S.; Ichimaru, N.; Wang, J.D.; Itoh, Y.; Otsuki, Y.; Morimoto, J.; Fukui, R.; Hoshiga, M.; Ishihara, T.; et al. Marked Prevention of Tumor Growth and Metastasis by a Novel Immunosuppressive Agent, FTY720, in Mouse Breast Cancer Models. Cancer Res. 2002, 62, 1410–1419. [Google Scholar] [PubMed]
- Woo, S.M.; Seo, B.R.; Min, K.; Kwon, T.K. FTY720 Enhances TRAIL-Mediated Apoptosis by up-Regulating DR5 and down-Regulating Mcl-1 in Cancer Cells. Oncotarget 2015, 6, 11614–11626. [Google Scholar] [CrossRef]
- Rupp, T.; Genest, L.; Babin, D.; Legrand, C.; Hunault, M.; Froget, G.; Castagné, V. Anti-CTLA-4 and Anti-PD-1 Immunotherapies Repress Tumor Progression in Preclinical Breast and Colon Model with Independent Regulatory T Cells Response. Transl. Oncol. 2022, 20, 101405. [Google Scholar] [CrossRef] [PubMed]
- Bederson, J.B.; Pitts, L.H.; Germano, S.M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H.M. Evaluation of 2, 3, 5-Triphenyltetrazolium Chloride as a Stain for Detection and Quantification of Experimental Cerebral Infarction in Rats. Stroke 1986, 17, 1304–1308. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D Tumor Spheroid Models for in Vitro Therapeutic Screening: A Systematic Approach to Enhance the Biological Relevance of Data Obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef] [PubMed]
- Phung, Y.T.; Barbone, D.; Broaddus, V.C.; Ho, M. Rapid Generation of In Vitro Multicellular Spheroids for the Study of Monoclonal Antibody Therapy. J. Cancer 2011, 2, 507–514. [Google Scholar] [CrossRef]
- Sant, S.; Johnston, P.A. The Production of 3D Tumor Spheroids for Cancer Drug Discovery. Drug Discov. Today Technol. 2017, 23, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bien-Möller, S.; Chen, F.; Xiao, Y.; Köppe, H.; Jedlitschky, G.; Meyer, U.; Tolksdorf, C.; Grube, M.; Marx, S.; Tzvetkov, M.V.; et al. The Putative S1PR1 Modulator ACT-209905 Impairs Growth and Migration of Glioblastoma Cells In Vitro. Cancers 2023, 15, 4273. [Google Scholar] [CrossRef]
- Bien-Möller, S.; Lange, S.; Holm, T.; Böhm, A.; Paland, H.; Küpper, J.; Herzog, S.; Weitmann, K.; Havemann, C.; Vogelgesang, S.; et al. Expression of S1P Metabolizing Enzymes and Receptors Correlate with Survival Time and Regulate Cell Migration in Glioblastoma Multiforme. Oncotarget 2016, 7, 13031–13046. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Lachambre, M.-P.; Plouffe, K.; Sartelet, H.; Béliveau, R. Modulation of Invasive Properties of CD133+ Glioblastoma Stem Cells: A role for MT1-MMP in Bioactive Lysophospholipid Signaling. Mol. Carcinog. 2009, 48, 910–919. [Google Scholar] [CrossRef]
- Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A.; et al. Interference with Distinct Steps of Sphingolipid Synthesis and Signaling Attenuates Proliferation of U87MG Glioma Cells. Biochem. Pharmacol. 2015, 96, 119–130. [Google Scholar] [CrossRef]
- Xiao, Y. Influence of ACT-209905 on Glioblastoma Growth In Vitro. Ph.D. Thesis, Universität Greifswald, Mecklenburg-Vorpommern, Germany, 2018. [Google Scholar]
- Lee, H.; Kim, D.; Youn, B. Targeting Oncogenic Rewiring of Lipid Metabolism for Glioblastoma Treatment. Int. J. Mol. Sci. 2022, 23, 13818. [Google Scholar] [CrossRef]
- Stepanovska, B.; Huwiler, A. Targeting the S1P Receptor Signaling Pathways as a Promising Approach for Treatment of Autoimmune and Inflammatory Diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A Metabolic Shift Favoring Sphingosine 1-Phosphate at the Expense of Ceramide Controls Glioblastoma Angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Min, K.; Kwon, T.K. Induction of Lysosomal Membrane Permeabilization Is a Major Event of FTY720-Mediated Non-Apoptotic Cell Death in Human Glioma Cells. Cancers 2020, 12, 3388. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, N.; Seeboeck, R.; Hofmann, E.; Schweiger, H.; Smolinska, V.; Mohr, T.; Boyer, A.; Sommergruber, W.; Lechner, P.; Pichler-Huebschmann, C.; et al. Organotypic Three-Dimensional Cancer Cell Cultures Mirror Drug Responses in Vivo: Lessons Learned from the Inhibition of EGFR Signaling. Oncotarget 2017, 8, 107423. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, V.; Yakisich, J.S.; Kulkarni, Y.; Azad, N.; Iyer, A.K.V. Chemoresistance of Lung Cancer Cells: 2D and 3D In Vitro Models for Anticancer Drug Screening; IntechOpen: London, UK, 2018; ISBN 978-1-78984-350-7. [Google Scholar]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The Effect of 2D and 3D Cell Cultures on Treatment Response, EMT Profile and Stem Cell Features in Head and Neck Cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, M.; Sterzynska, K.; Andrzejewska, M.; Nowicki, M.; Januchowski, R. Drug Resistance Evaluation in Novel 3D in Vitro Model. Biomed. Pharmacother. 2021, 138, 111536. [Google Scholar] [CrossRef] [PubMed]
- Sahu, U.; Barth, R.F.; Otani, Y.; McCormack, R.; Kaur, B. Rat and Mouse Brain Tumor Models for Experimental Neuro-Oncology Research. J. Neuropathol. Exp. Neurol. 2022, 81, 312–329. [Google Scholar] [CrossRef]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.-Y.; Okada, H.; Walker, P.R. Responsiveness to Anti-PD-1 and Anti-CTLA-4 Immune Checkpoint Blockade in SB28 and GL261 Mouse Glioma Models. Oncoimmunology 2018, 7, e1501137. [Google Scholar] [CrossRef]
- Yoshii, F.; Moriya, Y.; Ohnuki, T.; Ryo, M.; Takahashi, W. Neurological Safety of Fingolimod: An Updated Review. Clin. Exp. Neuroimmunol. 2017, 8, 233–243. [Google Scholar] [CrossRef]
- Snelder, N.; Ploeger, B.A.; Luttringer, O.; Stanski, D.R.; Danhof, M. Translational Pharmacokinetic Modeling of Fingolimod (FTY720) as a Paradigm Compound Subject to Sphingosine Kinase-Mediated Phosphorylation. Drug Metab. Dispos. 2014, 42, 1367–1378. [Google Scholar] [CrossRef]
- Nirogi, R.; Padala, N.P.; Ajjala, D.R.; Boggavarapu, R.K.; Kunduru, P. Incurred Sample Reanalysis of Fingolimod and Fingolimod Phosphate in Blood: Stability Evaluation and Application to a Rat Pharmacokinetic Study. Bioanalysis 2017, 9, 565–577. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research [CDER]. Application Number: 22-527. Clinical Pharmacology and Biopharmaceutics Review(s). Available online: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022527Orig1s000clinpharmr.pdf (accessed on 8 November 2020).
- Stuart, A. Grossman The Feasibility of Combining Brain Radiation and Fingolimod in Healthy Rodents and in Humans with Newly Diagnosed Glioblastoma. Available online: https://www.researchsquare.com (accessed on 18 January 2023).
- Pchejetski, D.; Bohler, T.; Brizuela, L.; Sauer, L.; Doumerc, N.; Golzio, M.; Salunkhe, V.; Teissié, J.; Malavaud, B.; Waxman, J.; et al. FTY720 (Fingolimod) Sensitizes Prostate Cancer Cells to Radiotherapy by Inhibition of Sphingosine Kinase-1. Cancer Res. 2010, 70, 8651–8661. [Google Scholar] [CrossRef]
- Ho, J.W.Y.; Man, K.; Sun, C.K.; Lee, T.K.; Poon, R.T.P.; Fan, S.T. Effects of a Novel Immunomodulating Agent, FTY720, on Tumor Growth and Angiogenesis in Hepatocellular Carcinoma. Mol. Cancer Ther. 2005, 4, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Huang, R.; Zheng, Y.; Zhang, Z.; Liang, A. Glioma-Derived Mutations in Isocitrate Dehydrogenase 2 Beneficial to Traditional Chemotherapy. Biochem. Biophys. Res. Commun. 2011, 410, 218–223. [Google Scholar] [CrossRef]
- Stessin, A.M.; Banu, M.A.; Clausi, M.G.; Berry, N.; Boockvar, J.A.; Ryu, S. FTY720/Fingolimod, an Oral S1PR Modulator, Mitigates Radiation Induced Cognitive Deficits. Neurosci. Lett. 2017, 658, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Halfon, M.J.; Jack, D.; Rubstein, A.; Villa, A. Acting Centrally or Peripherally: A Renewed Interest in the Central Nervous System Penetration of Disease-Modifying Drugs in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2021, 56, 103264. [Google Scholar] [CrossRef]
- Langlois, B.; Saupe, F.; Rupp, T.; Arnold, C.; van der Heyden, M.; Orend, G.; Hussenet, T. AngioMatrix, a Signature of the Tumor Angiogenic Switch-Specific Matrisome, Correlates with Poor Prognosis for Glioma and Colorectal Cancer Patients. Oncotarget 2014, 5, 10529–10545. [Google Scholar] [CrossRef] [PubMed]
- Riboni, L.; Abdel Hadi, L.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef]
- Mathivet, T.; Bouleti, C.; Van Woensel, M.; Stanchi, F.; Verschuere, T.; Phng, L.-K.; Dejaegher, J.; Balcer, M.; Matsumoto, K.; Georgieva, P.B.; et al. Dynamic Stroma Reorganization Drives Blood Vessel Dysmorphia during Glioma Growth. EMBO Mol. Med. 2017, 9, 1629–1645. [Google Scholar] [CrossRef]
- Mousseau, Y.; Mollard, S.; Faucher-Durand, K.; Richard, L.; Nizou, A.; Cook-Moreau, J.; Baaj, Y.; Qiu, H.; Plainard, X.; Fourcade, L.; et al. Fingolimod Potentiates the Effects of Sunitinib Malate in a Rat Breast Cancer Model. Breast Cancer Res. Treat. 2012, 134, 31–40. [Google Scholar] [CrossRef]
- Schmid, G.; Guba, M.; Ischenko, I.; Papyan, A.; Joka, M.; Schrepfer, S.; Bruns, C.J.; Jauch, K.-W.; Heeschen, C.; Graeb, C. The Immunosuppressant FTY720 Inhibits Tumor Angiogenesis via the Sphingosine 1-Phosphate Receptor 1. J. Cell. Biochem. 2007, 101, 259–270. [Google Scholar] [CrossRef]
- Shang, K.; He, J.; Zou, J.; Qin, C.; Lin, L.; Zhou, L.-Q.; Yang, L.-L.; Wu, L.-J.; Wang, W.; Zhan, K.-B.; et al. Fingolimod Promotes Angiogenesis and Attenuates Ischemic Brain Damage via Modulating Microglial Polarization. Brain Res. 2020, 1726, 146509. [Google Scholar] [CrossRef]
- Zou, Y.; Hu, J.; Huang, W.; Ye, S.; Han, F.; Du, J.; Shao, M.; Guo, R.; Lin, J.; Zhao, Y.; et al. Non-Mitogenic Fibroblast Growth Factor 1 Enhanced Angiogenesis Following Ischemic Stroke by Regulating the Sphingosine-1-Phosphate 1 Pathway. Front. Pharmacol. 2020, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Ng, K.T.; Man, K.; Ho, J.W.; Sun, C.K.; Lee, T.K.; Zhao, Y.; Lo, C.M.; Poon, R.T.; Fan, S.T. Marked Suppression of Tumor Growth by FTY720 in a Rat Liver Tumor Model: The Significance of Down-Regulation of Cell Survival Akt Pathway. Int. J. Oncol. 2007, 30, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Pineda, E.T.; Nerem, R.M.; Ahsan, T. Differentiation patterns of embryonic stem cells in two versus three dimensional culture. Cells Tissues Organs 2013, 197, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Hum, N.R.; Sebastian, A.; Gilmore, S.F.; He, W.; Martin, K.A.; Hinckley, A.; Dubbin, K.R.; Moya, M.L.; Wheeler, E.K.; Coleman, M.A.; et al. Comparative Molecular Analysis of Cancer Behavior Cultured In Vitro, In Vivo, and Ex Vivo. Cancers 2020, 12, 690. [Google Scholar] [CrossRef]
- Gstalder, C.; Ader, I.; Cuvillier, O. FTY720 (Fingolimod) Inhibits HIF1 and HIF2 Signaling, Promotes Vascular Remodeling, and Chemosensitizes in Renal Cell Carcinoma Animal Model. Mol. Cancer Ther. 2016, 15, 2465–2474. [Google Scholar] [CrossRef]
- Ota, K.; Okuma, T.; Lorenzo, A.D.; Yokota, A.; Hino, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Miyazawa, K. Fingolimod Sensitizes EGFR Wild-type Non-small Cell Lung Cancer Cells to Lapatinib or Sorafenib and Induces Cell Cycle Arrest. Oncol. Rep. 2019, 42, 231–242. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Luo, D.-D.; Wan, S.-B.; Qu, X.-J. S1PR2 Inhibitors Potently Reverse 5-FU Resistance by Downregulating DPD Expression in Colorectal Cancer. Pharmacol. Res. 2020, 155, 104717. [Google Scholar] [CrossRef]
- Li, M.-H.; Swenson, R.; Harel, M.; Jana, S.; Stolarzewicz, E.; Hla, T.; Shapiro, L.H.; Ferrer, F. Antitumor Activity of a Novel Sphingosine-1-Phosphate 2 Antagonist, AB1, in Neuroblastoma. J. Pharmacol. Exp. Ther. 2015, 354, 261–268. [Google Scholar] [CrossRef]
- Olesch, C.; Sirait-Fischer, E.; Berkefeld, M.; Fink, A.F.; Susen, R.M.; Ritter, B.; Michels, B.E.; Steinhilber, D.; Greten, F.R.; Savai, R.; et al. S1PR4 Ablation Reduces Tumor Growth and Improves Chemotherapy via CD8+ T Cell Expansion. J. Clin. Investig. 2020, 130, 5461. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davy, M.; Genest, L.; Legrand, C.; Pelouin, O.; Froget, G.; Castagné, V.; Rupp, T. Evaluation of Temozolomide and Fingolimod Treatments in Glioblastoma Preclinical Models. Cancers 2023, 15, 4478. https://doi.org/10.3390/cancers15184478
Davy M, Genest L, Legrand C, Pelouin O, Froget G, Castagné V, Rupp T. Evaluation of Temozolomide and Fingolimod Treatments in Glioblastoma Preclinical Models. Cancers. 2023; 15(18):4478. https://doi.org/10.3390/cancers15184478
Chicago/Turabian StyleDavy, Mélodie, Laurie Genest, Christophe Legrand, Océane Pelouin, Guillaume Froget, Vincent Castagné, and Tristan Rupp. 2023. "Evaluation of Temozolomide and Fingolimod Treatments in Glioblastoma Preclinical Models" Cancers 15, no. 18: 4478. https://doi.org/10.3390/cancers15184478
APA StyleDavy, M., Genest, L., Legrand, C., Pelouin, O., Froget, G., Castagné, V., & Rupp, T. (2023). Evaluation of Temozolomide and Fingolimod Treatments in Glioblastoma Preclinical Models. Cancers, 15(18), 4478. https://doi.org/10.3390/cancers15184478