
BRAF Non-V600 Mutations in Metastatic Colorectal Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
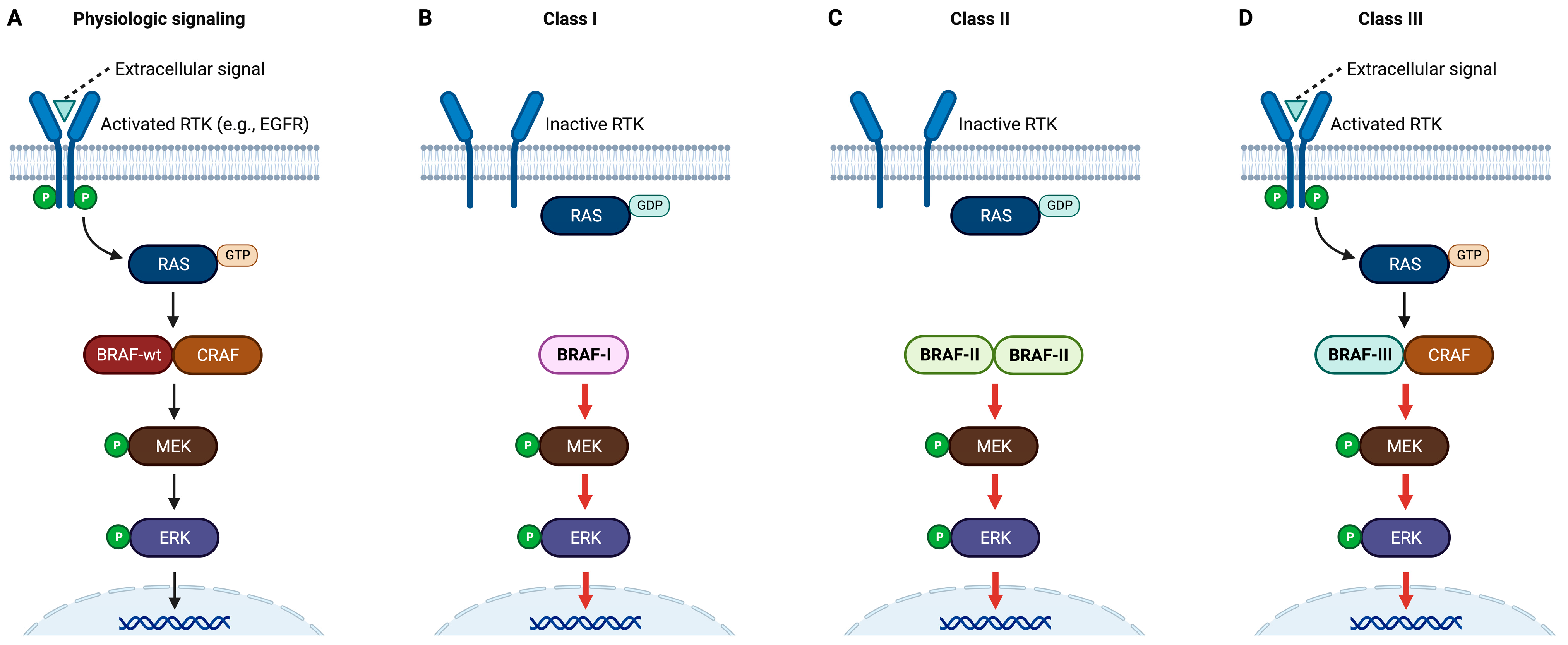
2. Classification of BRAF Mutations
3. BRAF Non-V600 Mutations in CRC
4. Ongoing Clinical Trials in BRAF Non-V600-Mutant CRC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Cancer Stat Facts: Colorectal Cancer. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 1 June 2023).
- Tournigand, C.; André, T.; Achille, E.; Lledo, G.; Flesh, M.; Mery-Mignard, D.; Quinaux, E.; Couteau, C.; Buyse, M.; Ganem, G.; et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J. Clin. Oncol. 2004, 22, 229–237. [Google Scholar] [CrossRef]
- Falcone, A.; Ricci, S.; Brunetti, I.; Pfanner, E.; Allegrini, G.; Barbara, C.; Crinò, L.; Benedetti, G.; Evangelista, W.; Fanchini, L.; et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: The Gruppo Oncologico Nord Ovest. J. Clin. Oncol. 2007, 25, 1670–1676. [Google Scholar] [PubMed]
- Fuchs, C.S.; Marshall, J.; Mitchell, E.; Wierzbicki, R.; Ganju, V.; Jeffery, M.; Schulz, J.; Richards, D.; Soufi-Mahjoubi, R.; Wang, B.; et al. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: Results from the BICC-C Study. J. Clin. Oncol. 2007, 25, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Muro, K.; Shitara, K.; Yamazaki, K.; Shiozawa, M.; Ohori, H.; Takashima, A.; Yokota, M.; Makiyama, A.; Akazawa, N.; et al. Panitumumab vs Bevacizumab Added to Standard First-line Chemotherapy and Overall Survival Among Patients with RAS Wild-type, Left-Sided Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2023, 329, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hazar-Rethinam, M.; Kleyman, M.; Han, G.C.; Liu, D.; Ahronian, L.G.; Shahzade, H.A.; Chen, L.; Parikh, A.R.; Allen, J.N.; Clark, J.W.; et al. Convergent Therapeutic Strategies to Overcome the Heterogeneity of Acquired Resistance in BRAFV600E Colorectal Cancer. Cancer Discov. 2018, 8, 417–427. [Google Scholar] [CrossRef]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef]
- Tol, J.; Nagtegaal, I.D.; Punt, C.J. BRAF mutation in metastatic colorectal cancer. N. Engl. J. Med. 2009, 361, 98–99, Erratum in N. Engl. J. Med. 2011, 365, 869. [Google Scholar] [CrossRef]
- Bando, H.; Ohtsu, A.; Yoshino, T. Therapeutic landscape and future direction of metastatic colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-mutant metastatic colorectal cancer: A review of treatment options and evidence-based guidelines. Ann. Oncol. 2021, 32, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Strickler, J.H.; Bekaii-Saab, T.S.; Yaeger, R. BRAF-Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022, 40, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Bardelli, A.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418, 934. [Google Scholar] [CrossRef]
- Yuen, S.T.; Davies, H.; Chan, T.L.; Ho, J.W.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Tsui, W.W.; Chan, A.S.; et al. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002, 62, 6451–6455. [Google Scholar]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar]
- Chan, T.L.; Zhao, W.; Leung, S.Y.; Yuen, S.T. Cancer Genome Project. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003, 63, 4878–4881. [Google Scholar]
- Wang, L.; Cunningham, J.M.; Winters, J.L.; Guenther, J.C.; French, A.J.; Boardman, L.A.; Burgart, L.J.; McDonnell, S.K.; Schaid, D.J.; Thibodeau, S.N. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003, 63, 5209–5212. [Google Scholar] [PubMed]
- Dankner, M. Targeted Therapy for Colorectal Cancers with Non-V600 BRAF Mutations: Perspectives for Precision Oncology. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Owsley, J.; Stein, M.K.; Porter, J.; In, G.K.; Salem, M.; O’Day, S.; Elliott, A.; Poorman, K.; Gibney, G.; VanderWalde, A. Prevalence of class I-III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp. Biol. Med. 2021, 246, 31–39. [Google Scholar] [CrossRef] [PubMed]
- The cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/ (accessed on 23 June 2023).
- Molina-Cerrillo, J.; San Román, M.; Pozas, J.; Alonso-Gordoa, T.; Pozas, M.; Conde, E.; Rosas, M.; Grande, E.; García-Bermejo, M.L.; Carrato, A. BRAF Mutated Colorectal Cancer: New Treatment Approaches. Cancers 2020, 12, 1571. [Google Scholar] [CrossRef] [PubMed]
- Tejpar, S.; Bertagnolli, M.; Bosman, F.; Lenz, H.J.; Garraway, L.; Waldman, F.; Warren, R.; Bild, A.; Collins-Brennan, D.; Hahn, H.; et al. Prognostic and predictive biomarkers in resected colon cancer: Current status and future perspectives for integrating genomics into biomarker discovery. Oncologist 2010, 15, 390–404. [Google Scholar] [CrossRef]
- Shitara, K.; Yonesaka, K.; Denda, T.; Yamazaki, K.; Moriwaki, T.; Tsuda, M.; Takano, T.; Okuda, H.; Nishina, T.; Sakai, K.; et al. Randomized study of FOLFIRI plus either panitumumab or bevacizumab for wild-type KRAS colorectal cancer-WJOG 6210G. Cancer Sci. 2016, 107, 1843–1850. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Schirripa, M.; Biason, P.; Lonardi, S.; Pella, N.; Pino, M.S.; Urbano, F.; Antoniotti, C.; Cremolini, C.; Corallo, S.; Pietrantonio, F.; et al. Class 1, 2, and 3 BRAF-Mutated Metastatic Colorectal Cancer: A Detailed Clinical, Pathologic, and Molecular Characterization. Clin. Cancer Res. 2019, 25, 3954–3961. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, J.; Han, X.; Yang, H.; Wang, S.; Lin, D.; Shi, Y. Effectors of epidermal growth factor receptor pathway: The genetic profiling ofKRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS ONE 2013, 8, e81628. [Google Scholar] [CrossRef]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Durrant, D.E.; Morrison, D.K. Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br. J. Cancer 2018, 118, 3–8. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef]
- Shinozaki, E.; Yoshino, T.; Yamazaki, K.; Muro, K.; Yamaguchi, K.; Nishina, T.; Yuki, S.; Shitara, K.; Bando, H.; Mimaki, S.; et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: The Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br. J. Cancer. 2017, 117, 1450–1458. [Google Scholar]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.; Kelly, R.J.; et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients with Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef]
- Herr, R.; Halbach, S.; Heizmann, M.; Busch, H.; Boerries, M.; Brummer, T. BRAF inhibition upregulates a variety of receptor tyrosine kinases and their downstream effector Gab2 in colorectal cancer cell lines. Oncogene 2018, 37, 1576–1593. [Google Scholar] [CrossRef]
- Binimetinib and Encorafenib for the Treatment of Advanced Solid Tumors with Non-V600E BRAF Mutations (BEAVER). Updated 26 September 2022. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03839342 (accessed on 9 June 2023).
- Rose, A.A.N.; Ayodele, O.; Genta, S.; Muniz, T.P.; Kelly, D.; Hodgson, K.; King, I.; Stockley, T.; Pugh, T.J.; Kamil, Z.S.; et al. Preliminary results of BEAVER: An investigator-initiated phase II study of binimetinib and encorafenib for the treatment of advanced solid tumors with non-V600E BRAF mutations (mts). J. Clin. Oncol. 2021, 39 (Suppl. S15), e15038. [Google Scholar] [CrossRef]
- A Study of Binimetinib and Encorafenib in Advanced BRAF Mutant Cancers. Updated 8 February 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT03843775 (accessed on 9 June 2023).
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer. 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Testing Trametinib as a Potential Targeted Treatment in Cancers with BRAF Genetic Changes (MATCH-Subprotocol R). Updated 20 March 2023. 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04439279 (accessed on 9 June 2023).
- A Study of ASN007 in Patients with Advanced Solid Tumors. Updated 9 July 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03415126 (accessed on 18 June 2023).
- Tolcher, A.W.; Sullivan, R.J.; Rasco, D.W.; Eroglu, Z.; Lakhani, N.; Kessler, D.; Usansky, H.; Reddy, S.; Denis, L.J.; Janku, F. Phase 1 clinical safety and efficacy of ASN007, a novel oral ERK1/2 inhibitor, in patients with RAS, RAF or MEK mutant advanced solid tumors. Mol. Cancer Ther. 2019, 18 (Suppl. S12), PR09. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Study of Ulixertinib for Patients with Advanced Malignancies Harboring MEK or Atypical BRAF Alterations. Updated 21 December 2022. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04488003 (accessed on 9 June 2023).
- Burkard, M.E.; McKean, M.; Ahnert, J.R.; Mettu, N.B.; Jones, J.C.; Misleh, J.G.; Ma, W.W.; Lim, K.H.; Chiorean, E.G.; Pishvaian, M.J.; et al. A two-part, phase II, multi-center study of the ERK inhibitor ulixertinib (BVD-523) for patients with advanced malignancies harboring MEK or atypical BRAF alterations (BVD-523-ABC). J. Clin. Oncol. 2022, 40 (Suppl. S16), TPS3172. [Google Scholar] [CrossRef]
- Franovic, A.; Miller, N.; Severson, P.; Kanouni, T.; Timple, N.; Jiang, P.; Murphy, E.; Martin, E. The next-generation pan-RAF inhibitor, KIN-2787, is active in class II and class III BRAF mutant models. J. Clin. Oncol. 2021, 39 (Suppl. S15), 3116. [Google Scholar] [CrossRef]
- A Study to Evaluate KIN-2787 in Participants with BRAF and/or NRAS Mutation Positive Solid Tumors. Updated 9 June 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04913285 (accessed on 18 June 2023).
- A Study of BDTX-4933 in Patients with BRAF and Select RAS/MAPK Mutation-Positive Cancers. Updated 26 April 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05786924 (accessed on 18 June 2023).

{kind=link}
| Class | Site | Alterations | Occurrences in CRC Cohorts | |
|---|---|---|---|---|
| GENIE | MSK-IMPACT | |||
| Class I | Activation segment | V600D | ||
| V600E | 1228 | 75 | ||
| V600K | ||||
| V600L | 1 | |||
| V600M | ||||
| V600R | ||||
| Class II | Activation segment | L597Q | 1 | |
| L597R | 3 | |||
| L597S | ||||
| L597V | ||||
| T599R | ||||
| T599dup | 4 | 3 | ||
| K601E | 14 | 2 | ||
| K601N | 5 | |||
| K601Q | ||||
| K601T | ||||
| P-loop | G464A | |||
| G464E | ||||
| G464R | 2 | |||
| G464V | 2 | |||
| G469A | 16 | 3 | ||
| G469R | 12 | |||
| G469V | 10 | |||
| V471F | 2 | |||
| Miscellaneous | Q257R | |||
| I463S | ||||
| L485F | 2 | |||
| L485_P490delinsY | ||||
| N486_P490del | ||||
| V487_P492delinsA | ||||
| K499E | ||||
| L505F | ||||
| L505H | ||||
| E586K | ||||
| V600_K601delinsE | 1 | |||
| V600_K601delinsEN | ||||
| V600_S605delinsEISRWR | ||||
| Class III | Activation segment | T599A | ||
| P-loop | G466A | 1 | ||
| G466E | 6 | |||
| G466R | 4 | |||
| G466V | 8 | 2 | ||
| S467L | 1 | |||
| G469E | 6 | 2 | ||
| DFG motif | D594A | 3 | ||
| D594E | 2 | |||
| D594G | 92 | 6 | ||
| D594H | ||||
| D594N | 21 | 3 | ||
| D594V | 3 | 1 | ||
| F595L | 6 | |||
| G596C | 1 | |||
| G596D | ||||
| G596R | 2 | |||
| Catalytic loop | N581I | 5 | 1 | |
| N581K | ||||
| N581S | 10 | 2 | ||
| N581Y | ||||
| Miscellaneous | D287H | |||
| V459L | ||||
| K483E | 3 | |||
| Clinical Trial Number | Inhibitor(s) | Target(s) | Clinical Phase | Sponsor | |
|---|---|---|---|---|---|
| NCT03843775 | Combination of | Encorafenib | BRAF | Phase I/II | MSKCC (in collaboration with Array BioPharma) |
| Binimetinib | MEK | ||||
| NCT03839342 | Combination of | Encorafenib | BRAF | Phase II | University Health Network, Toronto |
| Binimetinib | MEK | ||||
| NCT04439279 | Trametinib | MEK | Phase II | NCI | |
| NCT04488003 | Ulixertinib | ERK | Phase II | BioMed Valley Discoveries, Inc. | |
| NCT03415126 | ASN007 | ERK | Phase I | Asana BioSciences | |
| NCT04913285 | Exarafenib | BRAF | Phase I | Kinnate Biopharma | |
| NCT05786924 | BDTX-4933 | BRAF | Phase I | Black Diamond Therapeutics, Inc. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Xie, H. BRAF Non-V600 Mutations in Metastatic Colorectal Cancer. Cancers 2023, 15, 4604. https://doi.org/10.3390/cancers15184604
Liu J, Xie H. BRAF Non-V600 Mutations in Metastatic Colorectal Cancer. Cancers. 2023; 15(18):4604. https://doi.org/10.3390/cancers15184604
Chicago/Turabian StyleLiu, Junjia, and Hao Xie. 2023. "BRAF Non-V600 Mutations in Metastatic Colorectal Cancer" Cancers 15, no. 18: 4604. https://doi.org/10.3390/cancers15184604
APA StyleLiu, J., & Xie, H. (2023). BRAF Non-V600 Mutations in Metastatic Colorectal Cancer. Cancers, 15(18), 4604. https://doi.org/10.3390/cancers15184604