
Exploring the Molecular Landscape of Myelofibrosis, with a Focus on Ras and Mitogen-Activated Protein (MAP) Kinase Signaling

Abstract
:Simple Summary
Abstract
1. Introduction
2. Driver Mutations in Myelofibrosis
2.1. JAK2
2.2. MPL
2.3. CALR
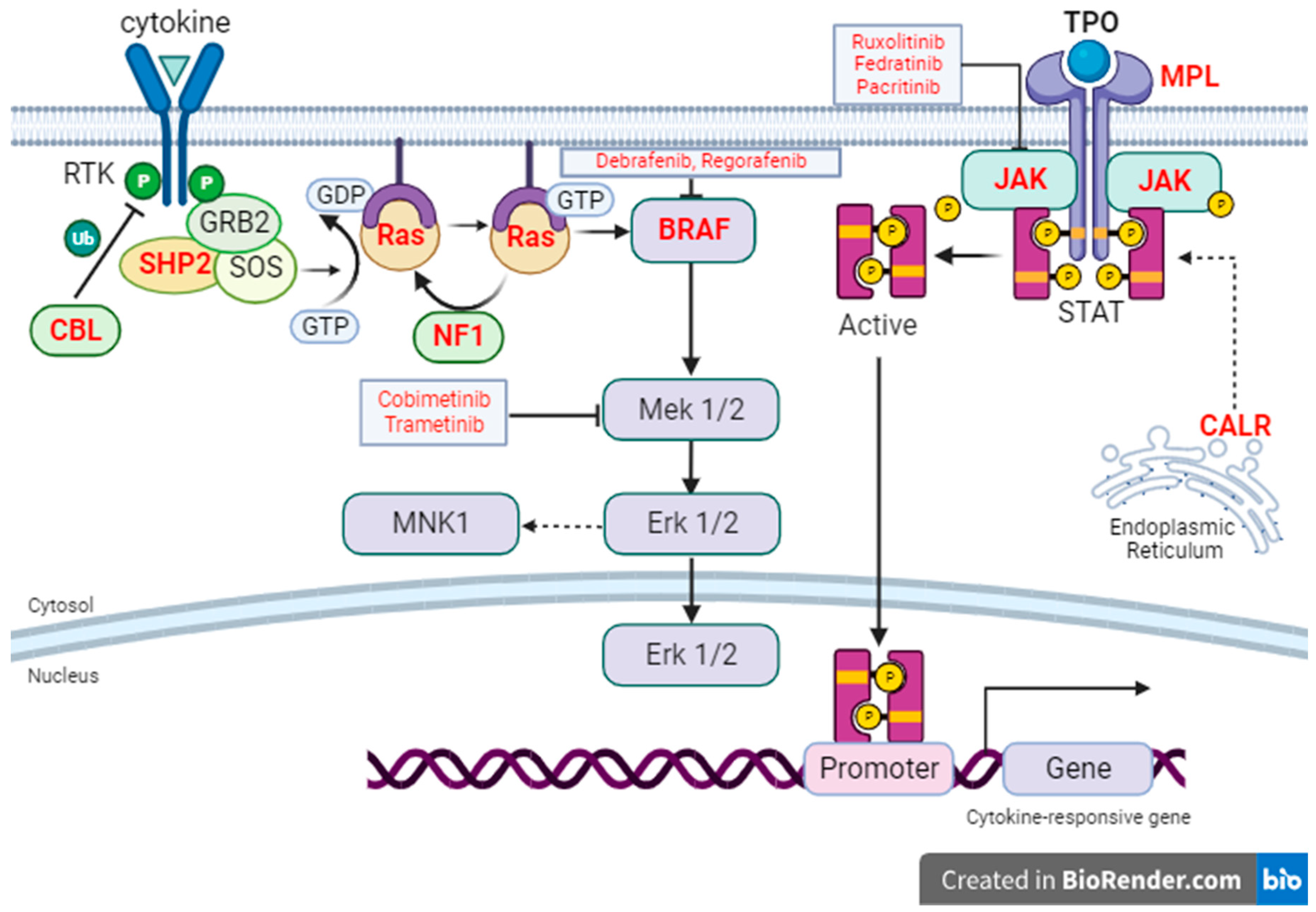
3. Triple-Negative Myelofibrosis
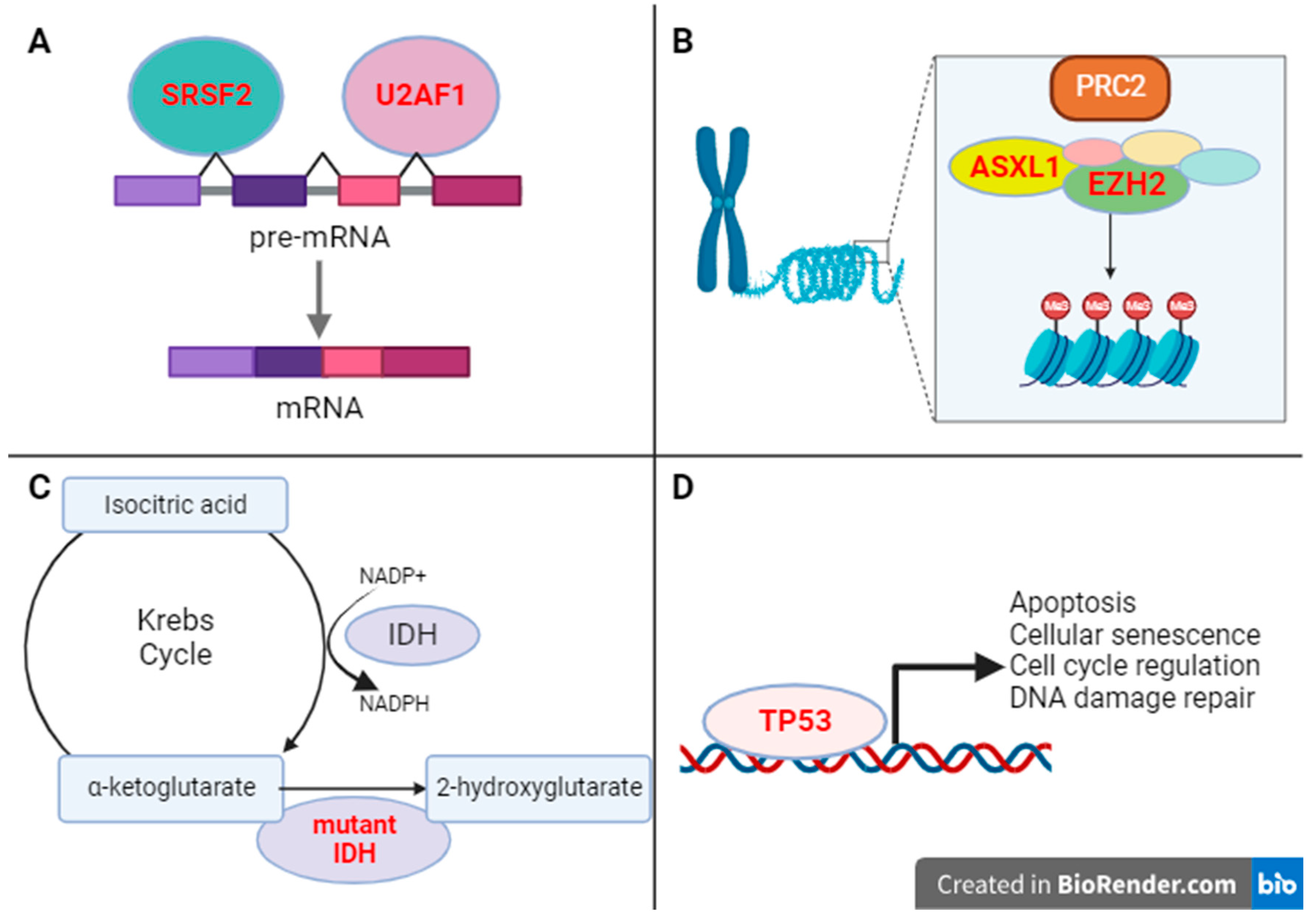
4. Mutations in Epigenetic Regulators
4.1. ASXL1
4.2. EZH2
5. Mutations in Splicing Factors
5.1. SRSF2
5.2. U2AF1
6. Mutations in IDH-Regulators of Cellular Metabolism
7. Mutations in TP53 Tumor Suppressor Gene
8. Expanding the Molecular Horizon in Myelofibrosis
9. Ras/MAP Kinase Signaling in Myelofibrosis
10. Targeting Ras Signaling in Myeloid Neoplasms
11. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Zhou, T.; Georgeon, S.; Moser, R.; Moore, D.J.; Caflisch, A.; Hantschel, O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014, 28, 404–407. [Google Scholar] [CrossRef]
- Singer, J.W.; Al-Fayoumi, S.; Ma, H.; Komrokji, R.S.; Mesa, R.; Verstovsek, S. Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor. J. Exp. Pharmacol. 2016, 8, 11–19. [Google Scholar] [CrossRef]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.-J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef]
- Verstovsek, S.; for the COMFORT-I Investigators; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 2017, 10, 55. [Google Scholar]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs. best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef]
- Tefferi, A.; Gangat, N.; Pardanani, A.; Crispino, J.D. Myelofibrosis: Genetic Characteristics and the Emerging Therapeutic Landscape. Cancer Res. 2022, 82, 749–763. [Google Scholar] [CrossRef]
- Santos, F.P.S.; Getta, B.; Masarova, L.; Famulare, C.; Schulman, J.; Datoguia, T.S.; Puga, R.D.; Paiva, R.d.M.A.; Arcila, M.E.; Hamerschlak, N.; et al. Prognostic impact of RAS-pathway mutations in patients with myelofibrosis. Leukemia 2020, 34, 799–810. [Google Scholar] [CrossRef]
- Gianelli, U.; Thiele, J.; Orazi, A.; Gangat, N.; Vannucchi, A.M.; Tefferi, A.; Kvasnicka, H.M. International Consensus Classification of myeloid and lymphoid neoplasms: Myeloproliferative neoplasms. Virchows Arch. Int. J. Pathol. 2023, 482, 53–68. [Google Scholar] [CrossRef]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E., 3rd; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef]
- Moliterno, A.R.; Kaizer, H.; Reeves, B.N. JAK2V617F allele burden in polycythemia vera: Burden of proof. Blood 2023, 141, 1934–1942. [Google Scholar] [CrossRef]
- Broséus, J.; Park, J.-H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Gangat, N.; Coltro, G.; Lasho, T.L.; Loscocco, G.G.; Finke, C.M.; Morsia, E.; Sordi, B.; Szuber, N.; Hanson, C.A.; et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, A.G.; Ranavaya, A.; Jamil, M.O. MPL Y252H anMd PL F126fs mutations in essential thrombocythemia: Case series and review of literature. Hematol. Rep. 2019, 11, 7868. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Elala, Y.; Barraco, D.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Targeted Next-Generation Sequencing in Polycythemia Vera and Essential Thrombocythemia. Blood 2015, 126, 354. [Google Scholar] [CrossRef]
- Shide, K.; Shimoda, H.K.; Kumano, T.; Karube, K.; Kameda, T.; Takenaka, K.; Oku, S.; Abe, H.; Katayose, K.S.; Kubuki, Y.; et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia 2008, 22, 87–95. [Google Scholar] [CrossRef]
- Rozovski, U.; Verstovsek, S.; Manshouri, T.; Dembitz, V.; Bozinovic, K.; Newberry, K.; Zhang, Y.; Bove, J.E.; Pierce, S.; Kantarjian, H.; et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and MPL mutation status for patients with primary myelofibrosis. Haematologica 2017, 102, 79–84. [Google Scholar] [CrossRef]
- Chen, E.; Mullally, A. How does JAK2V617F contribute to the pathogenesis of myeloproliferative neoplasms? Hematology 2014, 2014, 268–276. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Maddali, M.; Kulkarni, U.P.; Ravindra, N.; Jajodia, E.; Arunachalam, A.K.; Suresh, H.; Venkatraman, A.; George, B.; Mathews, V.; Balasubramanian, P. JAK2 exon 12 mutations in cases with JAK2V617F-negative polycythemia vera and primary myelofibrosis. Ann. Hematol. 2020, 99, 983–989. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Calabresi, L. The MPL mutation. Int. Rev. Cell Mol. Biol. 2021, 365, 163–178. [Google Scholar]
- Mejía-Ochoa, M.; Acevedo Toro, P.A.; Cardona-Arias, J.A. Systematization of analytical studies of polycythemia vera, essential thrombocythemia and primary myelofibrosis, and a meta-analysis of the frequency of JAK2, CALR and MPL mutations: 2000–2018. BMC Cancer 2019, 19, 590. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Tefferi, A. Infrequent occurrence of MPL exon 10 mutations in polycythemia vera and post-polycythemia vera myelofibrosis. Am. J. Hematol. 2011, 86, 701–702. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef]
- Li, N.; Yao, Q.M.; Gale, R.P.; Li, J.L.; Li, L.D.; Zhao, X.S.; Jiang, H.; Jiang, Q.; Jiang, B.; Shi, H.X.; et al. Frequency and allele burden of CALR mutations in Chinese with essential thrombocythemia and primary myelofibrosis without JAK2(V617F) or MPL mutations. Leuk. Res. 2015, 39, 510–514. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Luo, W.; Yu, Z. Calreticulin (CALR) mutation in myeloproliferative neoplasms (MPNs). Stem Cell Investig. 2015, 2, 16. [Google Scholar]
- How, J.; Hobbs, G.S.; Mullally, A. Mutant calreticulin in myeloproliferative neoplasms. Blood 2019, 134, 2242–2248. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Li, J.; Prins, D.; Park, H.J.; Grinfeld, J.; Gonzalez-Arias, C.; Loughran, S.; Dovey, O.M.; Klampfl, T.; Bennett, C.; Hamilton, T.L.; et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood 2018, 131, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Yzaguirre, A.D.; Padmanabhan, A.; de Groh, E.D.; Engleka, K.A.; Li, J.; Speck, N.A.; Epstein, J.A. Loss of neurofibromin Ras-GAP activity enhances the formation of cardiac blood islands in murine embryos. eLife 2015, 4, e07780. [Google Scholar] [CrossRef]
- Chou, Y.-T.; Bivona, T.G. Chapter Seven—Inhibition of SHP2 as an approach to block RAS-driven cancers. In Advances in Cancer Research; O’Bryan, J.P., Piazza, G.A., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 153, pp. 205–236. [Google Scholar]
- Merlinsky, T.R.; Levine, R.L.; Pronier, E. Unfolding the Role of Calreticulin in Myeloproliferative Neoplasm Pathogenesis. Clin. Cancer Res. 2019, 25, 2956–2962. [Google Scholar] [CrossRef]
- Reynolds, S.B.; Pettit, K. New approaches to tackle cytopenic myelofibrosis. Hematology 2022, 2022, 235–244. [Google Scholar] [CrossRef]
- Aguirre, L.E.; Jain, A.G.; Ball, S.; Al Ali, N.; Tinsley-Vance, S.M.; Sallman, D.A.; Sweet, K.; Lancet, J.E.; Padron, E.; Kuykendall, A.T.; et al. Triple-Negative Myelofibrosis: Disease Features, Response to Treatment and Outcomes. Blood 2021, 138 (Suppl. 1), 1494. [Google Scholar] [CrossRef]
- Antonioli, E.; Guglielmelli, P.; Poli, G.; Bogani, C.; Pancrazzi, A.; Longo, G.; Ponziani, V.; Tozzi, L.; Pieri, L.; Santini, V.; et al. Influence of JAK2V617F allele burden on phenotype in essential thrombocythemia. Haematologica 2008, 93, 41–48. [Google Scholar] [CrossRef]
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the Mutational Profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, W.; Wang, M.; Li, Y.; Wang, X.; Yang, E.; Ming, J.; Quan, R.; Hu, X. Prognostic value of ASXL1 mutations in patients with primary myelofibrosis and its relationship with clinical features: A meta-analysis. Ann. Hematol. 2021, 100, 465–479. [Google Scholar] [CrossRef]
- Andréasson, B.; Pettersson, H.; Wasslavik, C.; Johansson, P.; Palmqvist, L.; Asp, J. ASXL1 mutations, previous vascular complications and age at diagnosis predict survival in 85 WHO-defined polycythaemia vera patients. Br. J. Haematol. 2020, 189, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Akada, H.; Nath, D.; Hutchison, R.E.; Mohi, G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood 2016, 127, 3410–3423. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Mullally, A. The Molecular Genetics of Myeloproliferative Neoplasms. Cold Spring Harb. Perspect. Med. 2020, 10, a034876. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Screening for ASXL1 and SRSF2 mutations is imperative for treatment decision-making in otherwise low or intermediate-1 risk patients with myelofibrosis. Br. J. Haematol. 2018, 183, 678–681. [Google Scholar] [CrossRef]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. SRSF2 mutations in primary myelofibrosis: Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Li, B.; Gale, R.P.; Xu, Z.; Qin, T.; Song, Z.; Zhang, P.; Bai, J.; Zhang, L.; Zhang, Y.; Liu, J.; et al. Non-driver mutations in myeloproliferative neoplasm-associated myelofibrosis. J. Hematol. Oncol. 2017, 10, 99. [Google Scholar] [CrossRef]
- Tefferi, A.; Jimma, T.; Sulai, N.H.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; McClure, R.F.; Pardanani, A. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: Clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia 2012, 26, 475–480. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef]
- Gagelmann, N.; Badbaran, A.; Salit, R.B.; Schroeder, T.; Gurnari, C.; Pagliuca, S.; Panagiota, V.; Rautenberg, C.; Cassinat, B.; Thol, F.; et al. Impact of TP53 on outcome of patients with myelofibrosis undergoing hematopoietic stem cell transplantation. Blood 2023, 141, 2901. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, M.O.; Song, J.; Komrokji, R.S.; Sallman, D.A.; Kuykendall, A.; Padron, E.; Mirza, S.; Zhang, L. TP53 Mutations Are a Rare Event in Primary Myelofibrosis, Associated with TET2 Mutations, and Suggest Poor Clinical Outcome. Blood 2017, 130 (Suppl. 1), 5270. [Google Scholar]
- Ogawa, F.; Walters, M.S.; Shafquat, A.; O’Beirne, S.L.; Kaner, R.J.; Mezey, J.G.; Zhang, H.; Leopold, P.L.; Crystal, R.G. Role of KRAS in regulating normal human airway basal cell differentiation. Respir. Res. 2019, 20, 181. [Google Scholar] [CrossRef]
- Malathi Kandarpa, T.Q.; Robinson, D.; Wu, Y.-M.; Pettit, K.; Li, Q.; Sartor, M.; Chinnaiyan, A.; Talpaz, M. Broad next generation integrated sequencing of myelofibrosis identifies disease-specific and age-related genomic alterations. Clin. Cancer Res. 2023. accepted for publication. [Google Scholar]
- Liyasova, M.S.; Ma, K.; Lipkowitz, S. Molecular Pathways: Cbl Proteins in Tumorigenesis and Antitumor Immunity—Opportunities for Cancer Treatment. Clin. Cancer Res. 2015, 21, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Bartalucci, N.; Ravenda, E.; Gelli, E.; Sant’Antonio, E.; et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020, 4, 3677–3687. [Google Scholar] [CrossRef]
- Singh, N.R. Genomic diversity in myeloproliferative neoplasms: Focus on myelofibrosis. Transl. Pediatr. 2015, 4, 107–115. [Google Scholar] [PubMed]
- Molosh, A.I.; Shekhar, A. Chapter 2—Neurofibromatosis type 1 as a model system to study molecular mechanisms of autism spectrum disorder symptoms. In Progress in Brain Research; Shekhar, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 241, pp. 37–62. [Google Scholar]
- Hussain, M.R.M.; Baig, M.; Mohamoud, H.S.A.; Ulhaq, Z.; Hoessli, D.C.; Khogeer, G.S.; Al-Sayed, R.R.; Al-Aama, J.Y. BRAF gene: From human cancers to developmental syndromes. Saudi J. Biol. Sci. 2015, 22, 359–373. [Google Scholar] [CrossRef]
- Wan, Z.; Han, B. Comparison and Implications of Mutational Profiles of Myelodysplastic Syndromes, Myeloproliferative Neoplasms, and Myelodysplastic/Myeloproliferative Neoplasms: A Meta-Analysis. Front. Oncol. 2020, 10, 579221. [Google Scholar] [CrossRef]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. CMLS 2019, 76, 2511–2523. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Coltro, G.; Mannelli, F.; Rotunno, G.; Loscocco, G.G.; Mannarelli, C.; Maccari, C.; Paoli, C.; Romagnoli, S.; Bartalucci, N.; et al. ASXL1 mutations are prognostically significant in PMF, but not MF following essential thrombocythemia or polycythemia vera. Blood Adv. 2022, 6, 2927–2931. [Google Scholar] [CrossRef]
- Tan, J.-Z.; Yan, Y.; Wang, X.-X.; Jiang, Y.; Xu, H.E. EZH2: Biology, disease, and structure-based drug discovery. Acta Pharmacol. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Kropp, E.M.; Li, Q. Mechanisms of resistance to targeted therapies for relapsed or refractory acute myeloid leukemia. Exp. Hematol. 2022, 111, 13–24. [Google Scholar] [CrossRef]
- Li, K.; Wang, Z. Splicing factor SRSF2-centric gene regulation. Int. J. Biol. Sci. 2021, 17, 1708–1715. [Google Scholar] [CrossRef]
- Dutta, A.; Yang, Y.; Le, B.T.; Zhang, Y.; Abdel-Wahab, O.; Zang, C.; Mohi, G. U2af1 is required for survival and function of hematopoietic stem/progenitor cells. Leukemia 2021, 35, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.X.; Zhao, W.C.; Huang, H.B.; Wang, J.Q.; Zhang, H.; Lu, J.Y.; Wang, R.N.; Li, W.; Cheng, Z.; et al. Identification and characterization of isocitrate dehydrogenase 1 (IDH1) as a functional target of marine natural product grincamycin B. Acta Pharmacol. Sin. 2021, 42, 801–813. [Google Scholar] [CrossRef]
- Salati, M.; Caputo, F.; Baldessari, C.; Galassi, B.; Grossi, F.; Dominici, M.; Ghidini, M. IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers 2020, 12, 3310. [Google Scholar] [CrossRef]
- Bar-Natan, M. Ruxolitinib and Enasidenib for the Treatment of Accelerated or Blast-Phase Myeloproliferative Neoplasm or Chronic-Phase Myelofibrosis with an IDH2 Mutation. ClinicalTrialsgov Identifier: NCT04281498. 2023. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2021-00636&r=1#:~:text=Ruxolitinib%20is%20a%20treatment%20that,grow%20normal%20mature%20blood%20cells (accessed on 18 September 2023).
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Kuykendall, A.T.; Reuther, G.W. JAK2 inhibitor persistence in MPN: Uncovering a central role of ERK activation. Blood Cancer J. 2022, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Melo-Cardenas, J.; Bezavada, L.; Crawford, J.C.; Gurbuxani, S.; Cotton, A.; Kang, G.; Gossett, J.; Marinaccio, C.; Weinberg, R.; Hoffman, R.; et al. IL-13/IL-4 signaling contributes to fibrotic progression of the myeloproliferative neoplasms. Blood 2022, 140, 2805–2817. [Google Scholar] [CrossRef]
- Thatcher, J.D. The Ras-MAPK signal transduction pathway. Sci. Signal 2010, 3, tr1. [Google Scholar] [CrossRef]
- Pudewell, S.; Wittich, C.; Kazemein Jasemi, N.S.; Bazgir, F.; Ahmadian, M.R. Accessory proteins of the RAS-MAPK pathway: Moving from the side line to the front line. Commun. Biol. 2021, 4, 696. [Google Scholar] [CrossRef]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 2013, 1833, 122–139. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes. Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Satou, R.; Gonzalez-Villalobos, R.A. JAK-STAT and the renin-angiotensin system: The role of the JAK-STAT pathway in blood pressure and intrarenal renin-angiotensin system regulation. Jakstat 2012, 1, 250–256. [Google Scholar] [CrossRef]
- England, J.T.; McNamara, C.J.; Kennedy, J.A.; Capo-Chichi, J.-M.; Huang, J.; Arruda, A.; Nye, T.; Cheung, V.; Claudio, J.O.; Maze, D.; et al. Clinical and molecular correlates of JAK-inhibitor therapy failure in myelofibrosis: Long-term data from a molecularly annotated cohort. Leukemia 2022, 36, 1689–1692. [Google Scholar] [CrossRef]
- Tridente, G. (Ed.) Chapter 17—Ruxolitinib. In Adverse Events and Oncotargeted Kinase Inhibitors; Academic Press: Cambridge, MA, USA, 2017; pp. 375–393. [Google Scholar]
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, S.; Lee, R.; Fusi, A.; Lorigan, P. Dabrafenib and its use in the treatment of metastatic melanoma. Melanoma Manag. 2015, 2, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Khunger, A.; Khunger, M.; Velcheti, V. Dabrafenib in combination with trametinib in the treatment of patients with BRAF V600-positive advanced or metastatic non-small cell lung cancer: Clinical evidence and experience. Ther. Adv. Respir. Dis. 2018, 12, 1753466618767611. [Google Scholar] [CrossRef]
- Gentry, L.; Samatar, A.A.; Der, C.J. Chapter Four—Inhibitors of the ERK Mitogen-Activated Protein Kinase Cascade for Targeting RAS Mutant Cancers. In The Enzymes; Tamanoi, F., Der, C.J., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 34, pp. 67–106. [Google Scholar]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K. Chapter 11—Molecular modeling studies of fused pyrimidine derivatives at various receptors. In Fused Pyrimidine-Based Drug Discovery; Kumar, R., Vardanyan, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 273–332. [Google Scholar]
- Janku, F.; Patel, H.; Raghavan, V.K.; Goodman, A.; Barnes, T.G.; Kurzrock, R. MEK Inhibition with Trametinib in Patients with Non-Langerhans Cell Histiocytosis. Blood 2019, 134 (Suppl. 1), 2319. [Google Scholar] [CrossRef]
- Maiti, A.; Naqvi, K.; Kadia, T.M.; Borthakur, G.; Takahashi, K.; Bose, P.; Daver, N.G.; Patel, A.; Alvarado, Y.; Ohanian, M.; et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 142–148.e1. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 Inhibitor Trametinib in Combination with AKT Inhibitor GSK2141795 in Patients with Acute Myeloid Leukemia with RAS Mutations: A Phase II Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e13. [Google Scholar] [CrossRef]
- Bianchi, E.; Rontauroli, S.; Tavernari, L.; Mirabile, M.; Pedrazzi, F.; Genovese, E.; Sartini, S.; Dall’Ora, M.; Grisendi, G.; Fabbiani, L.; et al. Inhibition of ERK1/2 signaling prevents bone marrow fibrosis by reducing osteopontin plasma levels in a myelofibrosis mouse model. Leukemia 2023, 37, 1068–1079. [Google Scholar] [CrossRef]
- Ross, B.D.; Jang, Y.; Welton, A.; Bonham, C.A.; Palagama, D.S.; Heist, K.; Boppisetti, J.; Imaduwage, K.P.; Robison, T.; King, L.R.; et al. A lymphatic-absorbed multi-targeted kinase inhibitor for myelofibrosis therapy. Nat. Commun. 2022, 13, 4730. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Ito, Y. 4.19 Growth Factors and Protein-Modified Surfaces and Interfaces. In Comprehensive Biomaterials II; Ducheyne, P., Ed.; Elsevier: Oxford, UK, 2017; pp. 321–359. [Google Scholar]

{kind=link}
{kind=link}
| Mutation | Normal Function | Incidence in MF (%) | Incidence in PV (%) | Incidence in ET (%) |
|---|---|---|---|---|
| Driver mutations in myeloproliferative neoplasms | ||||
| JAK2 | Nonreceptor tyrosine kinase mediating normal hematopoiesis, cell growth [19,20,21] | 35–57 [19,20,21] | 96–99 [11,12] | 50–60 [13,14,40] |
| MPL | Protooncogene encoding for thrombopoietin receptor (TPOR) [23] | 0–7 [24,27] | 0–1 [24,25] | 5 [13,14] |
| CALR | Chaperone ER protein maintain calcium homeostasis and protein folding [31,32] | 25–53 [27,28] | 0 [12,24] | 25–27 [13,14] |
| “Triple negative” | - | 10–15 [41] | Rare | 35–52 [15] |
| Mutations in Regulators of Epigenetic Regulators | ||||
| ASXL1 | Epigenetic regulator of genes involved in chromatin remodeling [42] | 13–38 [42] | 8.2 [43] | 7–20 [13] |
| EZH2 | Epigenetic regulator of posttranslational histone modification [44] | 6–13 [44] | 0–3 [45] | 2–4 [13,15] |
| Mutations in Regulators of Splicing Factors | ||||
| SRSF2 | Splicing of pre-mRNA [46] | 3–17 [46,47] | <2 [15] | 2–3 [13] |
| U2AF1 | Splicing factor involved in pre-mRNA splicing, promotion of HSPC survival [48] | 16–22 [48,49] | <2 [15] | 1 [13] |
| Mutations in Regulators of Cellular Metabolism | ||||
| IDH | Catalyzation of isocitrate decarboxylation to yield 2-oxoglutarate (2-OG) [50] | 4 [50] | <2 [15] | 1 [13] |
| Mutations in Tumor Suppressor Genes | ||||
| TP53 | Tumor suppression, cell cycle regulation [51] | 1–13 [52,53] | <2 [15] | 1–4 [13,45] |
| Ras/MAP Kinase Pathway Mutations | ||||
| KRAS/NRAS | Proto-oncogene with GTPase activity; K/Nras are two different Ras isoforms [8,54] | 2–15/4.4–14 [8,55] | <2 (NRAS) [15] | <2 (NRAS) [15] |
| CBL | E3 ubiquitin ligase; negatively regulates various receptor tyrosine kinases [56] | 5–13 [55,57,58] | <2 [15] | 1–2 [13] |
| NF1 | Inactivation of Ras-GTP through stimulation of constitutional GTPase [59] | 0–6 [55,58] | Rare | Rare |
| BRAF | Cytosolic serine/threonine kinase regulating MEK, ERK [60] | 0–1 [61] | Rare | Rare |
| PTPN11 (SHP2) | PTPN11 encodes for SHP2, a tyrosine phosphatase activating Ras/MAP Kinase [36] | 1 [8] | <2 [15] | <2 [15] |
| Inhibitor | Mechanism/Targets | FDA-Approved Treatment Indications |
|---|---|---|
| Ruxolitinib | Jak1; Jak2 [84] | Proliferative myelofibrosis; Glucocorticoid-refractory GVHD [38,85] |
| Fedratinib | Jak2; FLT3; BRD4 [38] | Symptomatic, severe cytopenic MF [38] |
| Pacritinib | Jak2; IRAK1 [38] | Proliferative and moderately cytopenic MF [38] |
| Dabrafenib | BRAF, CRAF [86] | BRAF V600E-mutated NSCLC; melanoma [87] |
| Vemurafenib | BRAF [86] | BRAF V600E-mutated NSCLC; melanoma [87] |
| Cobimetinib | Mek 1/2 [88] | Melanoma; histiocytic disorders [87,89] |
| Trametinib | Mek 1/2 [90] | Melanoma; histiocytic disorders [87,91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reynolds, S.B.; Pettit, K.; Kandarpa, M.; Talpaz, M.; Li, Q. Exploring the Molecular Landscape of Myelofibrosis, with a Focus on Ras and Mitogen-Activated Protein (MAP) Kinase Signaling. Cancers 2023, 15, 4654. https://doi.org/10.3390/cancers15184654
Reynolds SB, Pettit K, Kandarpa M, Talpaz M, Li Q. Exploring the Molecular Landscape of Myelofibrosis, with a Focus on Ras and Mitogen-Activated Protein (MAP) Kinase Signaling. Cancers. 2023; 15(18):4654. https://doi.org/10.3390/cancers15184654
Chicago/Turabian StyleReynolds, Samuel B., Kristen Pettit, Malathi Kandarpa, Moshe Talpaz, and Qing Li. 2023. "Exploring the Molecular Landscape of Myelofibrosis, with a Focus on Ras and Mitogen-Activated Protein (MAP) Kinase Signaling" Cancers 15, no. 18: 4654. https://doi.org/10.3390/cancers15184654
APA StyleReynolds, S. B., Pettit, K., Kandarpa, M., Talpaz, M., & Li, Q. (2023). Exploring the Molecular Landscape of Myelofibrosis, with a Focus on Ras and Mitogen-Activated Protein (MAP) Kinase Signaling. Cancers, 15(18), 4654. https://doi.org/10.3390/cancers15184654