
Estrogen Receptor Signaling in Breast Cancer
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Estrogen Receptors
3. Estrogen Signaling
3.1. Genomic Action of ER
3.2. Non-Genomic Action of ER
4. Coactivators and Corepressors of ER in Breast Cancer
5. Post-Translational Modifications of Estrogen Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site of Modification | Type of Modification | Enzymes | Functions | Reference |
|---|---|---|---|---|
| Y52 | phosphorylation | c-Abl | transcription activation, stability maintenance | [181] |
| Y219 | phosphorylation | c-Abl | DNA binding and dimerization | [181] |
| S102 | phosphorylation | GSK3 | transcription activation | [182] |
| S104/106 | phosphorylation | cyclin A-Cdk2, MAPK | transcription activation, dimerization | [182] |
| S118 | phosphorylation | ND, Cdk7, IKKα | RNA splicing, dimerization, transcription activation | [182,183] |
| S167 | phosphorylation | Akt, p90 RSK, S6K1 | stability maintenance | [184] |
| S236 | phosphorylation | PKA | dimerization inhibition | [146] |
| R260 | methylation | PRMT1 | non-genomic signaling | [185] |
| K266 | acetylation | p300 | transcription activation, DNA binding | [146] |
| K266 K268 | sumoylation | Ubc9, PIAS1, PIAS3 | transcription activation, DNA binding | [179] |
| S282 S559 | phosphorylation | CK2 | transcription inhibition | [186] |
| K302 K303 | ubiquitylation | CHIP | proteasomal degradation | [171] |
| K302 | acetylation | p300 | transcription inhibition | [151] |
| K302 | methylation | SET7 | regulation of ER turnover | [164] |
| K303 | acetylation | p300 | transcription inhibition | [151] |
| K303 | sumoylation | Ubc9, PIAS1, PIAS3 | transcription activation, DNA binding | [179] |
| S305 | phosphorylation | PAK1 | resistance to aromatase inhibitor, transcription activation | [187,188] |
| T311 | phosphorylation | p38-MAPK | nuclear localization | [147] |
| C447 | palmitoylation | PAT | plasma membrane localization | [189,190] |
| Y537 | phosphorylation | calf uterine kinase, SRC, EGFR | DNA binding, dimerization, proliferation | [191,192] |
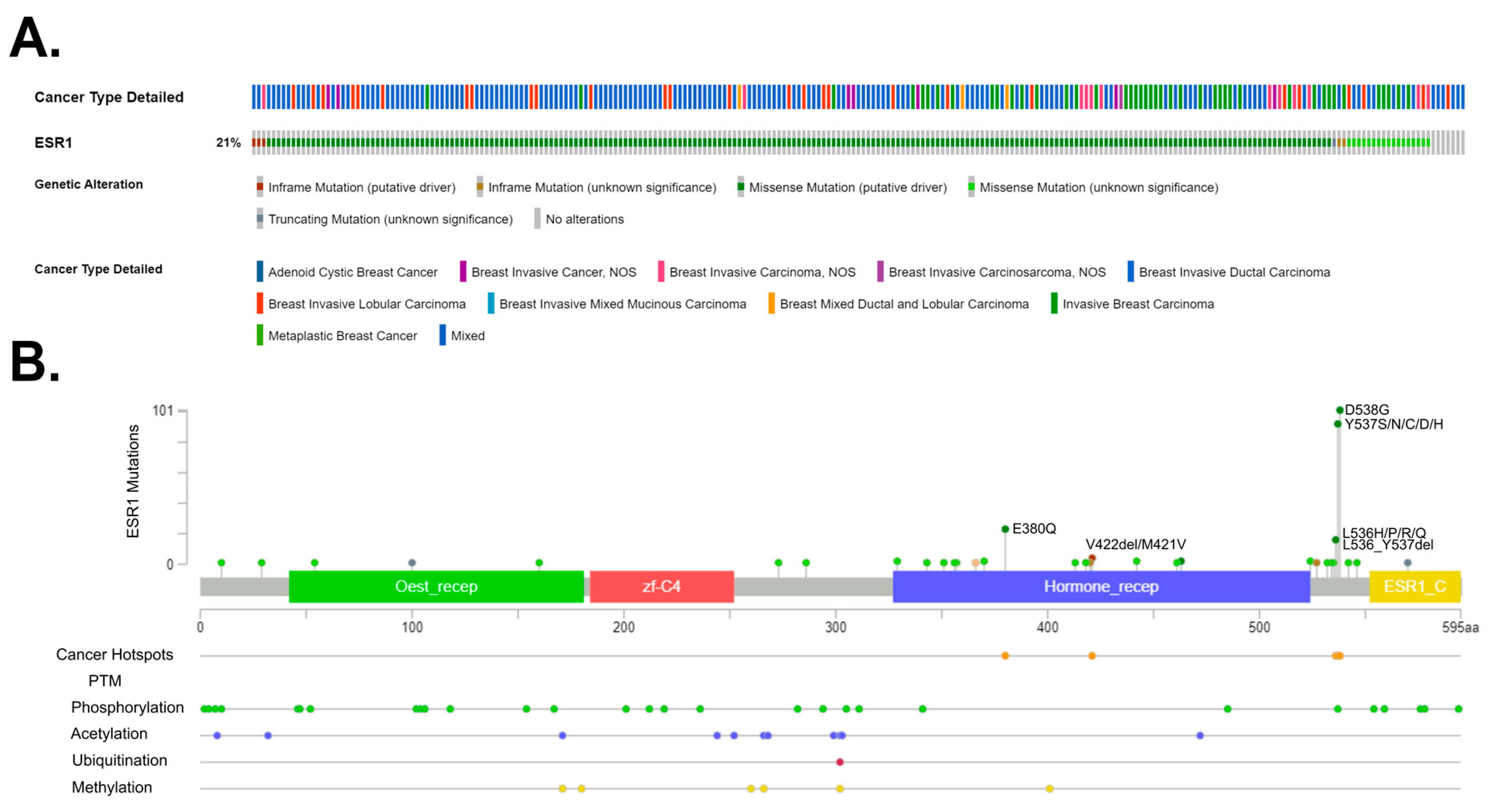
6. Estrogen Receptor Mutations
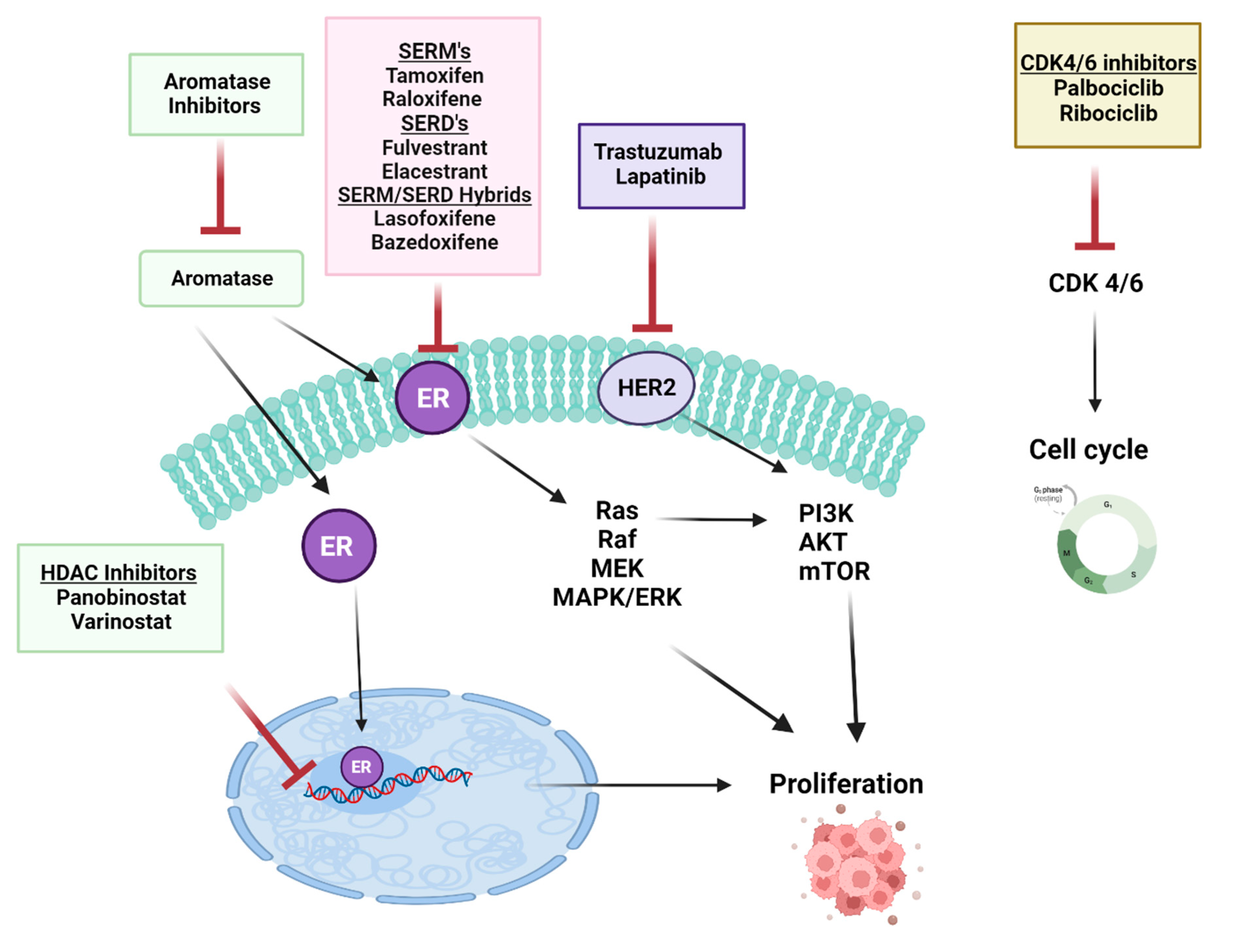
7. Therapeutic Targeting of ERs Pathways for Metastatic Control
8. The Summary of the Latest Developments
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Baehner, F.; Dabbs, D.J.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Breast Cancer Prognostic Classification in the Molecular Era: The Role of Histological Grade. Breast Cancer Res. 2010, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. ESMO Guidelines Committee Early Breast Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2019, 30, 1674. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; André, F.; Bergh, J.; et al. Personalizing the Treatment of Women with Early Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Cao, S.-S.; Lu, C.-T. Recent Perspectives of Breast Cancer Prognosis and Predictive Factors. Oncol. Lett. 2016, 12, 3674–3678. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Dias, K.; Dvorkin-Gheva, A.; Hallett, R.M.; Wu, Y.; Hassell, J.; Pond, G.R.; Levine, M.; Whelan, T.; Bane, A.L. Claudin-Low Breast Cancer; Clinical & Pathological Characteristics. PLoS ONE 2017, 12, e0168669. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nuñez, P.; Acuña-Aguilar, L.E.; Gómez-Valles, F.O.; Ramírez-Valdespino, C.A. Subtypes of Breast Cancer; Exon Publications: Brisbane, Australia, 2022. [Google Scholar]
- Nolan, E.; Lindeman, G.J.; Visvader, J.E. Deciphering Breast Cancer: From Biology to the Clinic. Cell 2023, 186, 1708–1728. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Wang, J.-P.; Li, Y.; Fan, P.; Liu, G.; Zhang, N.; Conaway, M.; Wang, H.; Korach, K.S.; Bocchinfuso, W.; et al. Effects of Estrogen on Breast Cancer Development: Role of Estrogen Receptor Independent Mechanisms. Int. J. Cancer 2010, 127, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef]
- Langdon, S.P.; Herrington, C.S.; Hollis, R.L.; Gourley, C. Estrogen Signaling and Its Potential as a Target for Therapy in Ovarian Cancer. Cancers 2020, 12, 1647. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The Necessary Evil for Human Health, and Ways to Tame It. Biomed. Pharmacother. 2018, 102, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Gallez, A.; Dias Da Silva, I.; Wuidar, V.; Foidart, J.-M.; Péqueux, C. Estetrol and Mammary Gland: Friends or Foes? J. Mammary Gland Biol. Neoplasia 2021, 26, 297–308. [Google Scholar] [CrossRef]
- Baker, M.E. What Are the Physiological Estrogens? Steroids 2013, 78, 337–340. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Miller, W.L. Steroidogenesis: Unanswered Questions. Trends Endocrinol. Metab. 2017, 28, 771–793. [Google Scholar] [CrossRef]
- Savolainen-Peltonen, H.; Vihma, V.; Leidenius, M.; Wang, F.; Turpeinen, U.; Hämäläinen, E.; Tikkanen, M.J.; Mikkola, T.S. Breast Adipose Tissue Estrogen Metabolism in Postmenopausal Women with or without Breast Cancer. J. Clin. Endocrinol. Metab. 2014, 99, E2661–E2667. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R.A. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.P.A.; Gustafsson, J.-Å. Estrogen Receptors and the Metabolic Network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Lee, A.R. Estrogen and the Cardiovascular System. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef]
- Cersosimo, M.G.; Benarroch, E.E. Estrogen Actions in the Nervous System: Complexity and Clinical Implications. Neurology 2015, 85, 263–273. [Google Scholar] [CrossRef]
- Bernelot Moens, S.J.; Schnitzler, G.R.; Nickerson, M.; Guo, H.; Ueda, K.; Lu, Q.; Aronovitz, M.J.; Nickerson, H.; Baur, W.E.; Hansen, U.; et al. Rapid Estrogen Receptor Signaling Is Essential for the Protective Effects of Estrogen against Vascular Injury. Circulation 2012, 126, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Schierbeck, L.L.; Rejnmark, L.; Tofteng, C.L.; Stilgren, L.; Eiken, P.; Mosekilde, L.; Køber, L.; Jensen, J.-E.B. Effect of Hormone Replacement Therapy on Cardiovascular Events in Recently Postmenopausal Women: Randomised Trial. BMJ 2012, 345, e6409. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; Henderson, V.W.; Shoupe, D.; Budoff, M.J.; Hwang-Levine, J.; Li, Y.; Feng, M.; Dustin, L.; Kono, N.; et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N. Engl. J. Med. 2016, 374, 1221–1231. [Google Scholar] [CrossRef]
- Manson, J.E.; Hsia, J.; Johnson, K.C.; Rossouw, J.E.; Assaf, A.R.; Lasser, N.L.; Trevisan, M.; Black, H.R.; Heckbert, S.R.; Detrano, R.; et al. Estrogen plus Progestin and the Risk of Coronary Heart Disease. N. Engl. J. Med. 2003, 349, 523–534. [Google Scholar] [CrossRef]
- Kurtanović, N.; Tomašević, N.; Matić, S.; Proia, E.; Sabatino, M.; Antonini, L.; Mladenović, M.; Ragno, R. Human Estrogen Receptor Alpha Antagonists, Part 3: 3-D Pharmacophore and 3-D QSAR Guided Brefeldin A Hit-to-Lead Optimization toward New Breast Cancer Suppressants. Molecules 2022, 27, 2823. [Google Scholar] [CrossRef]
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone Receptor Status, Tumor Characteristics, and Prognosis: A Prospective Cohort of Breast Cancer Patients. Breast Cancer Res. 2007, 9, R6. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Thomas, P. Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Korach, K.S. The Physiological Role of Estrogen Receptor Functional Domains. Essays Biochem. 2021, 65, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Bihani, T. Selective Estrogen Receptor Modulators (SERMs) and Selective Estrogen Receptor Degraders (SERDs) in Cancer Treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Schubert, M.; Brunet, F.; Paris, M.; Bertrand, S.; Benoit, G.; Laudet, V. Nuclear Hormone Receptor Signaling in Amphioxus. Dev. Genes Evol. 2008, 218, 651–665. [Google Scholar] [CrossRef]
- Holland, L.Z.; Albalat, R.; Azumi, K.; Benito-Gutiérrez, E.; Blow, M.J.; Bronner-Fraser, M.; Brunet, F.; Butts, T.; Candiani, S.; Dishaw, L.J.; et al. The Amphioxus Genome Illuminates Vertebrate Origins and Cephalochordate Biology. Genome Res. 2008, 18, 1100–1111. [Google Scholar] [CrossRef]
- Keay, J.; Bridgham, J.T.; Thornton, J.W. The Octopus Vulgaris Estrogen Receptor Is a Constitutive Transcriptional Activator: Evolutionary and Functional Implications. Endocrinology 2006, 147, 3861–3869. [Google Scholar] [CrossRef]
- Matsumoto, T.; Nakamura, A.M.; Mori, K.; Akiyama, I.; Hirose, H.; Takahashi, Y. Oyster Estrogen Receptor: cDNA Cloning and Immunolocalization. Gen. Comp. Endocrinol. 2007, 151, 195–201. [Google Scholar] [CrossRef]
- Keay, J.; Thornton, J.W. Hormone-Activated Estrogen Receptors in Annelid Invertebrates: Implications for Evolution and Endocrine Disruption. Endocrinology 2009, 150, 1731–1738. [Google Scholar] [CrossRef]
- Barnett, D.H. Identification and Characterization of Estrogen Receptor-Regulated Gene Expression Programs; University of Illinois: Urbana, IL, USA, 2010. [Google Scholar]
- Callard, G.V.; Tarrant, A.M.; Novillo, A.; Yacci, P.; Ciaccia, L.; Vajda, S.; Chuang, G.-Y.; Kozakov, D.; Greytak, S.R.; Sawyer, S.; et al. Evolutionary Origins of the Estrogen Signaling System: Insights from Amphioxus. J. Steroid Biochem. Mol. Biol. 2011, 127, 176–188. [Google Scholar] [CrossRef]
- Thornton, J.W.; Need, E.; Crews, D. Resurrecting the Ancestral Steroid Receptor: Ancient Origin of Estrogen Signaling. Science 2003, 301, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human Oestrogen Receptor cDNA: Sequence, Expression and Homology to v-Erb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a Novel Receptor Expressed in Rat Prostate and Ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.W. Evolution of Vertebrate Steroid Receptors from an Ancestral Estrogen Receptor by Ligand Exploitation and Serial Genome Expansions. Proc. Natl. Acad. Sci. USA 2001, 98, 5671–5676. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Chambon, P.; White, J.H. Defining a Minimal Estrogen Receptor DNA Binding Domain. Nucleic Acids Res. 1993, 21, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Zwart, W.; de Leeuw, R.; Rondaij, M.; Neefjes, J.; Mancini, M.A.; Michalides, R. The Hinge Region of the Human Estrogen Receptor Determines Functional Synergy between AF-1 and AF-2 in the Quantitative Response to Estradiol and Tamoxifen. J. Cell Sci. 2010, 123, 1253–1261. [Google Scholar] [CrossRef]
- Montano, M.M.; Müller, V.; Trobaugh, A.; Katzenellenbogen, B.S. The Carboxy-Terminal F Domain of the Human Estrogen Receptor: Role in the Transcriptional Activity of the Receptor and the Effectiveness of Antiestrogens as Estrogen Antagonists. Mol. Endocrinol. 1995, 9, 814–825. [Google Scholar] [CrossRef]
- Koide, A.; Zhao, C.; Naganuma, M.; Abrams, J.; Deighton-Collins, S.; Skafar, D.F.; Koide, S. Identification of Regions within the F Domain of the Human Estrogen Receptor Alpha That Are Important for Modulating Transactivation and Protein-Protein Interactions. Mol. Endocrinol. 2007, 21, 829–842. [Google Scholar] [CrossRef]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and Characterization of Human Estrogen Receptor Beta Isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a New Isoform of the Human Estrogen Receptor-Alpha (hER-Alpha) That Is Encoded by Distinct Transcripts and That Is Able to Repress hER-Alpha Activation Function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, Cloning, and Expression of Human Estrogen Receptor-α36, a Novel Variant of Human Estrogen Receptor-α66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef]
- Warner, M.; Fan, X.; Strom, A.; Wu, W.; Gustafsson, J.-Å. 25 Years of ERβ: A Personal Journey. J. Mol. Endocrinol. 2021, 68, R1–R9. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.H.; Al-Azzawi, F. Immunolocalisation of Oestrogen Receptor Beta in Human Tissues. J. Mol. Endocrinol. 2000, 24, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Huang, Z.-Y.; Xu, X.-L.; Li, J.; Fu, X.-W.; Deng, S.-L. Estrogen Receptor Function: Impact on the Human Endometrium. Front. Endocrinol. 2022, 13, 827724. [Google Scholar] [CrossRef]
- Lindberg, M.K.; Movérare, S.; Skrtic, S.; Gao, H.; Dahlman-Wright, K.; Gustafsson, J.-A.; Ohlsson, C. Estrogen Receptor (ER)-Beta Reduces ERalpha-Regulated Gene Transcription, Supporting a “Ying Yang” Relationship between ERalpha and ERbeta in Mice. Mol. Endocrinol. 2003, 17, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Wihlén, B.; Tujague, M.; Wan, J.; Ström, A.; Gustafsson, J.-A. Estrogen Receptor (ER) β Modulates ERα-Mediated Transcriptional Activation by Altering the Recruitment of c-Fos and c-Jun to Estrogen-Responsive Promoters. Mol. Endocrinol. 2006, 20, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Bresson, D.; Lucas, A.; Chauveau, C.; Vignon, F. ER Beta Inhibits Proliferation and Invasion of Breast Cancer Cells. Endocrinology 2001, 142, 4120–4130. [Google Scholar] [CrossRef]
- Sotoca, A.M.C.; van den Berg, H.; Vervoort, J.; van der Saag, P.; Ström, A.; Gustafsson, J.-A.; Rietjens, I.; Murk, A.J. Influence of Cellular ERalpha/ERbeta Ratio on the ERalpha-Agonist Induced Proliferation of Human T47D Breast Cancer Cells. Toxicol. Sci. 2008, 105, 303–311. [Google Scholar] [CrossRef]
- Heo, K.-S. Regulation of Post-Translational Modification in Breast Cancer Treatment. BMB Rep. 2019, 52, 113–118. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, S.; Gustafsson, J.-Å. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Shanle, E.K.; Xu, W. Selectively Targeting Estrogen Receptors for Cancer Treatment. Adv. Drug Deliv. Rev. 2010, 62, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a Gene (GPR30) with Homology to the G-Protein-Coupled Receptor Superfamily Associated with Estrogen Receptor Expression in Breast Cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- Lafferty, A.R.; Torpy, D.J.; Stowasser, M.; Taymans, S.E.; Lin, J.P.; Huggard, P.; Gordon, R.D.; Stratakis, C.A. A Novel Genetic Locus for Low Renin Hypertension: Familial Hyperaldosteronism Type II Maps to Chromosome 7 (7p22). J. Med. Genet. 2000, 37, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef]
- Olde, B.; Leeb-Lundberg, L.M.F. GPR30/GPER1: Searching for a Role in Estrogen Physiology. Trends Endocrinol. Metab. 2009, 20, 409–416. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Sabo, E. Association of the Membrane Estrogen Receptor, GPR30, with Breast Tumor Metastasis and Transactivation of the Epidermal Growth Factor Receptor. Steroids 2008, 73, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The Many Faces of Estrogen Signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef]
- Safe, S.; Kim, K. Non-Classical Genomic Estrogen Receptor (ER)/specificity Protein and ER/activating Protein-1 Signaling Pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Björnström, L.; Sjöberg, M. Estrogen Receptor-Dependent Activation of AP-1 via Non-Genomic Signalling. Nucl. Recept. 2004, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.; Wang, Y.; Shapiro, D.J.; Xu, W. Differential Requirements of Hsp90 and DNA for the Formation of Estrogen Receptor Homodimers and Heterodimers. J. Biol. Chem. 2010, 285, 16125–16134. [Google Scholar] [CrossRef]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef]
- Dhamad, A.E.; Zhou, Z.; Zhou, J.; Du, Y. Systematic Proteomic Identification of the Heat Shock Proteins (Hsp) That Interact with Estrogen Receptor Alpha (ERα) and Biochemical Characterization of the ERα-Hsp70 Interaction. PLoS ONE 2016, 11, e0160312. [Google Scholar] [CrossRef]
- Eckert, R.L.; Mullick, A.; Rorke, E.A.; Katzenellenbogen, B.S. Estrogen Receptor Synthesis and Turnover in MCF-7 Breast Cancer Cells Measured by a Density Shift Technique. Endocrinology 1984, 114, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguère, V. An Acetylation Switch Modulates the Transcriptional Activity of Estrogen-Related Receptor Alpha. Mol. Endocrinol. 2010, 24, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Yu, L.-R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.-E.; Wang, C.; Brindle, P.K.; Dent, S.Y.R.; Ge, K. Distinct Roles of GCN5/PCAF-Mediated H3K9ac and CBP/p300-Mediated H3K18/27ac in Nuclear Receptor Transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef]
- Murakami, S.; Nagari, A.; Kraus, W.L. Dynamic Assembly and Activation of Estrogen Receptor α Enhancers through Coregulator Switching. Genes Dev. 2017, 31, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Buxant, F.; Engohan-Aloghe, C.; Noël, J.-C. Estrogen Receptor, Progesterone Receptor, and Glucocorticoid Receptor Expression in Normal Breast Tissue, Breast in Situ Carcinoma, and Invasive Breast Cancer. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 254–257. [Google Scholar] [CrossRef]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the Glucocorticoid Receptor Is Associated with Poor Prognosis in Estrogen Receptor-Negative Breast Cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone Receptor Modulates ERα Action in Breast Cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Gordon, M.A.; Babbs, B.; Spoelstra, N.S.; Carson Butterfield, K.T.; Torkko, K.C.; Phan, V.T.; Barton, V.N.; Rogers, T.J.; Sartorius, C.A.; et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol. Cancer Res. 2016, 14, 1054–1067. [Google Scholar] [CrossRef]
- Hu, D.G.; Selth, L.A.; Tarulli, G.A.; Meech, R.; Wijayakumara, D.; Chanawong, A.; Russell, R.; Caldas, C.; Robinson, J.L.L.; Carroll, J.S.; et al. Androgen and Estrogen Receptors in Breast Cancer Coregulate Human UDP-Glucuronosyltransferases 2B15 and 2B17. Cancer Res. 2016, 76, 5881–5893. [Google Scholar] [CrossRef] [PubMed]
- Singhal, H.; Greene, M.E.; Tarulli, G.; Zarnke, A.L.; Bourgo, R.J.; Laine, M.; Chang, Y.-F.; Ma, S.; Dembo, A.G.; Raj, G.V.; et al. Genomic Agonism and Phenotypic Antagonism between Estrogen and Progesterone Receptors in Breast Cancer. Sci. Adv. 2016, 2, e1501924. [Google Scholar] [CrossRef] [PubMed]
- West, D.C.; Pan, D.; Tonsing-Carter, E.Y.; Hernandez, K.M.; Pierce, C.F.; Styke, S.C.; Bowie, K.R.; Garcia, T.I.; Kocherginsky, M.; Conzen, S.D. GR and ER Coactivation Alters the Expression of Differentiation Genes and Associates with Improved ER+ Breast Cancer Outcome. Mol. Cancer Res. 2016, 14, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Tangen, I.L.; Veneris, J.T.; Halle, M.K.; Werner, H.M.; Trovik, J.; Akslen, L.A.; Salvesen, H.B.; Conzen, S.D.; Fleming, G.F.; Krakstad, C. Expression of Glucocorticoid Receptor Is Associated with Aggressive Primary Endometrial Cancer and Increases from Primary to Metastatic Lesions. Gynecol. Oncol. 2017, 147, 672–677. [Google Scholar] [CrossRef]
- Vahrenkamp, J.M.; Yang, C.-H.; Rodriguez, A.C.; Almomen, A.; Berrett, K.C.; Trujillo, A.N.; Guillen, K.P.; Welm, B.E.; Jarboe, E.A.; Janat-Amsbury, M.M.; et al. Clinical and Genomic Crosstalk between Glucocorticoid Receptor and Estrogen Receptor α In Endometrial Cancer. Cell Rep. 2018, 22, 2995–3005. [Google Scholar] [CrossRef]
- Jeong, K.W.; Lee, Y.-H.; Stallcup, M.R. Recruitment of the SWI/SNF Chromatin Remodeling Complex to Steroid Hormone-Regulated Promoters by Nuclear Receptor Coactivator Flightless-I. J. Biol. Chem. 2009, 284, 29298–29309. [Google Scholar] [CrossRef]
- Ballaré, C.; Castellano, G.; Gaveglia, L.; Althammer, S.; González-Vallinas, J.; Eyras, E.; Le Dily, F.; Zaurin, R.; Soronellas, D.; Vicent, G.P.; et al. Nucleosome-Driven Transcription Factor Binding and Gene Regulation. Mol. Cell 2013, 49, 67–79. [Google Scholar] [CrossRef]
- Le Dily, F.; Vidal, E.; Cuartero, Y.; Quilez, J.; Nacht, A.S.; Vicent, G.P.; Carbonell-Caballero, J.; Sharma, P.; Villanueva-Cañas, J.L.; Ferrari, R.; et al. Hormone-Control Regions Mediate Steroid Receptor-Dependent Genome Organization. Genome Res. 2019, 29, 29–39. [Google Scholar] [CrossRef]
- Kolyvas, E.A.; Caldas, C.; Kelly, K.; Ahmad, S.S. Androgen Receptor Function and Targeted Therapeutics across Breast Cancer Subtypes. Breast Cancer Res. 2022, 24, 79. [Google Scholar] [CrossRef]
- Yang, F.; Ma, Q.; Liu, Z.; Li, W.; Tan, Y.; Jin, C.; Ma, W.; Hu, Y.; Shen, J.; Ohgi, K.A.; et al. Glucocorticoid Receptor:MegaTrans Switching Mediates the Repression of an ERα-Regulated Transcriptional Program. Mol. Cell 2017, 66, 321–331.e6. [Google Scholar] [CrossRef]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.R.; Harkless, R.V.; Oh, A.; Bowie, K.R.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid Receptor Modulation Decreases ER-Positive Breast Cancer Cell Proliferation and Suppresses Wild-Type and Mutant ER Chromatin Association. Breast Cancer Res. 2019, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Plasma Membrane Estrogen Receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Pupo, M.; Maggiolini, M.; Musti, A.M. GPER Mediates Non-Genomic Effects of Estrogen. Methods Mol. Biol. 2016, 1366, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; de Falco, A.; Bontempo, P.; Nola, E.; Auricchio, F. Tyrosine kinase/p21ras/MAP-Kinase Pathway Activation by Estradiol-Receptor Complex in MCF-7 Cells. EMBO J. 1996, 15, 1292–1300. [Google Scholar] [CrossRef]
- Watters, J.J.; Campbell, J.S.; Cunningham, M.J.; Krebs, E.G.; Dorsa, D.M. Rapid Membrane Effects of Steroids in Neuroblastoma Cells: Effects of Estrogen on Mitogen Activated Protein Kinase Signalling Cascade and c-Fos Immediate Early Gene Transcription. Endocrinology 1997, 138, 4030–4033. [Google Scholar] [CrossRef]
- Chen, Z.; Yuhanna, I.S.; Galcheva-Gargova, Z.; Karas, R.H.; Mendelsohn, M.E.; Shaul, P.W. Estrogen Receptor Alpha Mediates the Nongenomic Activation of Endothelial Nitric Oxide Synthase by Estrogen. J. Clin. Invest. 1999, 103, 401–406. [Google Scholar] [CrossRef]
- Zivadinovic, D.; Watson, C.S. Membrane Estrogen Receptor-Alpha Levels Predict Estrogen-Induced ERK1/2 Activation in MCF-7 Cells. Breast Cancer Res. 2005, 7, R130–R144. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A Variant of Estrogen Receptor-{alpha}, hER-{alpha}36: Transduction of Estrogen- and Antiestrogen-Dependent Membrane-Initiated Mitogenic Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef]
- Rocca, A.; Braga, L.; Volpe, M.C.; Maiocchi, S.; Generali, D. The Predictive and Prognostic Role of RAS-RAF-MEK-ERK Pathway Alterations in Breast Cancer: Revision of the Literature and Comparison with the Analysis of Cancer Genomic Datasets. Cancers 2022, 14, 5306. [Google Scholar] [CrossRef]
- Ciruelos Gil, E.M. Targeting the PI3K/AKT/mTOR Pathway in Estrogen Receptor-Positive Breast Cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef]
- Saczko, J.; Michel, O.; Chwiłkowska, A.; Sawicka, E.; Mączyńska, J.; Kulbacka, J. Estrogen Receptors in Cell Membranes: Regulation and Signaling. In Transport Across Natural and Modified Biological Membranes and Its Implications in Physiology and Therapy; Kulbacka, J., Satkauskas, S., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 93–105. ISBN 9783319568959. [Google Scholar]
- Heery, D.M.; Kalkhoven, E.; Hoare, S.; Parker, M.G. A Signature Motif in Transcriptional Co-Activators Mediates Binding to Nuclear Receptors. Nature 1997, 387, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Torres, K.; Liu, X.; Liu, C.-G.; Pollock, R.E. An Overview of Chromatin-Regulating Proteins in Cells. Curr. Protein Pept. Sci. 2016, 17, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Triki, M.; Lapierre, M.; Cavailles, V.; Mokdad-Gargouri, R. Expression and Role of Nuclear Receptor Coregulators in Colorectal Cancer. World J. Gastroenterol. 2017, 23, 4480–4490. [Google Scholar] [CrossRef]
- Qin, L.; Liao, L.; Redmond, A.; Young, L.; Yuan, Y.; Chen, H.; O’Malley, B.W.; Xu, J. The AIB1 Oncogene Promotes Breast Cancer Metastasis by Activation of PEA3-Mediated Matrix Metalloproteinase 2 (MMP2) and MMP9 Expression. Mol. Cell. Biol. 2008, 28, 5937–5950. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.-F.; Chan, Y.-K.; Grills, C.; McCrudden, C.M.; Gunasekharan, V.; Shi, Z.; Wong, A.S.-Y.; Lappin, T.R.; Chan, K.-W.; Fennell, D.A.; et al. Polyomavirus Enhancer Activator 3 Protein Promotes Breast Cancer Metastatic Progression through Snail-Induced Epithelial-Mesenchymal Transition. J. Pathol. 2011, 224, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Keeton, E.K.; McDonnell, D.P.; Brown, M. Coactivator AIB1 Links Estrogen Receptor Transcriptional Activity and Stability. Proc. Natl. Acad. Sci. USA 2004, 101, 11599–11604. [Google Scholar] [CrossRef]
- Gururaj, A.E.; Peng, S.; Vadlamudi, R.K.; Kumar, R. Estrogen Induces Expression of BCAS3, a Novel Estrogen Receptor-Alpha Coactivator, through Proline-, Glutamic Acid-, and Leucine-Rich Protein-1 (PELP1). Mol. Endocrinol. 2007, 21, 1847–1860. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, S.-H.; Yu, E.J.; Seo, W.-Y.; Kim, J.H. A Positive Role of DBC1 in PEA3-Mediated Progression of Estrogen Receptor-Negative Breast Cancer. Oncogene 2015, 34, 4500–4508. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, Y.; Sha, S.; Kong, X.; Zhu, H.; Xu, B.; Li, Y.; Wu, K. DBC1 Is over-Expressed and Associated with Poor Prognosis in Colorectal Cancer. Int. J. Clin. Oncol. 2014, 19, 106–112. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, X.; Zhang, Y.; Qiao, J.; Sasano, H.; McNamara, K.; Zhao, B.; Zhang, D.; Fan, Y.; Liu, L.; et al. Estradiol-Induced MMP-9 Expression via PELP1-Mediated Membrane-Initiated Signaling in ERα-Positive Breast Cancer Cells. Horm. Cancer 2020, 11, 87–96. [Google Scholar] [CrossRef]
- Altwegg, K.A.; Viswanadhapalli, S.; Mann, M.; Chakravarty, D.; Krishnan, S.; Liu, Z.; Liu, J.; Pratap, U.P.; Ebrahimi, B.; Sanchez, J.R.; et al. A First-in-Class Inhibitor of ER Coregulator PELP1 Targets ER+ Breast Cancer. Cancer Res. 2022, 82, 3830–3844. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Vadlamudi, R.K. PELP1: Structure, Biological Function and Clinical Significance. Gene 2016, 585, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Rajhans, R.; Chakravarty, D.; Nair, B.C.; Nair, S.S.; Evans, D.B.; Chen, S.; Tekmal, R.R. Regulation of Aromatase Induction by Nuclear Receptor Coregulator PELP1. J. Steroid Biochem. Mol. Biol. 2010, 118, 211–218. [Google Scholar] [CrossRef]
- Girard, B.J.; Daniel, A.R.; Lange, C.A.; Ostrander, J.H. PELP1: A Review of PELP1 Interactions, Signaling, and Biology. Mol. Cell. Endocrinol. 2014, 382, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Słowikowski, B.K.; Gałęcki, B.; Dyszkiewicz, W.; Jagodziński, P.P. Increased Expression of Proline-, Glutamic Acid- and Leucine-Rich Protein PELP1 in Non-Small Cell Lung Cancer. Biomed. Pharmacother. 2015, 73, 97–101. [Google Scholar] [CrossRef]
- Wang, X.; Tsang, J.Y.S.; Lee, M.A.; Ni, Y.-B.; Tong, J.H.; Chan, S.-K.; Cheung, S.-Y.; To, K.F.; Tse, G.M. The Clinical Value of PELP1 for Breast Cancer: A Comparison with Multiple Cancers and Analysis in Breast Cancer Subtypes. Cancer Res. Treat. 2019, 51, 706–717. [Google Scholar] [CrossRef]
- den Hollander, P.; Rayala, S.K.; Coverley, D.; Kumar, R. Ciz1, a Novel DNA-Binding Coactivator of the Estrogen Receptor Alpha, Confers Hypersensitivity to Estrogen Action. Cancer Res. 2006, 66, 11021–11029. [Google Scholar] [CrossRef]
- Liu, Q.; Niu, N.; Wada, Y.; Liu, J. The Role of Cdkn1A-Interacting Zinc Finger Protein 1 (CIZ1) in DNA Replication and Pathophysiology. Int. J. Mol. Sci. 2016, 17, 212. [Google Scholar] [CrossRef]
- Hörlein, A.J.; Näär, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Söderström, M.; Glass, C.K. Ligand-Independent Repression by the Thyroid Hormone Receptor Mediated by a Nuclear Receptor Co-Repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef]
- Heinzel, T.; Lavinsky, R.M.; Mullen, T.-M.; Söderström, M.; Laherty, C.D.; Torchia, J.; Yang, W.-M.; Brard, G.; Ngo, S.D.; Davie, J.R.; et al. A Complex Containing N-CoR, mSln3 and Histone Deacetylase Mediates Transcriptional Repression. Nature 1997, 387, 43–48. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both Corepressor Proteins SMRT and N-CoR Exist in Large Protein Complexes Containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone Acetyltransferases: Challenges in Targeting Bi-Substrate Enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Légaré, S.; Basik, M. Minireview: The Link Between ERα Corepressors and Histone Deacetylases in Tamoxifen Resistance in Breast Cancer. Mol. Endocrinol. 2016, 30, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Noblejas-López, M.D.M.; Morcillo-García, S.; Nieto-Jiménez, C.; Nuncia-Cantarero, M.; Győrffy, B.; Galan-Moya, E.M.; Pandiella, A.; Ocaña, A. Evaluation of Transcriptionally Regulated Genes Identifies NCOR1 in Hormone Receptor Negative Breast Tumors and Lung Adenocarcinomas as a Potential Tumor Suppressor Gene. PLoS ONE 2018, 13, e0207776. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, S.-H.; Lee, J.; Sung, G.-J.; Song, J.-H.; Kwak, S.; Jeong, J.-H.; Kong, M.-J.; Hwang, J.-T.; Choi, H.-K.; et al. TNFα Enhances Tamoxifen Sensitivity through Dissociation of ERα-p53-NCOR1 Complexes in ERα-Positive Breast Cancer. Cancers 2021, 13, 2601. [Google Scholar] [CrossRef]
- Ritter, M.J.; Amano, I.; Imai, N.; Soares De Oliveira, L.; Vella, K.R.; Hollenberg, A.N. Nuclear Receptor CoRepressors, NCOR1 and SMRT, Are Required for Maintaining Systemic Metabolic Homeostasis. Mol. Metab. 2021, 53, 101315. [Google Scholar] [CrossRef]
- Aylon, Y.; Furth, N.; Mallel, G.; Friedlander, G.; Nataraj, N.B.; Dong, M.; Hassin, O.; Zoabi, R.; Cohen, B.; Drendel, V.; et al. Breast Cancer Plasticity Is Restricted by a LATS1-NCOR1 Repressive Axis. Nat. Commun. 2022, 13, 7199. [Google Scholar] [CrossRef]
- Eakin, C.M.; Maccoss, M.J.; Finney, G.L.; Klevit, R.E. Estrogen Receptor Alpha Is a Putative Substrate for the BRCA1 Ubiquitin Ligase. Proc. Natl. Acad. Sci. USA 2007, 104, 5794–5799. [Google Scholar] [CrossRef]
- Wang, C.; Bai, F.; Zhang, L.-H.; Scott, A.; Li, E.; Pei, X.-H. Estrogen Promotes Estrogen Receptor Negative BRCA1-Deficient Tumor Initiation and Progression. Breast Cancer Res. 2018, 20, 74. [Google Scholar] [CrossRef]
- Popov, V.M.; Zhou, J.; Shirley, L.A.; Quong, J.; Yeow, W.-S.; Wright, J.A.; Wu, K.; Rui, H.; Vadlamudi, R.K.; Jiang, J.; et al. The Cell Fate Determination Factor DACH1 Is Expressed in Estrogen Receptor-Alpha-Positive Breast Cancer and Represses Estrogen Receptor-Alpha Signaling. Cancer Res. 2009, 69, 5752–5760. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, Y.; Zhang, W.; Popov, V.M.; Wang, M.; Pattabiraman, N.; Suñé, C.; Cvekl, A.; Wu, K.; Jiang, J.; et al. Transcription Elongation Regulator 1 Is a Co-Integrator of the Cell Fate Determination Factor Dachshund Homolog 1. J. Biol. Chem. 2010, 285, 40342–40350. [Google Scholar] [CrossRef] [PubMed]
- Aman, S.; Li, Y.; Cheng, Y.; Yang, Y.; Lv, L.; Li, B.; Xia, K.; Li, S.; Wu, H. DACH1 Inhibits Breast Cancer Cell Invasion and Metastasis by down-Regulating the Transcription of Matrix Metalloproteinase 9. Cell Death Discov. 2021, 7, 351. [Google Scholar] [CrossRef]
- Wu, K.; Jiao, X.; Li, Z.; Katiyar, S.; Casimiro, M.C.; Yang, W.; Zhang, Q.; Willmarth, N.E.; Chepelev, I.; Crosariol, M.; et al. Cell Fate Determination Factor Dachshund Reprograms Breast Cancer Stem Cell Function. J. Biol. Chem. 2011, 286, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, A.; Blatt, K.; Cerny-Reiterer, S.; Sadovnik, I.; Herrmann, H.; Marian, B.; Grunt, T.W.; Zielinski, C.C.; Valent, P. Cancer Stem Cells in Basic Science and in Translational Oncology: Can We Translate into Clinical Application? J. Hematol. Oncol. 2015, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yu, S.; Yuan, X.; Xiong, J.; Kuang, D.; Pestell, R.G.; Wu, K. DACH1 Suppresses Breast Cancer as a Negative Regulator of CD44. Sci. Rep. 2017, 7, 4361. [Google Scholar] [CrossRef]
- Powe, D.G.; Dhondalay, G.K.R.; Lemetre, C.; Allen, T.; Habashy, H.O.; Ellis, I.O.; Rees, R.; Ball, G.R. DACH1: Its Role as a Classifier of Long Term Good Prognosis in Luminal Breast Cancer. PLoS ONE 2014, 9, e84428. [Google Scholar] [CrossRef]
- Lee, J.M.; Hammarén, H.M.; Savitski, M.M.; Baek, S.H. Control of Protein Stability by Post-Translational Modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef]
- Yuan, B.; Cheng, L.; Gupta, K.; Chiang, H.-C.; Gupta, H.B.; Sareddy, G.R.; Wang, D.; Lathrop, K.; Elledge, R.; Wang, P.; et al. Tyrosine Phosphorylation Regulates ERβ Ubiquitination, Protein Turnover, and Inhibition of Breast Cancer. Oncotarget 2016, 7, 42585–42597. [Google Scholar] [CrossRef]
- Atsriku, C.; Britton, D.J.; Held, J.M.; Schilling, B.; Scott, G.K.; Gibson, B.W.; Benz, C.C.; Baldwin, M.A. Systematic Mapping of Posttranslational Modifications in Human Estrogen Receptor-α with Emphasis on Novel Phosphorylation Sites. Mol. Cell. Proteomics 2009, 8, 467–480. [Google Scholar] [CrossRef]
- Ikeda, K.; Ogawa, S.; Tsukui, T.; Horie-Inoue, K.; Ouchi, Y.; Kato, S.; Muramatsu, M.; Inoue, S. Protein Phosphatase 5 Is a Negative Regulator of Estrogen Receptor-Mediated Transcription. Mol. Endocrinol. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Weitsman, G.E.; Li, L.; Skliris, G.P.; Davie, J.R.; Ung, K.; Niu, Y.; Curtis-Snell, L.; Tomes, L.; Watson, P.H.; Murphy, L.C. Estrogen Receptor-Alpha Phosphorylated at Ser118 Is Present at the Promoters of Estrogen-Regulated Genes and Is Not Altered due to HER-2 Overexpression. Cancer Res. 2006, 66, 10162–10170. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Pace, P.E.; Coombes, R.C.; Ali, S. Phosphorylation of Human Estrogen Receptor Alpha by Protein Kinase A Regulates Dimerization. Mol. Cell. Biol. 1999, 19, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Bai, W. Regulation of Estrogen Receptor Nuclear Export by Ligand-Induced and p38-Mediated Receptor Phosphorylation. Mol. Cell. Biol. 2002, 22, 5835–5845. [Google Scholar] [CrossRef]
- Masaki, T.; Habara, M.; Sato, Y.; Goshima, T.; Maeda, K.; Hanaki, S.; Shimada, M. Calcineurin Regulates the Stability and Activity of Estrogen Receptor α. Proc. Natl. Acad. Sci. USA 2021, 118, e2114258118. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Neven, P.; Loibl, S.; Andre, F. Advances in the Treatment of Advanced Oestrogen-Receptor-Positive Breast Cancer. Lancet 2017, 389, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Wang, C.; Fu, M.; Angeletti, R.H.; Siconolfi-Baez, L.; Reutens, A.T.; Albanese, C.; Lisanti, M.P.; Katzenellenbogen, B.S.; Kato, S.; Hopp, T.; et al. Direct Acetylation of the Estrogen Receptor Alpha Hinge Region by p300 Regulates Transactivation and Hormone Sensitivity. J. Biol. Chem. 2001, 276, 18375–18383. [Google Scholar] [CrossRef]
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.E.; Homenko, D.R.; Kraus, W.L. Acetylation of Estrogen Receptor Alpha by p300 at Lysines 266 and 268 Enhances the Deoxyribonucleic Acid Binding and Transactivation Activities of the Receptor. Mol. Endocrinol. 2006, 20, 1479–1493. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, L.; Han, Y.; Wu, F.; Yang, W.-S.; Zhang, Z.; Huo, T.; Zhu, Y.; Yu, C.; Kim, H.; et al. Acetylation of Histone H3K27 Signals the Transcriptional Elongation for Estrogen Receptor Alpha. Commun. Biol. 2020, 3, 165. [Google Scholar] [CrossRef]
- Vasquez, Y.; Setlem, R.; Murakami, S.; Kraus, W. SUN-028 Role of Estrogen Receptor Alpha (ERa) Acetylation in Estrogen-Dependent Gene Regulation in Breast Cancers. J. Endocr. Soc. 2019, 3, SUN-028. [Google Scholar] [CrossRef]
- Waddell, A.; Mahmud, I.; Ding, H.; Huo, Z.; Liao, D. Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer. Cancers 2021, 13, 2799. [Google Scholar] [CrossRef]
- Tsuboi, K.; Nagatomo, T.; Gohno, T.; Higuchi, T.; Sasaki, S.; Fujiki, N.; Kurosumi, M.; Takei, H.; Yamaguchi, Y.; Niwa, T.; et al. Single CpG Site Methylation Controls Estrogen Receptor Gene Transcription and Correlates with Hormone Therapy Resistance. J. Steroid Biochem. Mol. Biol. 2017, 171, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome—Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lee, K.-M.; Han, W.; Choi, J.-Y.; Lee, J.-Y.; Kang, G.H.; Park, S.K.; Noh, D.-Y.; Yoo, K.-Y.; Kang, D. Estrogen and Progesterone Receptor Status Affect Genome-Wide DNA Methylation Profile in Breast Cancer. Hum. Mol. Genet. 2010, 19, 4273–4277. [Google Scholar] [CrossRef] [PubMed]
- Fackler, M.J.; Umbricht, C.B.; Williams, D.; Argani, P.; Cruz, L.-A.; Merino, V.F.; Teo, W.W.; Zhang, Z.; Huang, P.; Visvananthan, K.; et al. Genome-Wide Methylation Analysis Identifies Genes Specific to Breast Cancer Hormone Receptor Status and Risk of Recurrence. Cancer Res. 2011, 71, 6195–6207. [Google Scholar] [CrossRef]
- Rønneberg, J.A.; Fleischer, T.; Solvang, H.K.; Nordgard, S.H.; Edvardsen, H.; Potapenko, I.; Nebdal, D.; Daviaud, C.; Gut, I.; Bukholm, I.; et al. Methylation Profiling with a Panel of Cancer Related Genes: Association with Estrogen Receptor, TP53 Mutation Status and Expression Subtypes in Sporadic Breast Cancer. Mol. Oncol. 2011, 5, 61–76. [Google Scholar] [CrossRef]
- Benevolenskaya, E.V.; Islam, A.B.M.M.K.; Ahsan, H.; Kibriya, M.G.; Jasmine, F.; Wolff, B.; Al-Alem, U.; Wiley, E.; Kajdacsy-Balla, A.; Macias, V.; et al. DNA Methylation and Hormone Receptor Status in Breast Cancer. Clin. Epigenetics 2016, 8, 17. [Google Scholar] [CrossRef]
- Umeh-Garcia, M.; O’Geen, H.; Simion, C.; Gephart, M.H.; Segal, D.J.; Sweeney, C.A. Aberrant Promoter Methylation Contributes to LRIG1 Silencing in Basal/triple-Negative Breast Cancer. Br. J. Cancer 2022, 127, 436–448. [Google Scholar] [CrossRef]
- Pradhan, S.; Chin, H.G.; Estève, P.-O.; Jacobsen, S.E. SET7/9 Mediated Methylation of Non-Histone Proteins in Mammalian Cells. Epigenetics 2009, 4, 383–387. [Google Scholar] [CrossRef]
- Subramanian, K.; Jia, D.; Kapoor-Vazirani, P.; Powell, D.R.; Collins, R.E.; Sharma, D.; Peng, J.; Cheng, X.; Vertino, P.M. Regulation of Estrogen Receptor Alpha by the SET7 Lysine Methyltransferase. Mol. Cell 2008, 30, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. Regulation of Mammalian DNA Replication via the Ubiquitin-Proteasome System. Adv. Exp. Med. Biol. 2017, 1042, 421–454. [Google Scholar] [CrossRef]
- Tripathi, V.; Agarwal, H.; Priya, S.; Batra, H.; Modi, P.; Pandey, M.; Saha, D.; Raghavan, S.C.; Sengupta, S. MRN Complex-Dependent Recruitment of Ubiquitylated BLM Helicase to DSBs Negatively Regulates DNA Repair Pathways. Nat. Commun. 2018, 9, 1016. [Google Scholar] [CrossRef]
- Komander, D. The Emerging Complexity of Protein Ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef]
- Han, D.; Wang, L.; Jiang, S.; Yang, Q. The Ubiquitin-Proteasome System in Breast Cancer. Trends Mol. Med. 2023, 29, 599–621. [Google Scholar] [CrossRef]
- Berry, N.B.; Fan, M.; Nephew, K.P. Estrogen Receptor-Alpha Hinge-Region Lysines 302 and 303 Regulate Receptor Degradation by the Proteasome. Mol. Endocrinol. 2008, 22, 1535–1551. [Google Scholar] [CrossRef]
- Helzer, K.T.; Hooper, C.; Miyamoto, S.; Alarid, E.T. Ubiquitylation of Nuclear Receptors: New Linkages and Therapeutic Implications. J. Mol. Endocrinol. 2015, 54, R151–R167. [Google Scholar] [CrossRef]
- Stanišić, V.; Malovannaya, A.; Qin, J.; Lonard, D.M.; O’Malley, B.W. OTU Domain-Containing Ubiquitin Aldehyde-Binding Protein 1 (OTUB1) Deubiquitinates Estrogen Receptor (ER) Alpha and Affects ERalpha Transcriptional Activity. J. Biol. Chem. 2009, 284, 16135–16145. [Google Scholar] [CrossRef]
- Tang, J.; Luo, Y.; Tian, Z.; Liao, X.; Cui, Q.; Yang, Q.; Wu, G. TRIM11 Promotes Breast Cancer Cell Proliferation by Stabilizing Estrogen Receptor α. Neoplasia 2020, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shen, Y.; Yin, L.; He, J.; Ni, X.; Luo, G.; Chen, X.; Zhu, W.; Zhong, J.; Liu, J.; et al. Knockdown of ZBTB7A Inhibits Cell Proliferation of Breast Cancer through Regulating the Ubiquitination of Estrogen Receptor Alpha. Life Sci. 2019, 239, 117042. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Deng, Q.; Zhao, H.; Xie, M.; Chen, L.; Yin, F.; Qin, X.; Zheng, W.; Zhao, Y.; Li, Z. Development of Stabilized Peptide-Based PROTACs against Estrogen Receptor α. ACS Chem. Biol. 2018, 13, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C.; Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Rojas-Ochoa, A. Proteolysis-Targeting Chimeras and Their Implications in Breast Cancer. Explor. Target Antitumor Ther. 2021, 2, 496–510. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in Sumoylation: A Decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.-C.; Corbo, L. Sumoylation of the Estrogen Receptor Alpha Hinge Region Regulates Its Transcriptional Activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Vallet, A.; El Ezzy, M.; Diennet, M.; Haidar, S.; Bouvier, M.; Mader, S. The AF-2 Cofactor Binding Region Is Key for the Selective SUMOylation of Estrogen Receptor Alpha by Antiestrogens. J. Biol. Chem. 2023, 299, 102757. [Google Scholar] [CrossRef]
- He, X.; Zheng, Z.; Song, T.; Wei, C.; Ma, H.; Ma, Q.; Zhang, Y.; Xu, Y.; Shi, W.; Ye, Q.; et al. C-Abl Regulates Estrogen Receptor Alpha Transcription Activity through Its Stabilization by Phosphorylation. Oncogene 2010, 29, 2238–2251. [Google Scholar] [CrossRef]
- Medunjanin, S.; Hermani, A.; De Servi, B.; Grisouard, J.; Rincke, G.; Mayer, D. Glycogen Synthase Kinase-3 Interacts with and Phosphorylates Estrogen Receptor Alpha and Is Involved in the Regulation of Receptor Activity. J. Biol. Chem. 2005, 280, 33006–33014. [Google Scholar] [CrossRef]
- Masuhiro, Y.; Mezaki, Y.; Sakari, M.; Takeyama, K.-I.; Yoshida, T.; Inoue, K.; Yanagisawa, J.; Hanazawa, S.; O’malley, B.W.; Kato, S. Splicing Potentiation by Growth Factor Signals via Estrogen Receptor Phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 8126–8131. [Google Scholar] [CrossRef]
- Arnold, S.F.; Obourn, J.D.; Jaffe, H.; Notides, A.C. Serine 167 Is the Major Estradiol-Induced Phosphorylation Site on the Human Estrogen Receptor. Mol. Endocrinol. 1994, 8, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Williams, C.C.; Basu, A.; El-Gharbawy, A.; Carrier, L.M.; Smith, C.L.; Rowan, B.G. Identification of Four Novel Phosphorylation Sites in Estrogen Receptor Alpha: Impact on Receptor-Dependent Gene Expression and Phosphorylation by Protein Kinase CK2. BMC Biochem. 2009, 10, 36. [Google Scholar] [CrossRef]
- Tharakan, R.; Lepont, P.; Singleton, D.; Kumar, R.; Khan, S. Phosphorylation of Estrogen Receptor Alpha, Serine Residue 305 Enhances Activity. Mol. Cell. Endocrinol. 2008, 295, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Iacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Andò, S.; Fuqua, S.A.W. Phosphorylation of the Mutant K303R Estrogen Receptor Alpha at Serine 305 Affects Aromatase Inhibitor Sensitivity. Oncogene 2010, 29, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-Dependent Estrogen Receptor Alpha Membrane Localization: Regulation by 17beta-Estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Deschenes, R.J.; Levin, E.R. DHHC-7 and -21 Are Palmitoylacyltransferases for Sex Steroid Receptors. Mol. Biol. Cell 2012, 23, 188–199. [Google Scholar] [CrossRef]
- Márquez, D.C.; Lee, J.; Lin, T.; Pietras, R.J. Epidermal Growth Factor Receptor and Tyrosine Phosphorylation of Estrogen Receptor. Endocrine 2001, 16, 73–81. [Google Scholar] [CrossRef]
- Simond, A.M.; Ling, C.; Moore, M.J.; Condotta, S.A.; Richer, M.J.; Muller, W.J. Point-Activated ESR1Y541S Has a Dramatic Effect on the Development of Sexually Dimorphic Organs. Genes Dev. 2020, 34, 1304–1309. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A Compendium of Mutational Cancer Driver Genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Krøigård, A.B.; Larsen, M.J.; Lænkholm, A.-V.; Knoop, A.S.; Jensen, J.D.; Bak, M.; Mollenhauer, J.; Thomassen, M.; Kruse, T.A. Identification of Metastasis Driver Genes by Massive Parallel Sequencing of Successive Steps of Breast Cancer Progression. PLoS ONE 2018, 13, e0189887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Y.; Chen, B.; Guo, L.; Cao, L.; Ren, C.; Wen, L.; Li, K.; Jia, M.; Li, C.; et al. Characterization of Frequently Mutated Cancer Genes in Chinese Breast Tumors: A Comparison of Chinese and TCGA Cohorts. Ann. Transl. Med. 2019, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Hermida-Prado, F.; Jeselsohn, R. The ESR1 Mutations: From Bedside to Bench to Bedside. Cancer Res. 2021, 81, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multi-dimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404, Erratum in Cancer Discov. 2012, 2, 960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An Estrogen Receptor Mutant with Strong Hormone-Independent Activity from a Metastatic Breast Cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of Constitutively Active Estrogen Receptor-α Mutations in Pretreated Advanced Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Fribbens, C.; Garcia Murillas, I.; Beaney, M.; Hrebien, S.; O’Leary, B.; Kilburn, L.; Howarth, K.; Epstein, M.; Green, E.; Rosenfeld, N.; et al. Tracking Evolution of Aromatase Inhibitor Resistance with Circulating Tumour DNA Analysis in Metastatic Breast Cancer. Ann. Oncol. 2018, 29, 145–153. [Google Scholar] [CrossRef]
- Liao, H.; Huang, W.; Pei, W.; Li, H. Detection of ESR1 Mutations Based on Liquid Biopsy in Estrogen Receptor-Positive Metastatic Breast Cancer: Clinical Impacts and Prospects. Front. Oncol. 2020, 10, 587671. [Google Scholar] [CrossRef]
- Najim, O.; Seghers, S.; Sergoynne, L.; Van Gaver, H.; Papadimitriou, K.; Wouters, K.; Trinh, X.B.; Huizing, M.T.; Tjalma, W. The Association between Type of Endocrine Therapy and Development of Estrogen Receptor-1 Mutation(s) in Patients with Hormone-Sensitive Advanced Breast Cancer: A Systematic Review and Meta-Analysis of Randomized and Non-Randomized Trials. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188315. [Google Scholar] [CrossRef]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen Receptor Alpha Somatic Mutations Y537S and D538G Confer Breast Cancer Endocrine Resistance by Stabilizing the Activating Function-2 Binding Conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen Receptor Mutations and Their Role in Breast Cancer Progression. Breast Cancer Res. 2014, 16, 494. [Google Scholar] [CrossRef]
- Gu, G.; Tian, L.; Gao, M.; Rechoum, Y.; Gelsomino, L.; Dustin, D.; Corona-Rodriguez, A.; Beyer, A.R.; Tsimelzon, A.; Zhang, X.; et al. Abstract 22: The Y537S ESR1 Mutation Is a Dominant Driver of Distant ER-Positive Breast Cancer Metastasis. Cancer Res. 2018, 78, 22. [Google Scholar] [CrossRef]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 Mutations in Breast Cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M.; Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Qiu, X.; Buchwalter, G.; et al. Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell 2018, 33, 173–186.e5. [Google Scholar] [CrossRef] [PubMed]
- Puyang, X.; Furman, C.; Zheng, G.Z.; Wu, Z.J.; Banka, D.; Aithal, K.; Agoulnik, S.; Bolduc, D.M.; Buonamici, S.; Caleb, B.; et al. Discovery of Selective Estrogen Receptor Covalent Antagonists for the Treatment of ERαWT and ERαMUT Breast Cancer. Cancer Discov. 2018, 8, 1176–1193. [Google Scholar] [CrossRef] [PubMed]
- Brett, J.O.; Spring, L.M.; Bardia, A.; Wander, S.A. ESR1 Mutation as an Emerging Clinical Biomarker in Metastatic Hormone Receptor-Positive Breast Cancer. Breast Cancer Res. 2021, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, S.; Blanchard, Z.; Williams, M.M.; Berrett, K.C.; Li, Z.; Oesterreich, S.; Richer, J.K.; Gertz, J. Estrogen Receptor Alpha Mutations in Breast Cancer Cells Cause Gene Expression Changes through Constant Activity and Secondary Effects. Cancer Res. 2021, 81, 539–551. [Google Scholar] [CrossRef]
- Martin, L.-A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of Naturally Occurring ESR1 Mutations in Breast Cancer Cell Lines Modelling Endocrine Resistance. Nat. Commun. 2017, 8, 1865. [Google Scholar] [CrossRef]
- Nardone, A.; De Angelis, C.; Trivedi, M.V.; Osborne, C.K.; Schiff, R. The Changing Role of ER in Endocrine Resistance. Breast 2015, 24, S60–S66. [Google Scholar] [CrossRef]
- Glück, S. Consequences of the Convergence of Multiple Alternate Pathways on the Estrogen Receptor in the Treatment of Metastatic Breast Cancer. Clin. Breast Cancer 2017, 17, 79–90. [Google Scholar] [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.-P.; Santen, R.J.; Kim, T.-H.; Park, H.; Fan, P.; Yue, W. Estrogen Stimulation of Cell Migration Involves Multiple Signaling Pathway Interactions. Endocrinology 2010, 151, 5146–5156. [Google Scholar] [CrossRef]
- He, T.; Yang, W.; Zhang, X.; Li, P.; Yang, D.; Wu, Y.; Fan, Y.; Xiang, M.; Huang, Q.; Chen, J.; et al. Comparative Effectiveness of Tamoxifen, Toremifene, Letrozole, Anastrozole, and Exemestane on Lipid Profiles in Breast Cancer Patients: A Network Meta-Analysis. Medicine 2020, 99, e18550. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, L.R.; Goa, K.L. Toremifene. A Review of Its Pharmacological Properties and Clinical Efficacy in the Management of Advanced Breast Cancer. Drugs 1997, 54, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Huang, J.; Shen, L.; Zhu, S.; Gao, W.; Wu, J.; Huang, O.; He, J.; Zhu, L.; Chen, W.; et al. A Prospective, Randomized Study of Toremifene vs. Tamoxifen for the Treatment of Premenopausal Breast Cancer: Safety and Genital Symptom Analysis. BMC Cancer 2020, 20, 663. [Google Scholar] [CrossRef]
- Wernli, K.J.; Knerr, S.; Li, T.; Leppig, K.; Ehrlich, K.; Farrell, D.; Gao, H.; Bowles, E.J.A.; Graham, A.L.; Luta, G.; et al. Effect of Personalized Breast Cancer Risk Tool on Chemoprevention and Breast Imaging: ENGAGED-2 Trial. JNCI Cancer Spectr. 2021, 5, pkaa114. [Google Scholar] [CrossRef]
- Robertson, J.F.R.; Bondarenko, I.M.; Trishkina, E.; Dvorkin, M.; Panasci, L.; Manikhas, A.; Shparyk, Y.; Cardona-Huerta, S.; Cheung, K.-L.; Philco-Salas, M.J.; et al. Fulvestrant 500 Mg versus Anastrozole 1 Mg for Hormone Receptor-Positive Advanced Breast Cancer (FALCON): An International, Randomised, Double-Blind, Phase 3 Trial. Lancet 2016, 388, 2997–3005. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef]
- Lainé, M.; Fanning, S.W.; Chang, Y.-F.; Green, B.; Greene, M.E.; Komm, B.; Kurleto, J.D.; Phung, L.; Greene, G.L. Lasofoxifene as a Potential Treatment for Therapy-Resistant ER-Positive Metastatic Breast Cancer. Breast Cancer Res. 2021, 23, 54. [Google Scholar] [CrossRef]
- Fanning, S.W.; Jeselsohn, R.; Dharmarajan, V.; Mayne, C.G.; Karimi, M.; Buchwalter, G.; Houtman, R.; Toy, W.; Fowler, C.E.; Han, R.; et al. The SERM/SERD Bazedoxifene Disrupts ESR1 Helix 12 to Overcome Acquired Hormone Resistance in Breast Cancer Cells. eLife 2018, 7, e37161. [Google Scholar] [CrossRef]
- Fanning, S.W.; Greene, G.L. Next-Generation ERα Inhibitors for Endocrine-Resistant ER+ Breast Cancer. Endocrinology 2019, 160, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Walsh, E.M.; Tao, J.J.; Chandarlapaty, S.; Jhaveri, K. Accelerating Drug Development in Breast Cancer: New Frontiers for ER Inhibition. Cancer Treat. Rev. 2022, 109, 102432. [Google Scholar] [CrossRef] [PubMed]
- Fernando, T.M.; Moore, H.M.; Wongchenko, M.J.; Metcalfe, C. Next-Generation Estrogen Receptor–Targeted Therapeutics. Annu. Rev. Cancer Biol. 2023, 7, 313–330. [Google Scholar] [CrossRef]
- Díaz-Cruz, E.S.; Sugimoto, Y.; Gallicano, G.I.; Brueggemeier, R.W.; Furth, P.A. Comparison of Increased Aromatase versus ERα in the Generation of Mammary Hyperplasia and Cancer. Cancer Res. 2011, 71, 5477–5487. [Google Scholar] [CrossRef]
- Mukhopadhyay, K.D.; Liu, Z.; Bandyopadhyay, A.; Kirma, N.B.; Tekmal, R.R.; Wang, S.; Sun, L.-Z. Aromatase Expression Increases the Survival and Malignancy of Estrogen Receptor Positive Breast Cancer Cells. PLoS ONE 2015, 10, e0121136. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Luo, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; Griffith, M.; Griffith, O.L.; Skidmore, Z.L.; Spies, N.C.; Ramu, A.; et al. A Phase I Trial of BKM120 (Buparlisib) in Combination with Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer. Agents Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Franco, S.X.; Florance, A.; O’Rourke, L.; Maltzman, J.; Johnston, S. Lapatinib plus Letrozole as First-Line Therapy for HER-2+ Hormone Receptor-Positive Metastatic Breast Cancer. Oncologist 2010, 15, 122–129. [Google Scholar] [CrossRef]
- Farahmand, L.; Merikhian, P.; Jalili, N.; Darvishi, B.; Majidzadeh-A, K. Significant Role of MUC1 in Development of Resistance to Currently Existing Anti-Cancer Therapeutic Agents. Curr. Cancer Drug Targets 2018, 18, 737–748. [Google Scholar] [CrossRef]
- Patel, A.; Tiwari, A.K.; Chufan, E.E.; Sodani, K.; Anreddy, N.; Singh, S.; Ambudkar, S.V.; Stephani, R.; Chen, Z.-S. PD173074, a Selective FGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells. Cancer Chemother. Pharmacol. 2013, 72, 189–199. [Google Scholar] [CrossRef]
- Seckl, M.; Badman, P.D.; Liu, X.; MacPherson, I.R.; Zubairi, I.H.; Baird, R.D.; Garcia-Corbacho, J.; Cresti, N.; Plummer, E.R.; Armstrong, A.C.; et al. RADICAL Trial: A Phase Ib/IIa Study to Assess the Safety and Efficacy of AZD4547 in Combination with Either Anastrozole or Letrozole in ER Positive Breast Cancer Patients Progressing on These Aromatase Inhibitors (AIs). J. Clin. Orthod. 2017, 35, 1059. [Google Scholar] [CrossRef]
- Kulkoyluoglu-Cotul, E.; Smith, B.P.; Wrobel, K.; Zhao, Y.C.; Chen, K.L.A.; Hieronymi, K.; Imir, O.B.; Duong, K.; O’Callaghan, C.; Mehta, A.; et al. Combined Targeting of Estrogen Receptor Alpha and XPO1 Prevent Akt Activation, Remodel Metabolic Pathways and Induce Autophagy to Overcome Tamoxifen Resistance. Cancers 2019, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Shafique, M.; Ismail-Khan, R.; Extermann, M.; Sullivan, D.; Goodridge, D.; Boulware, D.; Hogue, D.; Soliman, H.; Khong, H.; Han, H.S. A Phase II Trial of Selinexor (KPT-330) for Metastatic Triple-Negative Breast Cancer. Oncologist 2019, 24, 887-e416. [Google Scholar] [CrossRef] [PubMed]
- Thein, K.Z.; Piha-Paul, S.A.; Tsimberidou, A.; Karp, D.D.; Janku, F.; Fu, S.; Subbiah, V.; Hong, D.S.; Yap, T.A.; Shah, J.; et al. Selinexor in Combination with Standard Chemotherapy in Patients with Advanced or Metastatic Solid Tumors. Exp. Hematol. Oncol. 2021, 10, 59. [Google Scholar] [CrossRef]
- Cotul, E.K.; Zuo, Q.; Santaliz-Casiano, A.; Imir, O.B.; Mogol, A.N.; Tunc, E.; Duong, K.; Lee, J.K.; Ramesh, R.; Odukoya, E.; et al. Combined Targeting of Estrogen Receptor Alpha and Exportin 1 in Metastatic Breast Cancers. Cancers 2020, 12, 2397. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhai, L.; Wang, H.; Liu, C.; Zhang, J.; Chen, W.; Wei, Q. Downregulation of LncRNA GAS5 Causes Trastuzumab Resistance in Breast Cancer. Oncotarget 2016, 7, 27778–27786. [Google Scholar] [CrossRef]
- Ouyang, Y.X.; Feng, J.; Wang, Z.; Zhang, G.J.; Chen, M. miR-221/222 Sponge Abrogates Tamoxifen Resistance in ER-Positive Breast Cancer Cells through Restoring the Expression of ERα. Mol. Biomed. 2021, 2, 20. [Google Scholar] [CrossRef]
- Li, J.; Lai, Y.; Ma, J.; Liu, Y.; Bi, J.; Zhang, L.; Chen, L.; Yao, C.; Lv, W.; Chang, G.; et al. miR-17-5p Suppresses Cell Proliferation and Invasion by Targeting ETV1 in Triple-Negative Breast Cancer. BMC Cancer 2017, 17, 745. [Google Scholar] [CrossRef]
- Ljepoja, B.; García-Roman, J.; Sommer, A.-K.; Wagner, E.; Roidl, A. MiRNA-27a Sensitizes Breast Cancer Cells to Treatment with Selective Estrogen Receptor Modulators. Breast 2019, 43, 31–38. [Google Scholar] [CrossRef]
- Yin, K.; Yin, W.; Wang, Y.; Zhou, L.; Liu, Y.; Yang, G.; Wang, J.; Lu, J. MiR-206 Suppresses Epithelial Mesenchymal Transition by Targeting TGF-β Signaling in Estrogen Receptor Positive Breast Cancer Cells. Oncotarget 2016, 7, 24537–24548. [Google Scholar] [CrossRef]
- Nichols, M. New Directions for Drug-Resistant Breast Cancer: The CDK4/6 Inhibitors. Future Med. Chem. 2015, 7, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus Fulvestrant in PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer after a CDK4/6 Inhibitor (BYLieve): One Cohort of a Phase 2, Multicentre, Open-Label, Non-Comparative Study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and Clinical Significance of ESR1 Mutations in ER-Positive Metastatic Breast Cancer Patients Receiving Fulvestrant. Nat. Commun. 2016, 7, 11579. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 Mutations—A Mechanism for Acquired Endocrine Resistance in Breast Cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef]
- Ladd, B.; Mazzola, A.M.; Bihani, T.; Lai, Z.; Bradford, J.; Collins, M.; Barry, E.; Goeppert, A.U.; Weir, H.M.; Hearne, K.; et al. Effective Combination Therapies in Preclinical Endocrine Resistant Breast Cancer Models Harboring ER Mutations. Oncotarget 2016, 7, 54120–54136. [Google Scholar] [CrossRef]
- Garner, F.; Shomali, M.; Paquin, D.; Lyttle, C.R.; Hattersley, G. RAD1901: A Novel, Orally Bioavailable Selective Estrogen Receptor Degrader That Demonstrates Antitumor Activity in Breast Cancer Xenograft Models. Anticancer. Drugs 2015, 26, 948–956. [Google Scholar] [CrossRef]
- Bihani, T.; Patel, H.K.; Arlt, H.; Tao, N.; Jiang, H.; Brown, J.L.; Purandare, D.M.; Hattersley, G.; Garner, F. Elacestrant (RAD1901), a Selective Estrogen Receptor Degrader (SERD), Has Antitumor Activity in Multiple ER+ Breast Cancer Patient-Derived Xenograft Models. Clin. Cancer Res. 2017, 23, 4793–4804. [Google Scholar] [CrossRef]
- Bardia, A.; Aftimos, P.; Bihani, T.; Anderson-Villaluz, A.T.; Jung, J.; Conlan, M.G.; Kaklamani, V.G. EMERALD: Phase III Trial of Elacestrant (RAD1901) vs Endocrine Therapy for Previously Treated ER+ Advanced Breast Cancer. Future Oncol. 2019, 15, 3209–3218. [Google Scholar] [CrossRef]
- Patel, H.K.; Tao, N.; Lee, K.-M.; Huerta, M.; Arlt, H.; Mullarkey, T.; Troy, S.; Arteaga, C.L.; Bihani, T. Elacestrant (RAD1901) Exhibits Anti-Tumor Activity in Multiple ER+ Breast Cancer Models Resistant to CDK4/6 Inhibitors. Breast Cancer Res. 2019, 21, 146. [Google Scholar] [CrossRef]
- Hoy, S.M. Elacestrant: First Approval. Drugs 2023, 83, 555–561. [Google Scholar] [CrossRef]
- Ng, C.K.Y.; Schultheis, A.M.; Bidard, F.-C.; Weigelt, B.; Reis-Filho, J.S. Breast Cancer Genomics from Microarrays to Massively Parallel Sequencing: Paradigms and New Insights. J. Natl. Cancer Inst. 2015, 107, djv015. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H.-J.; Gelderblom, H. Histone Deacetylase Inhibitors: An Overview of the Clinical Studies in Solid Tumors. Anticancer Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-T.; Wang, Y.-W.; Chen, C.-T.; Ho, C.-M.; Su, W.-H.; Jou, Y.-S. HDAC Inhibitors Augmented Cell Migration and Metastasis through Induction of PKCs Leading to Identification of Low Toxicity Modalities for Combination Cancer Therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ye, J.; Kijima, I.; Evans, D. The HDAC Inhibitor LBH589 (panobinostat) Is an Inhibitory Modulator of Aromatase Gene Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 11032–11037. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized Phase II, Double-Blind, Placebo-Controlled Study of Exemestane with or without Entinostat in Postmenopausal Women with Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer Progressing on Treatment with a Nonsteroidal Aromatase Inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Schech, A.; Kazi, A.; Yu, S.; Shah, P.; Sabnis, G. Histone Deacetylase Inhibitor Entinostat Inhibits Tumor-Initiating Cells in Triple-Negative Breast Cancer Cells. Mol. Cancer Ther. 2015, 14, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-W.; Yeh, Y.-L.; Wang, Y.-C.; Huang, W.-J.; Ho, S.-Y.; Lin, P.; Wang, Y.-J. Combination of the Novel Histone Deacetylase Inhibitor YCW1 and Radiation Induces Autophagic Cell Death through the Downregulation of BNIP3 in Triple-Negative Breast Cancer Cells in Vitro and in an Orthotopic Mouse Model. Mol. Cancer 2016, 15, 46. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A.; Kim, D.K.; Song, S.-H.; Kim, H.-J.; Lee, K.-H.; Kim, T.-Y.; Han, S.-W.; Oh, D.-Y.; Kim, T.-Y.; et al. Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid (SAHA), Enhances Anti-Tumor Effects of the Poly (ADP-Ribose) Polymerase (PARP) Inhibitor Olaparib in Triple-Negative Breast Cancer Cells. Breast Cancer Res. 2015, 17, 33. [Google Scholar] [CrossRef]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of Interactions between Cisplatin and Two Histone Deacetylase Inhibitors in MCF7, T47D and MDA-MB-231 Human Breast Cancer Cell Lines—An Isobolographic Analysis. PLoS ONE 2015, 10, e0143013. [Google Scholar] [CrossRef]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M.H. Functional Activation of the Estrogen Receptor-α and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903. [Google Scholar] [CrossRef]
- Iwata, H.; Nakamura, R.; Masuda, N.; Yamashita, T.; Yamamoto, Y.; Kobayashi, K.; Tsurutani, J.; Iwasa, T.; Yonemori, K.; Tamura, K.; et al. Efficacy and Exploratory Biomarker Analysis of Entinostat plus Exemestane in Advanced or Recurrent Breast Cancer: Phase II Randomized Controlled Trial. Jpn. J. Clin. Oncol. 2023, 53, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef] [PubMed]
- Piggott, L.; Silva, A.; Robinson, T.; Santiago-Gómez, A.; Simões, B.M.; Becker, M.; Fichtner, I.; Andera, L.; Young, P.; Morris, C.; et al. Acquired Resistance of ER-Positive Breast Cancer to Endocrine Treatment Confers an Adaptive Sensitivity to TRAIL through Posttranslational Downregulation of c-FLIP. Clin. Cancer Res. 2018, 24, 2452–2463. [Google Scholar] [CrossRef] [PubMed]



| Coactivators | Influence on ERα |
| AIB1 | The AIB1 coactivator is one of the transcription factors that react with ERα in a ligand-dependent manner, and its coactivator activity is enhanced by the CIB1δ and PKCε-mediated phosphorylation of AIB1. This action results in an increased expression of target genes, e.g., those responsible for cell migration, such as PEA3 (the polyomavirus enhancer activator 3), MMP2 (metalloproteinase 2), and MMP9 (metalloproteinase 9), and it is therefore directly related to tumorigenesis and metastasis [107,108]. The AIB1 coactivator activates ERα-dependent transcription by recruiting HAT to the chromatin of the ESR1 gene. In addition, the AIB1 protein is involved in the regulation of the degradation of the ERα via the ubiquitin–proteasome system (UPS) [109]. |
| BCAS3 | The highly conserved BCAS3 (breast cancer-amplified sequence 3) coactivator, like AIB1, interacts with ERα’s transcriptional complex, in conjunction with PELP1’s (proline-, glutamic acid-, and leucine-rich protein 1) coactivator, causing the activation of ERα-encoding gene transcription [110]. |
| DBC1 | It has been demonstrated that the DBC1 (deleted in breast cancer 1) protein, a negative regulator of deacetylase SIRT1, functions as a nER coactivator, and it is essential for the formation of the ER transcription complex and the proliferation of estrogen-dependent breast cancer cells. The deletion of DBC1 from ER-negative breast cancer cells was shown to decrease cell proliferation in vivo and in vitro, and increased DBC1 expression resulted in a negative prognosis and shortened recurrence-free survival in the ER-negative patients [111]. In addition, DBC1 overexpression is observed in prostate, gastric, esophageal, and colorectal cancers and has led to a worsening of the predicted poor prognosis [112]. |
| PELP1 | PELP1 regulates the genomic and non-genomic ERα signaling. It interacts with many transcription factors, and its activity is observed in the cell nucleus, cytosol, and plasma membrane [113,114]. It has an important role in the remodeling of chromatin by interacting with histones and histone-modifying enzymes [115]. PELP1 causes the activation of tyrosine kinase SRC, resulting in the reorganization of the cell cytoskeleton [116,117]. Increased PELP1 expression has been observed as a result of enhanced tumor cell invasion [118]. The effect of PELP1 is an epigenetic modification, leading to ERα activation [113]. PELP1 has been proposed as a biomarker of hormone-dependent cancers, i.e., ovarian and breast cancer [115,119]. |
| CIZ1 | CIZ1 (Cip1-interacting zinc-finger protein), a DNA-binding protein, is implicated (as an ER coactivator) in the ER transactivation due to the cooperation of the ER to the chromatin target gene. In addition, the overexpression of CIZ1 causes an increase in sensitivity to estrogen, accelerating the growth rate of breast cancer cells [120]. The increased expression of CIZ1 is observable not only in breast cancer but also in cancers like colon, lung, gallbladder, prostate, and other diseases, e.g., rheumatoid arthritis [121]. |
| Corepressors | Influence on ERα |
| NCOR1 | NCOR1 (nuclear receptor corepressor 1) inhibits ERα expression, suppressing transcription through the ligand-binding domain of ERα [122]. NCOR1 regulates the availability of chromatin by activating histone deacetylase 3 (HDAC3) [123,124,125]. In addition, it acts antagonistic on histone acetyltransferase (HAT) and the HAT-activating enzyme, causing the inhibition of its expression, which promotes the formation of compact, inactive heterochromatin [126]. The loss of NCOR1 results in accelerating the development of breast cancer, and a decrease in its expression may be the result of acquiring resistance to tamoxifen [127,128]. Additionally, it has been shown that the association of NCOR1 with other corepressors such as SAFB1 (scaffold attachment factor B 1) and SAFB2 (scaffold attachment factor B 2) reduces the expression of ERα [129,130]. Recently, Aylon and colleagues reported [131] that NCOR1 repressive activity is enhanced by LAST1 (large tumor suppressor 1) and proposed that this axis may restrict breast cancer progression. |
| BRCA1 | BRCA1 is the corepressor of ERα that works by binding to the AF-2 ERα domain, thanks to which it leads to the monoubiquitylation of the ER together with BARD1 influencing ER activity [132]. In non-immortalized fibroblasts and breast cancer cells, BRCA1 deficiency has been shown to activate the PI3K/AKT pathway by accumulating AKT. This effect is reinforced by the fact that estrogen also activates the PI3K/AKT pathway in the ER-dependent and independent manner. Therefore, it has been shown that in the BRCA1-deficient breast cancer cells, estrogen causes the initiation of the carcinogenesis process by stimulating cell division via the AKT pathway and activating the epithelial–mesenchymal transition (EMT) [133]. |
| DACH1 | DACH1 (Dachshund 1) is one of the ER corepressors, which works by blocking its action [134]. It regulates gene expression by binding to DNA-binding transcription factors and by blocking DNA strands [135]. The downregulation of the transcription of MMP9 by DACH1 inhibits breast cancer tumor cells’ invasion and metastasis [136]. DACH1 also inhibits the growth of cancer stem cells (CSCs), resulting in the inhibition of metastasis [137,138]. Moreover, it was shown that DACH1 suppresses breast cancer via a negative regulation of CD44 (cluster of differentiation-44) [139]. DACH1 interacts with the ER by blocking the interaction between ESR1 and the activator, resulting in an increased activity of HDAC and reduced ER transcription [134]. DACH1 expression is upregulated in individuals who show longer disease-free survival, ER-positive breast cancer-free survival, and reduced metastasis [140]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miziak, P.; Baran, M.; Błaszczak, E.; Przybyszewska-Podstawka, A.; Kałafut, J.; Smok-Kalwat, J.; Dmoszyńska-Graniczka, M.; Kiełbus, M.; Stepulak, A. Estrogen Receptor Signaling in Breast Cancer. Cancers 2023, 15, 4689. https://doi.org/10.3390/cancers15194689
Miziak P, Baran M, Błaszczak E, Przybyszewska-Podstawka A, Kałafut J, Smok-Kalwat J, Dmoszyńska-Graniczka M, Kiełbus M, Stepulak A. Estrogen Receptor Signaling in Breast Cancer. Cancers. 2023; 15(19):4689. https://doi.org/10.3390/cancers15194689
Chicago/Turabian StyleMiziak, Paulina, Marzena Baran, Ewa Błaszczak, Alicja Przybyszewska-Podstawka, Joanna Kałafut, Jolanta Smok-Kalwat, Magdalena Dmoszyńska-Graniczka, Michał Kiełbus, and Andrzej Stepulak. 2023. "Estrogen Receptor Signaling in Breast Cancer" Cancers 15, no. 19: 4689. https://doi.org/10.3390/cancers15194689