The Landscape of Adoptive Cellular Therapies in Ovarian Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
Adoptive Cellular Therapy in the Era of Personalised Medicine
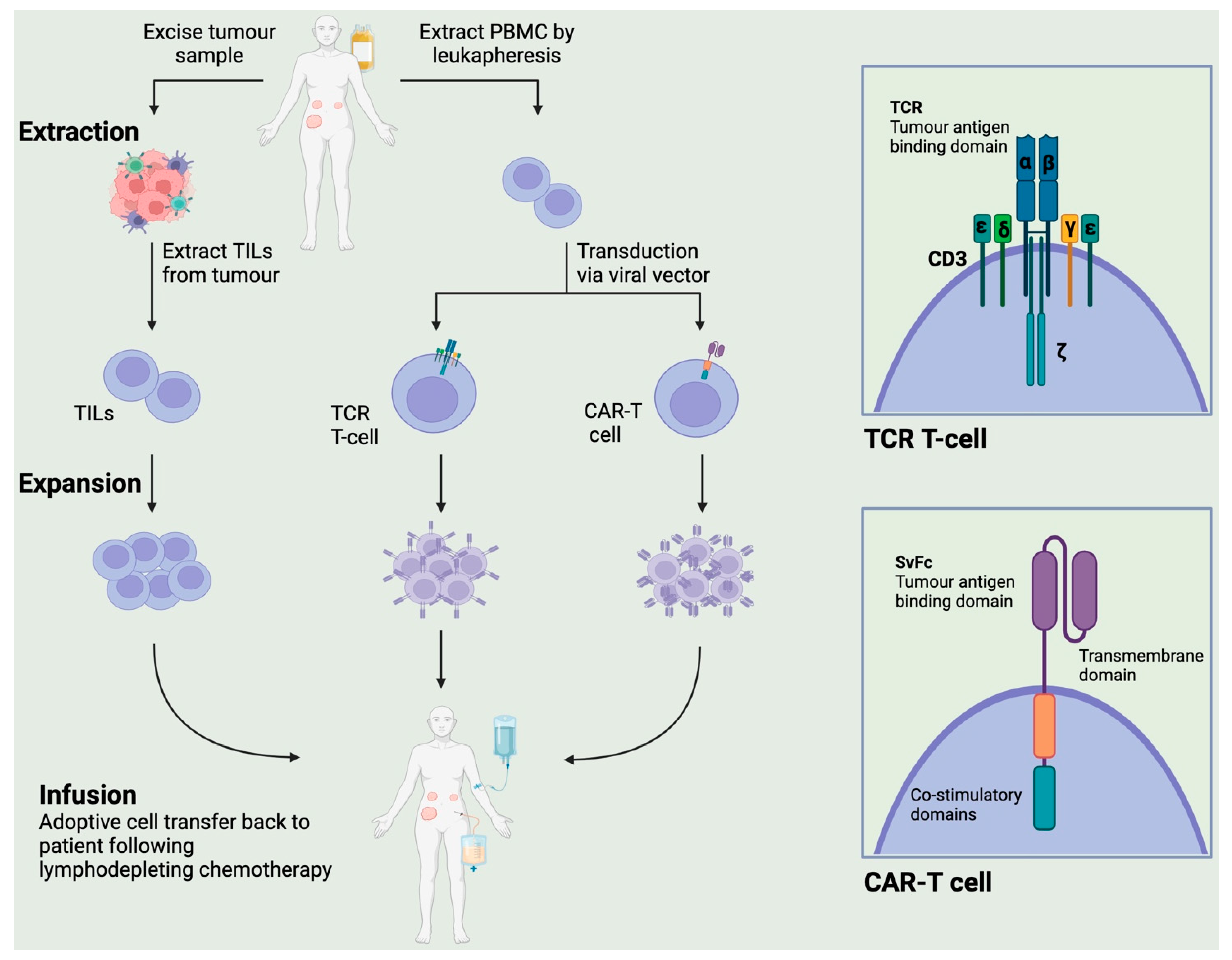
2. Types of Adoptive Cellular Therapy (ACT)
2.1. Chimeric Antigen Receptor T (CAR-T) Therapy
2.2. Genetically Engineered T Cell Receptor (TCR) Therapy
2.3. Tumour-Infiltrating Lymphocytes
3. Vaccines (Cell-Based)
4. Future Directions and Challenges
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Herold, C.; Gray, K.P.; Penson, R.T.; Horowitz, N.; Konstantinopoulos, P.A.; Castro, C.M.; Hill, S.J.; Curtis, J.; Luo, W.; et al. Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.S.; Ray-Coquard, I.L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab Alone or in Combination with Pegylated Liposomal Doxorubicin versus Pegylated Liposomal Doxorubicin Alone in Platinum-Resistant or Refractory Epithelial Ovarian Cancer: Primary and Biomarker Analysis of the Phase III JAVELIN Ovarian 200 Trial. Gynecol. Oncol. 2019, 154, 21–22. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). J. Clin. Oncol. 2021, 39, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.-W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II Study of Olaparib (O) plus Durvalumab (D) and Bevacizumab (B) (MEDIOLA): Initial Results in Patients (Pts) with Non-Germline BRCA-Mutated (Non-gBRCAm) Platinum Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2020, 31, S615–S616. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Shapira, R.; Santin, A.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Sehouli, J.; et al. Final Results from the KEYNOTE-100 Trial of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer. J. Clin. Oncol. 2020, 38 (Suppl. S15), 6005. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in Patients with Programmed Death Ligand 1–Positive Advanced Ovarian Cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef]
- Upadhaya, S.; Hubbard-Lucey, V.M.; Yu, J.X. Immuno-Oncology Drug Development Forges on despite COVID-19. Nat. Rev. Drug Discov. 2020, 19, 751–752. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Wong, Y.N.S.; Joshi, K.; Pule, M.; Peggs, K.S.; Swanton, C.; Quezada, S.A.; Linch, M. Evolving Adoptive Cellular Therapies in Urological Malignancies. Lancet Oncol. 2017, 18, e341–e353. [Google Scholar] [CrossRef] [PubMed]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for Gene-Engineered T Cell Immunotherapy for Solid Cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiu, J.; Sun, Y. The Basics of CAR T Design and Challenges in Immunotherapy of Solid Tumors—Ovarian Cancer as a Model. Hum. Vaccines Immunother. 2017, 13, 1548–1555. [Google Scholar] [CrossRef]
- Fowler, N.H.; Dickinson, M.; Dreyling, M.; Martinez-Lopez, J.; Kolstad, A.; Butler, J.; Ghosh, M.; Popplewell, L.; Chavez, J.C.; Bachy, E.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Follicular Lymphoma: The Phase 2 ELARA Trial. Nat. Med. 2022, 28, 325–332. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1–2 Trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, Z.; Yuan, Z.; Luo, Z.; Jin, H.; Qian, Q. New Strategies for the Treatment of Solid Tumors with CAR-T Cells. Int. J. Biol. Sci. 2016, 12, 718–729. [Google Scholar] [CrossRef]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming On-Target, off-Tumour Toxicity of CAR T Cell Therapy for Solid Tumours. Nat. Rev. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef]
- Ordóñez, N.G. Application of Mesothelin Immunostaining in Tumor Diagnosis. Am. J. Surg. Pathol. 2003, 27, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I Study of CAR-T Cells with PD-1 and TCR Disruption in Mesothelin-Positive Solid Tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- O’Cearbhaill, R.E.; Park, J.H.; Halton, E.F.; Diamonte, C.R.; Mead, E.; Lakhman, Y.; Kane, P.; Riviere, I.C.; Brentjens, R.J. A Phase I Clinical Trial of Autologous Chimeric Antigen Receptor (CAR) T Cells Genetically Engineered to Secrete IL-12 and to Target the MUC16ecto Antigen in Patients (Pts) with MUC16ecto+ Recurrent High-Grade Serous Ovarian Cancer (HGSOC). Gynecol. Oncol. 2020, 159, 42. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Van Tine, B.A.; Biswas, S.; McAlpine, C.; Johnson, M.L.; Olszanski, A.J.; Clarke, J.M.; Araujo, D.; Blumenschein, G.R.; Kebriaei, P.; et al. Autologous T Cell Therapy for MAGE-A4+ Solid Cancers in HLA-A*02+ Patients: A Phase 1 Trial. Nat. Med. 2023, 29, 104–114. [Google Scholar] [CrossRef]
- Butler, M.O.; Saibil, S.; Bonilla, L.; Franke, N.; Hakgor, S.; Boross-Harmer, S.; Majeed, H.; Nelles, M.; Ohashi, P.S.; Ross, K.; et al. Effect of Minimal Lymphodepletion Prior to ACT with TBI-1301, NY-ESO-1 Specific Gene-Engineered TCR-T Cells, on Clinical Responses and CRS. J. Clin. Oncol. 2019, 37 (Suppl. S15), 2537. [Google Scholar] [CrossRef]
- Odunsi, K.; Cristea, M.C.; Dorigo, O.; Jazaeri, A.A.; Slomovitz, B.M.; Chagin, K.; Van Winkle, E.; Kari, G.; Iyengar, M.; Norry, E.; et al. A Phase I/IIa, Open Label, Clinical Trial Evaluating the Safety and Efficacy of Autologous T Cells Expressing Enhanced TCRs Specific for NY-ESO-1 in Patients with Recurrent or Treatment Refractory Ovarian Cancer (NCT01567891). J. Clin. Oncol. 2017, 35 (Suppl. S15), TPS3094. [Google Scholar] [CrossRef]
- Aoki, Y.; Takakuwa, K.; Kodama, S.; Tanaka, K.; Takahashi, M.; Tokunaga, A.; Takahashi, T. Use of Adoptive Transfer of Tumor-Infiltrating Lymphocytes Alone or in Combination with Cisplatin-Containing Chemotherapy in Patients with Epithelial Ovarian Cancer. Cancer Res. 1991, 51, 1934–1939. [Google Scholar]
- Fujita, K.; Ikarashi, H.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T.; Tanaka, K. Prolonged Disease-Free Period in Patients with Advanced Epithelial Ovarian Cancer after Adoptive Transfer of Tumor-Infiltrating Lymphocytes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1995, 1, 501–507. [Google Scholar]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes in Patients with Metastatic Ovarian Cancer: A Pilot Study. OncoImmunology 2018, 7, e1502905. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; Burg, S.H.V.D.; Milne, K.; et al. Adoptive Cell Therapy in Combination with Checkpoint Inhibitors in Ovarian Cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Ethier, J.-L.; Bonilla, L.; Boross-Harmer, S.; Bouchard-Fortier, G.; Cipollone, J.; Clarke, B.A.; Dhani, N.C.; Easson, A.M.; Franke, N.; Goldstein, D.P.; et al. A Phase Ib Trial of Pembrolizumab (Pembro) Following Adoptive Cell Therapy (ACT) in Patients with Platinum-Resistant Ovarian Cancer; The ACTIVATE (Adoptive Cell Therapy InVigorated to Augment Tumor Eradication) Trial. J. Clin. Oncol. 2018, 36 (Suppl. S15), TPS5611. [Google Scholar] [CrossRef]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor Biomarker to Cancer Therapy, a Work in Progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Lu, Q.; Lou, T.; Cao, G.; Wang, S.; Zhang, Z. MUC16 Affects the Biological Functions of Ovarian Cancer Cells and Induces an Antitumor Immune Response by Activating Dendritic Cells. Ann. Transl. Med. 2020, 8, 1494. [Google Scholar] [CrossRef]
- Bax, H.J.; Chauhan, J.; Stavraka, C.; Santaolalla, A.; Osborn, G.; Khiabany, A.; Grandits, M.; López-Abente, J.; Palhares, L.C.G.F.; Chan Wah Hak, C.; et al. Folate Receptor Alpha in Ovarian Cancer Tissue and Patient Serum Is Associated with Disease Burden and Treatment Outcomes. Br. J. Cancer 2023, 128, 342–353. [Google Scholar] [CrossRef]
- Elnakat, H. Distribution, Functionality and Gene Regulation of Folate Receptor Isoforms: Implications in Targeted Therapy. Adv. Drug Deliv. Rev. 2004, 56, 1067–1084. [Google Scholar] [CrossRef]
- Cai, D.; Li, J.; Liu, D.; Hong, S.; Qiao, Q.; Sun, Q.; Li, P.; Lyu, N.; Sun, T.; Xie, S.; et al. Tumor-Expressed B7-H3 Mediates the Inhibition of Antitumor T-Cell Functions in Ovarian Cancer Insensitive to PD-1 Blockade Therapy. Cell. Mol. Immunol. 2020, 17, 227–236. [Google Scholar] [CrossRef]
- Gibbs, Z.A.; Whitehurst, A.W. Emerging Contributions of Cancer/Testis Antigens to Neoplastic Behaviors. Trends Cancer 2018, 4, 701–712. [Google Scholar] [CrossRef]
- Daudi, S.; Eng, K.H.; Mhawech-Fauceglia, P.; Morrison, C.; Miliotto, A.; Beck, A.; Matsuzaki, J.; Tsuji, T.; Groman, A.; Gnjatic, S.; et al. Expression and Immune Responses to MAGE Antigens Predict Survival in Epithelial Ovarian Cancer. PLoS ONE 2014, 9, e104099. [Google Scholar] [CrossRef]
- Szender, J.B.; Papanicolau-Sengos, A.; Eng, K.H.; Miliotto, A.J.; Lugade, A.A.; Gnjatic, S.; Matsuzaki, J.; Morrison, C.D.; Odunsi, K. NY-ESO-1 Expression Predicts an Aggressive Phenotype of Ovarian Cancer. Gynecol. Oncol. 2017, 145, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.; Zimmermann, S.; Arber, C.; Irving, M.; Trueb, L.; Coukos, G. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Saf. 2019, 42, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Rohaan, M.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Foppen, M.G.; Granhøj, J.S.; Nuijen, B.; Nijenhuis, C.; Beijnen, J.H.; et al. LBA3 Treatment with Tumor-Infiltrating Lymphocytes (TIL) versus Ipilimumab for Advanced Melanoma: Results from a Multicenter, Randomized Phase III Trial. Ann. Oncol. 2022, 33, S1406. [Google Scholar] [CrossRef]
- O’Malley, D.; Lee, S.; Psyrri, A.; Sukari, A.; Thomas, S.; Wenham, R.; Gogas, H.; Jazaeri, A.; Monk, B.; Rose, P.; et al. 492 Phase 2 Efficacy and Safety of Autologous Tumor-Infiltrating Lymphocyte (TIL) Cell Therapy in Combination with Pembrolizumab in Immune Checkpoint Inhibitor-Naïve Patients with Advanced Cancers. J. Immunother. Cancer 2021, 9 (Suppl. S2), A523. [Google Scholar] [CrossRef]
- Goode, E.L.; Block, M.S. Dose-Response Association of CD8 + Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Hamid, O.; Khushalani, N.I.; Lewis, K.D.; Medina, T.; Kluger, H.M.; Thomas, S.S.; Domingo-Musibay, E.; Pavlick, A.C.; Whitman, E.D.; et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J. Clin. Oncol. 2021, 39, 2656–2666. [Google Scholar] [CrossRef]
- Teng, F.; Mu, D.; Meng, X.; Kong, L.; Zhu, H.; Liu, S.; Zhang, J.; Yu, J. Tumor Infiltrating Lymphocytes (TILs) before and after Neoadjuvant Chemoradiotherapy and Its Clinical Utility for Rectal Cancer. Am. J. Cancer Res. 2015, 5, 2064–2074. [Google Scholar]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized Cancer Vaccine Effectively Mobilizes Antitumor T Cell Immunity in Ovarian Cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef]
- Zamarin, D.; Walderich, S.; Holland, A.; Zhou, Q.; Iasonos, A.E.; Torrisi, J.M.; Merghoub, T.; Chesebrough, L.F.; Mcdonnell, A.S.; Gallagher, J.M.; et al. Safety, Immunogenicity, and Clinical Efficacy of Durvalumab in Combination with Folate Receptor Alpha Vaccine TPIV200 in Patients with Advanced Ovarian Cancer: A Phase II Trial. J. Immunother. Cancer 2020, 8, e000829. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective Identification of Neoantigen-Specific Lymphocytes in the Peripheral Blood of Melanoma Patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.N.S.; Joshi, K.; Khetrapal, P.; Ismail, M.; Reading, J.L.; Sunderland, M.W.; Georgiou, A.; Furness, A.J.S.; Ben Aissa, A.; Ghorani, E.; et al. Urine-Derived Lymphocytes as a Non-Invasive Measure of the Bladder Tumor Immune Microenvironment. J. Exp. Med. 2018, 215, 2748–2759. [Google Scholar] [CrossRef]
- Weimer, P.; Wellbrock, J.; Sturmheit, T.; Oliveira-Ferrer, L.; Ding, Y.; Menzel, S.; Witt, M.; Hell, L.; Schmalfeldt, B.; Bokemeyer, C.; et al. Tissue-Specific Expression of TIGIT, PD-1, TIM-3, and CD39 by Γδ T Cells in Ovarian Cancer. Cells 2022, 11, 964. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Bellone, S.; Roman, J.J.; Smith, C.V.; Pecorelli, S.; Radominska-Pandya, A.; Cannon, M.J.; Parham, G.P. Phenotypic and Functional Analysis of Tumor-Infiltrating Lymphocytes Compared with Tumor-Associated Lymphocytes from Ascitic Fluid and Peripheral Blood Lymphocytes in Patients with Advanced Ovarian Cancer. Gynecol. Obstet. Investig. 2001, 51, 254–261. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Guercio, M.; Manni, S.; Boffa, I.; Caruso, S.; Di Cecca, S.; Sinibaldi, M.; Abbaszadeh, Z.; Camera, A.; Ciccone, R.; Polito, V.A.; et al. Inclusion of the Inducible Caspase 9 Suicide Gene in CAR Construct Increases Safety of CAR.CD19 T Cell Therapy in B-Cell Malignancies. Front. Immunol. 2021, 12, 755639. [Google Scholar] [CrossRef]
- Henry, J.; Oh, D.; Eskew, J.; Baranda, J.; Rodriguez Rivera, I.I.; Dumbrava, E.; Cohen, E.; Belani, R.; McCaigue, J.; Shedlock, D.; et al. 728 Phase 1 Study of P-MUC1C-ALLO1 Allogeneic CAR-T Cells in Patients with Epithelial-Derived Cancers. J. Immunother. Cancer 2022, 10 (Suppl. S2), A761. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T Cells with Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-Cell Intrinsic PD-1 Checkpoint Blockade: A Two-in-One Approach for Solid Tumor Immunotherapy. OncoImmunology 2017, 6, e1273302. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Wing, A.; Fajardo, C.A.; Posey, A.D., Jr.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus–Driven Production of a Bispecific T-Cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.-P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.A.; Han, S.W.; Lui, K.; Tchack, J.; Shapiro, R.; Berman, R.; Zhong, J.; Krogsgaard, M.; Osman, I.; Darvishian, F. Immunologic Heterogeneity of Tumor-Infiltrating Lymphocyte Composition in Primary Melanoma. Hum. Pathol. 2016, 57, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ Lymphocytes in Fresh Human Melanomas Enriches for Tumor-Reactive T Cells. J. Immunother. 2010, 33, 956. [Google Scholar] [CrossRef]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of Shared and Neoantigen-Reactive T Cells for Adoptive Cell Therapy Based on CD137 Separation. Front. Immunol. 2017, 8, 1211. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. “Final Common Pathway” of Human Cancer Immunotherapy: Targeting Random Somatic Mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced Detection of Neoantigen-Reactive T Cells Targeting Unique and Shared Oncogenes for Personalized Cancer Immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef]
- Chesney, J.; Wise-Draper, T.; Sarnaik, A.A.; Graf Finckenstein, F.; Hari, P.; Jagasia, M.; Desai, A.; Suzuki, A.; Wu, X.; Betof Warner, A. 883TiP A Phase I/II Open-Label Study (IOV-GM1-201) of TALEN-Mediated PD-1 Inactivated Autologous Tumor-Infiltrating Lymphocytes (TIL.; IOV-4001) in Patients with Advanced Melanoma and NSCLC. Ann. Oncol. 2022, 33, S952. [Google Scholar] [CrossRef]
- Ghorani, E.; Reading, J.L.; Henry, J.Y.; de Massy, M.R.; Rosenthal, R.; Turati, V.; Joshi, K.; Furness, A.J.S.; Aissa, A.B.; Saini, S.K.; et al. The T Cell Differentiation Landscape Is Shaped by Tumour Mutations in Lung Cancer. Nat. Cancer 2020, 1, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-Directed Immune Escape in Lung Cancer Evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Target | Phase | Size | Preconditioning | Observations |
|---|---|---|---|---|
| CAR-T Mesothelin [23] | Phase I | n = 15 (OVCA, n = 5) | Cyclophosphamide | No CRS was observed 1 incident of grade 4 AE–sepsis Best overall response was SD (n = 11/15; including all 5 OVCA patients) |
| CAR-T Mesothelin [24] | Phase I | n = 15 (OVCA, n = 1) | None | No CRS or autoimmune toxicity was observed in any of the patients Peak blood levels at 7–14 days and evidence of CAR-T cells in metastases at 2 weeks but not 3–4 weeks 7/15 achieved SD 3–4 weeks post infusion |
| CAR-T Mesothelin | Phase I/II | n = 15 | Cyclophosphamide/Fludarabine | Terminated due to slow accrual 14/15 patients had PD Result published on clinicaltrials.gov (NCT01583686) |
| CAR-T MUC16ecto [25] | Phase I | n = 18 | Cyclophosphamide or cyclophosphamide with fludarabine | Some degree of CRS was observed at all doses DLT observed in 2/3 of the patients who received LD Best response was SD |
| CAR-T FRα [26] | Phase I | n = 14 | None | Grade 3–4 toxicity in 5/8 patients who received IL-2 Grade 1–2 toxicity in group who received CAR-T cells without IL-2 T cells barely detectable in peripheral blood at 4 weeks in any patient No reduction in tumour burden in any patient |
| TCR MAGE-A4 [27] | Phase I | n = 38 (OVCA, n = 9) | Cyclophosphamide/Fludarabine | All patients experienced grade 3–4 haematological toxicity 55% patients had CRS across all tumour types 1 patient with OVCA had treatment-related death due to CVA after grade 3 neurotoxicity ORR was 24% 74% patients had SD or PR including 5 with OVCA Median duration of response was 25.6 weeks |
| TCR NY-ESO-1 [28] | Phase I | Estimated n = 22 | Cyclophosphamide/Fludarabine | No DLT observed 2 PR, 5 SD and 1 PD at 2 years |
| TCR NY-ESO-1 [29] | Phase I/IIb | n = 6 | Cyclophosphamide/Fludarabine | 5/6 participants had serious AE 100% all-cause mortality at 12 months |
| TIL [30] | Phase I | n = 17 | Cisplatin-based |
1/7 CR and 4/7 PR in the TIL group 7/10 CR and 2/10 PR in the cisplatin-based chemotherapy + TIL group |
| TIL [31] | Phase I | n = 24 | None | 100% DFS at 3 years in TIL group compared with 67.5% in control group |
| TIL [32] | Phase I | n = 6 | Cyclophosphamide |
6/6 patients had SD at 6 weeks High levels of LAG-3, PD-1 were identified in infused T-cells |
| TIL [33] | Phase I/II | n = 6 | Cyclophosphamide/Fludarabine |
1/6 PR at 12 months 5/6 SD at 12 months |
| Target | Phase | Size | Preconditioning | Status (Estimated Primary Completion Date) | Clinical Trial Number |
|---|---|---|---|---|---|
| CAR-T Mesothelin | Phase I | n = 10 | Cyclophosphamide/Fludarabine | Unknown (January 2023) | NCT03814447 |
| CAR-T Mesothelin | Phase I/II | n = 3 | None | Closed for accrual (February 2023) | NCT05372692 |
| CAR-T Mesothelin | Phase I/II | Estimated n = 20 | Cyclophosphamide/Fludarabine | Unknown (April 2023) | NCT03916679 |
| CAR-T Mesothelin | Phase I/II | Estimated n = 10 | Cyclophosphamide/Fludarabine | Open for accrual (May 2026) | NCT05963100 |
| CAR-T MUC16 | Phase I | Estimated n = 71 | Cyclophosphamide | Open for accrual (November 2028) | NCT03907527 |
| CAR-T MUC1-C | Phase 1 | Estimated n = 100 | Unknown | Open for accrual (April 2026) | NCT05239143 |
| CAR-T MOV19-BBZ | Phase I | Estimated n = 12–24 | Cyclophosphamide/Fludarabine | Open for accrual (October 2028) | NCT03585764 |
| CAR-T TAG-72 | Phase I | Estimated n = 33 | Cyclophosphamide/Fludarabine | Open for accrual (April 2027) | NCT05225363 |
| CAR-T B7H3 | Phase I | Estimated n = 25 | Cyclophosphamide/Fludarabine | Open for accrual (February 2025) | NCT04670068 |
| CAR-T B7H3 | Phase I/II | Estimated n = 15 | Cyclophosphamide/Fludarabine | Open for accrual (August 2026) | NCT05211557 |
| CAR-T ALPP | Phase I/II | Estimated n = 20 | Cyclophosphamide/Fludarabine | Closed for accrual (December 2022) | NCT04627740 |
| TCR MAGE-A4 | Phase II | Estimated n = 66 | Cyclophosphamide/Fludarabine | Open for accrual (October 2025) | NCT05601752 |
| TCR NY-ESO-1- | Phase I | n = 9 | Cyclophosphamide/Decitabine | Closed for accrual (March 2020) | NCT03017131 |
| TCR NY-ESO-1 | Phase I | n = 4 | Melphalan | Closed for accrual (January 2021) | NCT03691376 |
| TCR Neoantigens | Phase II | Estimated n = 270 | Cyclophosphamide/Fludarabine | Open for accrual (March 2027) | NCT03412877 |
| TIL | Phase II | Estimated n = 15 | Fludarabine Radiotherapy | Unknown (February 2021) | NCT03412526 |
| TIL | Phase I | n = 3 | Cyclophosphamide | Closed for accrual (June 2023) | NCT01883297 |
| TIL in combination with nivolumab, relatlimab and ipilimumab | Phase I/II | Estimated n = 18 | Cyclophosphamide/Fludarabine | Open for accrual (December 2022) | NCT04611126 |
| TIL [34] in combination with pembrolizumab | Phase I | n = 8 | Cyclophosphamide/Fludarabine | Closed for accrual (August 2020) | NCT03158935 |
| TIL in combination with IFNα | Phase I/II | Estimated n = 12 | Carboplatin/Paclitaxel | Unknown (August 2021) | NCT04072263 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, L.; Miller, R.E.; Wong, Y.N.S. The Landscape of Adoptive Cellular Therapies in Ovarian Cancer. Cancers 2023, 15, 4814. https://doi.org/10.3390/cancers15194814
Davis L, Miller RE, Wong YNS. The Landscape of Adoptive Cellular Therapies in Ovarian Cancer. Cancers. 2023; 15(19):4814. https://doi.org/10.3390/cancers15194814
Chicago/Turabian StyleDavis, Lucy, Rowan E Miller, and Yien Ning Sophia Wong. 2023. "The Landscape of Adoptive Cellular Therapies in Ovarian Cancer" Cancers 15, no. 19: 4814. https://doi.org/10.3390/cancers15194814