

Rilotumumab Resistance Acquired by Intracrine Hepatocyte Growth Factor Signaling
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Culture
2.2. Quantitative Immunoassays
2.3. Cell Proliferation and Anchorage Independent Growth Assays
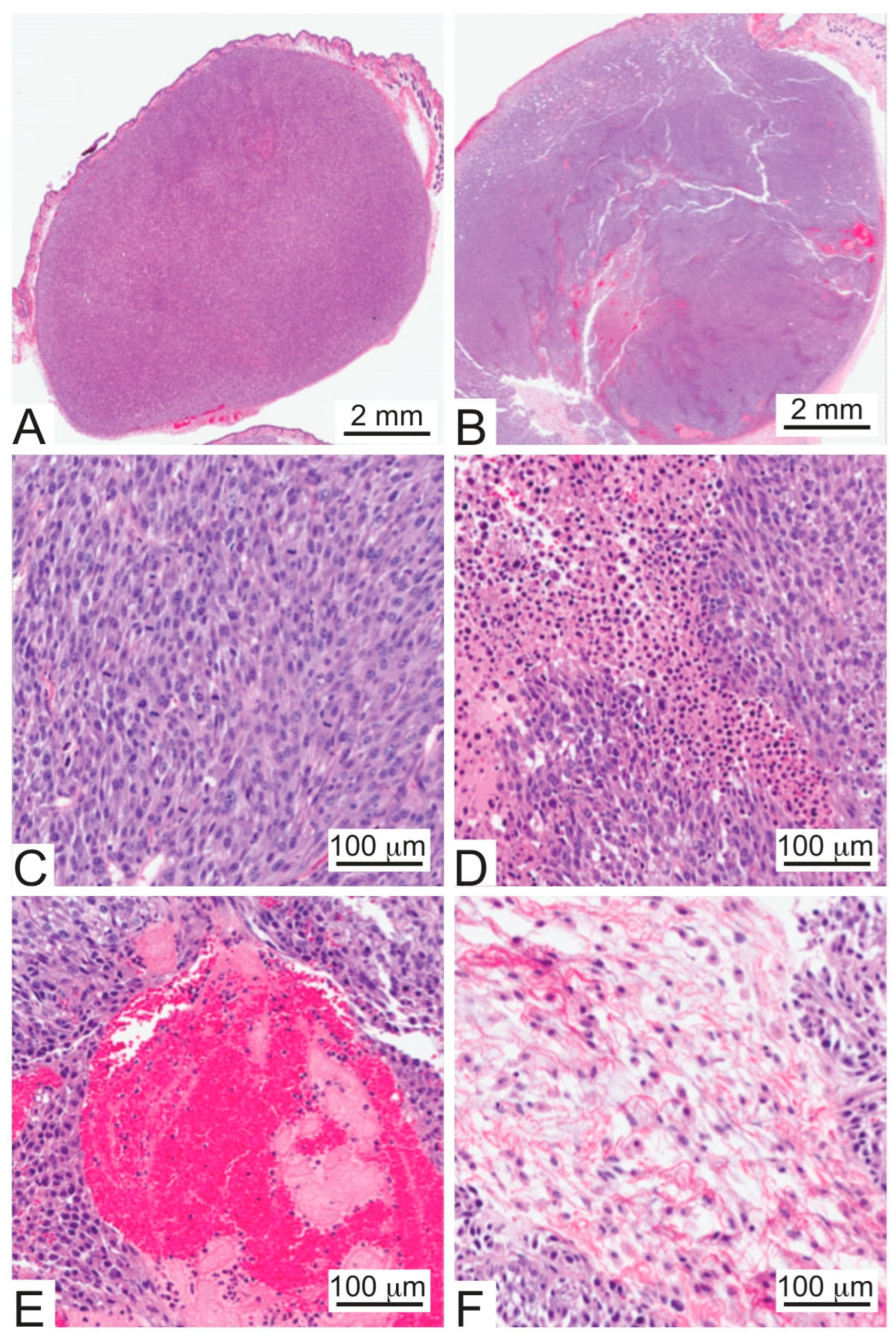
2.4. Tumorigenicity Assays
2.5. CGH and mRNA Profiling Arrays and Comparisons to TCGA Datasets
2.6. Fluorescence In Situ Hybridization (FISH) Karyotyping
2.7. Real-Time Quantitative PCR
2.8. SDS-PAGE, Immunoblotting, HGF Affinity Chromatography and Ultrafiltration
3. Results
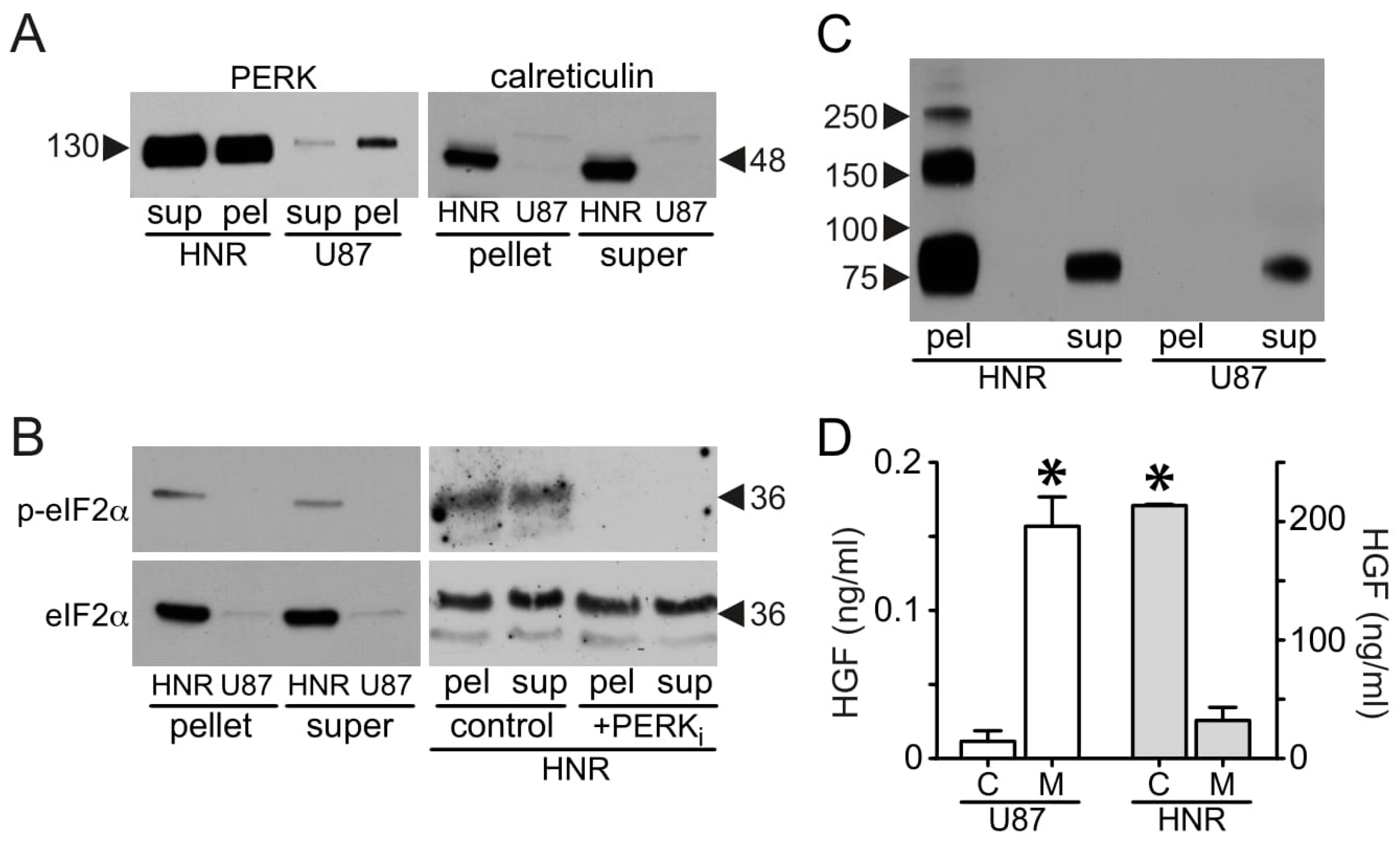
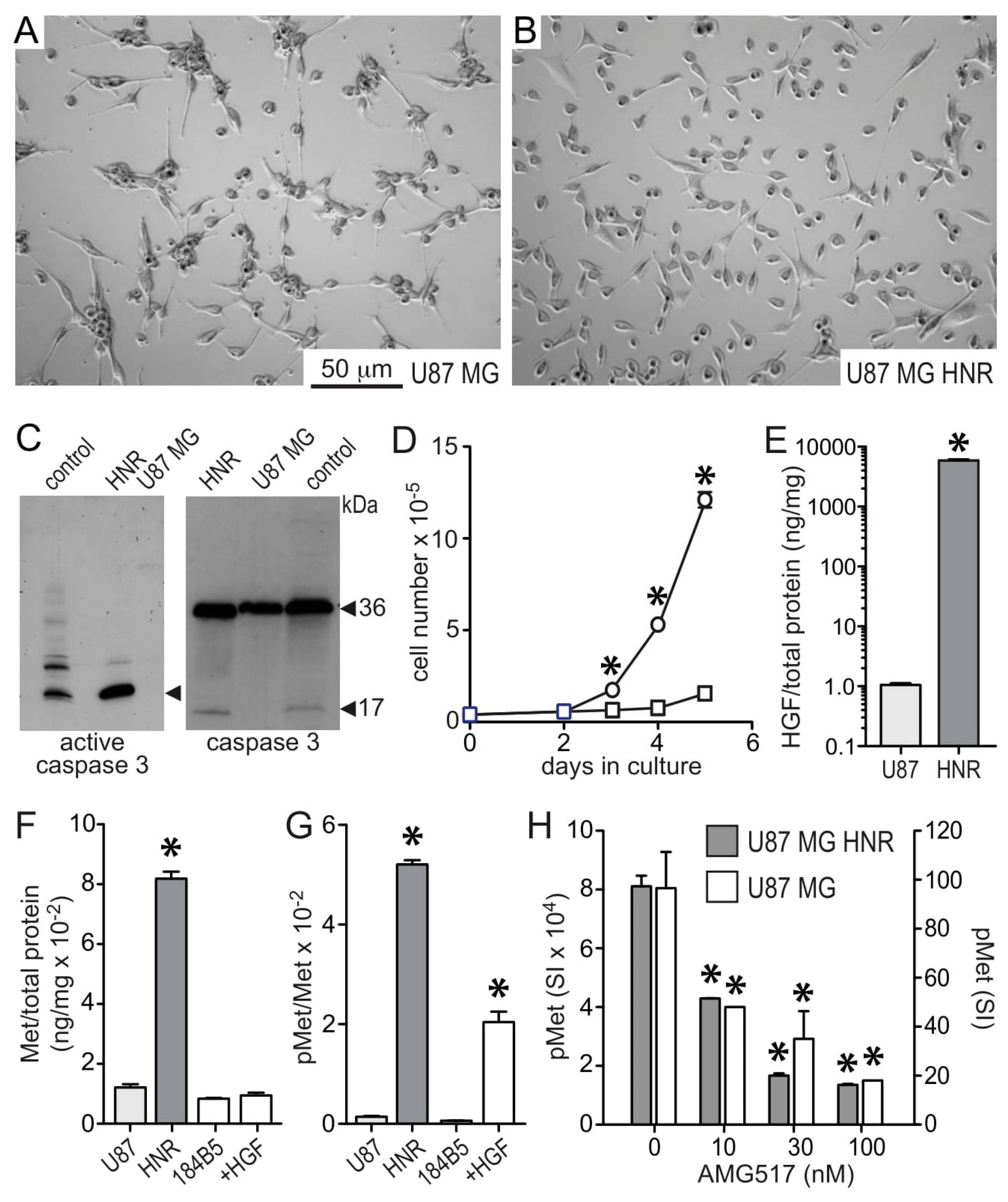
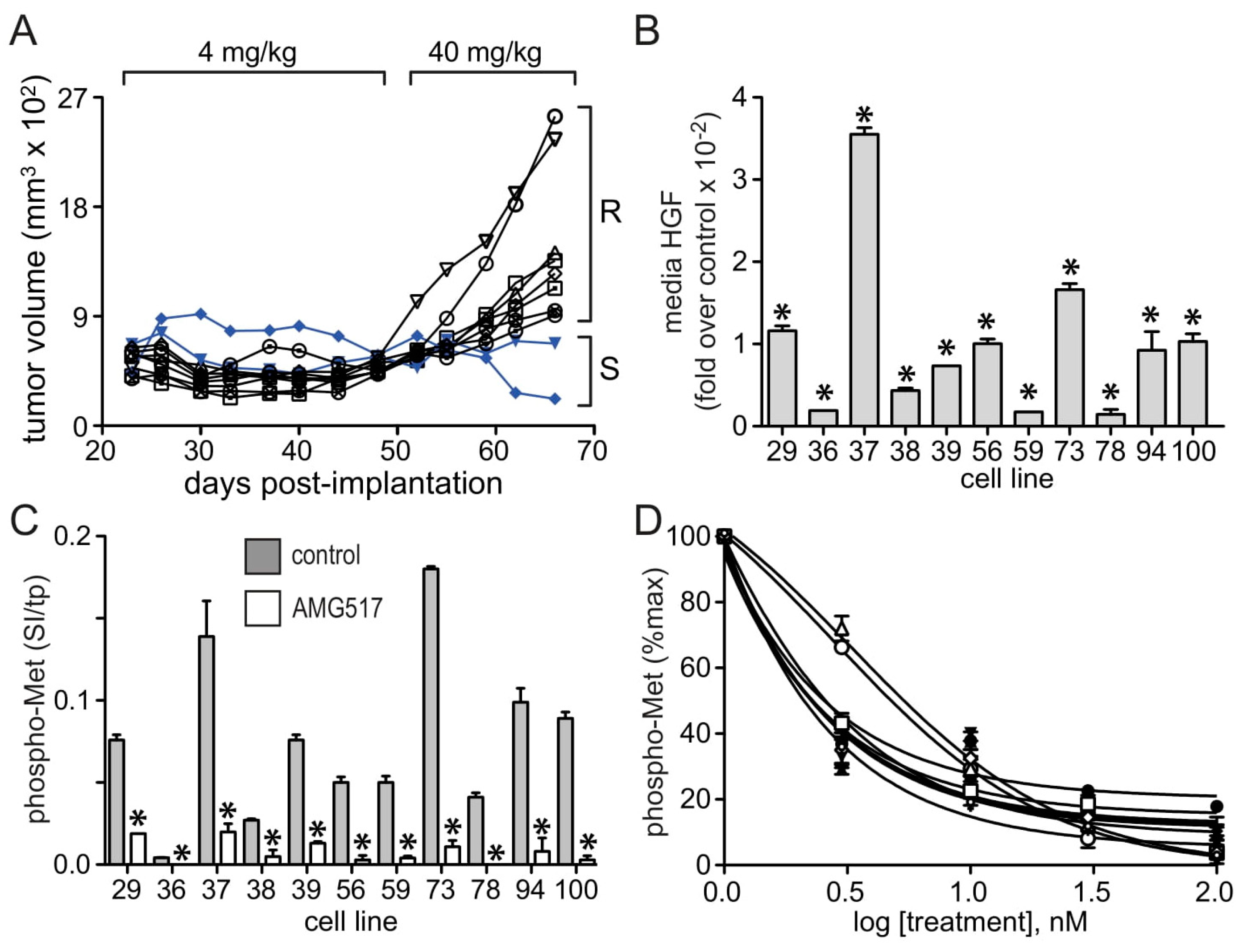
3.1. HGF and Met Superabundance in Rilotumumab-Resistant U87 MG Cells
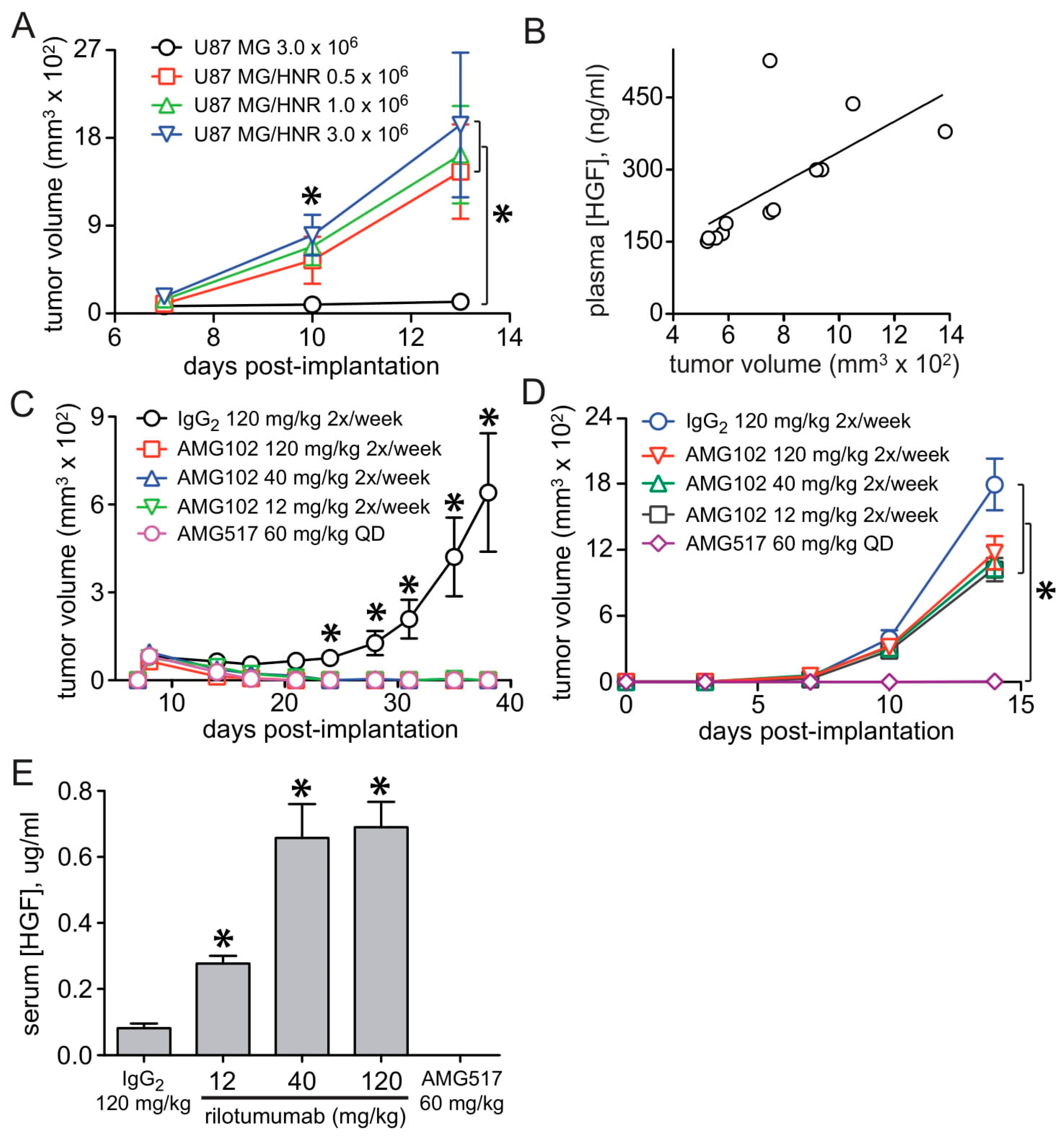
3.2. U87 MG/HNR Tumorigenesis Is Rilotumumab-Resistant Yet Remains MET-Pathway Dependent
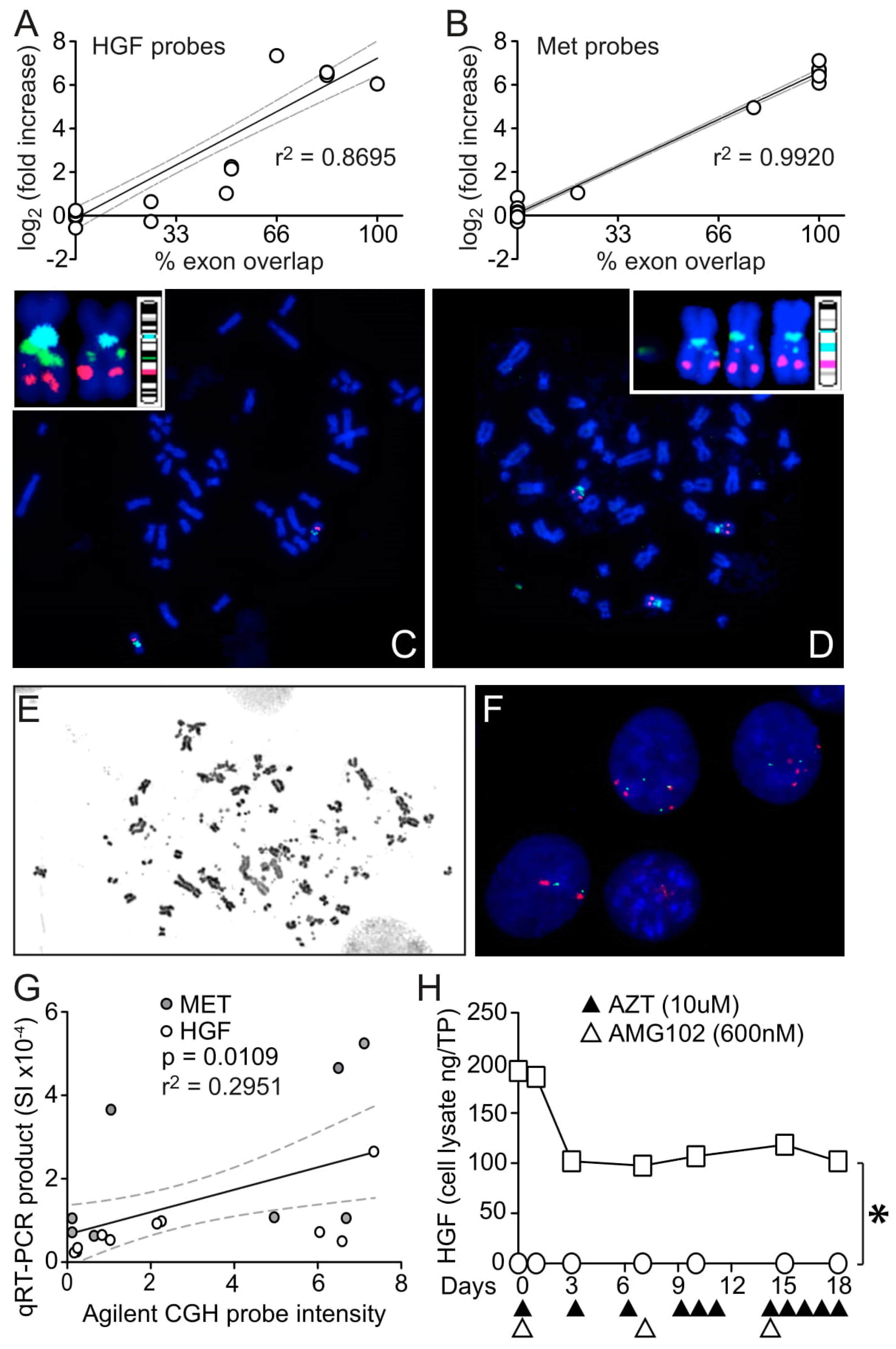
3.3. HGF and MET Gene Amplification in U87 MG/HNR
3.4. HGF Transcript Variant 5: A Possible but Unobserved Route to Resistance

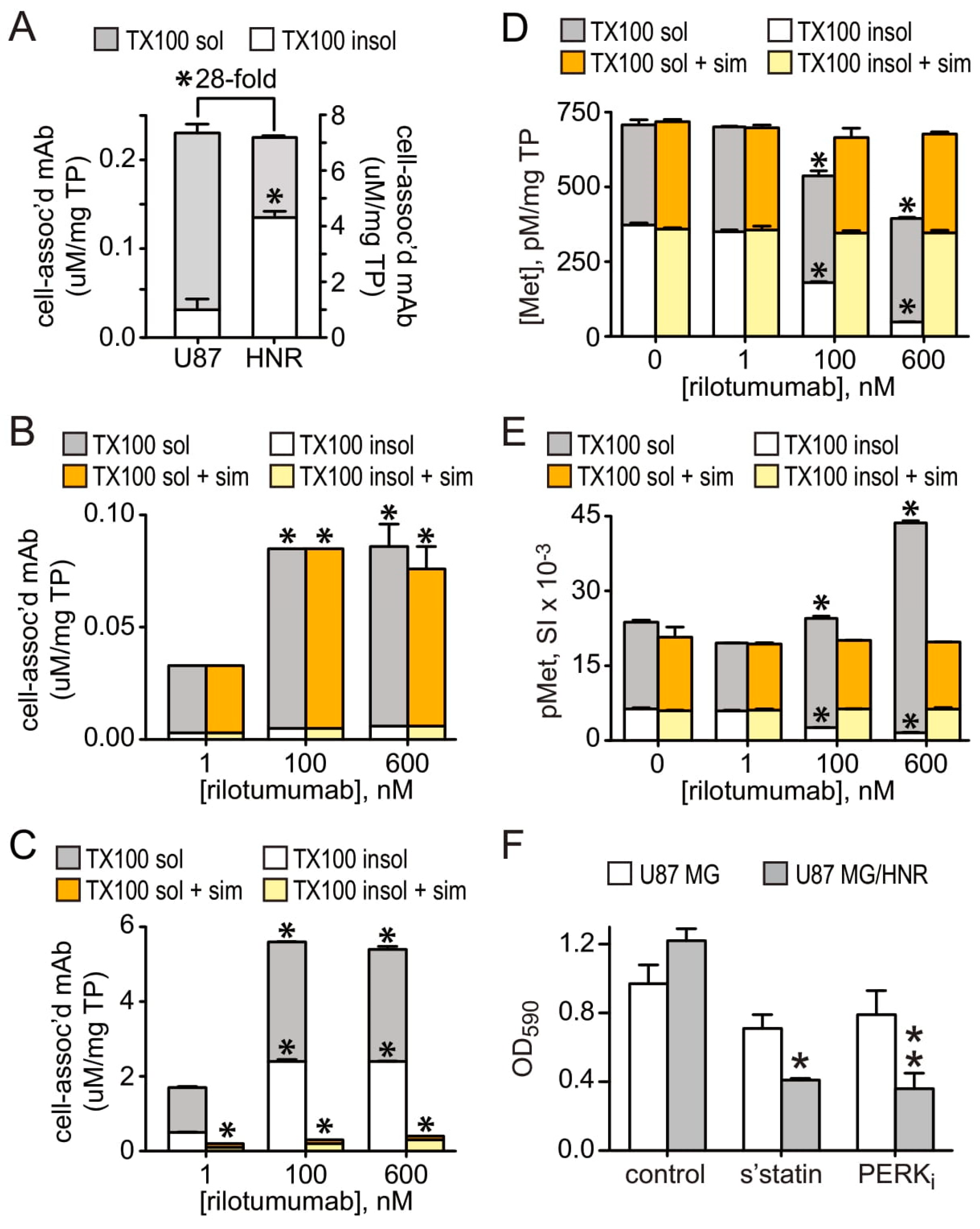
3.5. Mechanistic Insights from Gene Expression Profiling and Pathway Analysis
3.6. A Coordinated, Multiplex Route to Rilotumumab Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Murakami, I.; Katayama, T.; Tomizawa, K.; Osada, H.; Sekido, Y.; Maehara, Y.; Yatabe, Y.; Mitsudomi, T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin. Cancer Res. 2010, 16, 5489–5498. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef] [Green Version]
- Cappuzzo, F.; Jänne, P.A.; Skokan, M.; Finocchiaro, G.; Rossi, E.; Ligorio, C.; Zucali, P.A.; Terracciano, L.; Toschi, L.; Roncalli, M.; et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann. Oncol. 2009, 20, 298–304. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Romano, G.; Di Leva, G.; Nuovo, G.; Jeon, Y.J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2011, 18, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Takeuchi, S.; Kita, K.; Bando, H.; Nakamura, T.; Matsumoto, K.; Yano, S. Hepatocyte growth factor induces resistance to anti-epidermal growth factor receptor antibody in lung cancer. J. Thorac. Oncol. 2012, 7, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Garassino, M.C.; Mok, T.; Mitsudomi, T. Treatment strategies and outcomes for patients with EGFR-mutant non-small cell lung cancer resistant to EGFR tyrosine kinase inhibitors: Focus on novel therapies. Lung Cancer 2022, 21, 41–51. [Google Scholar] [CrossRef]
- Shah, M.P.; Neal, J.W. Targeting Acquired and Intrinsic Resistance Mechanisms in Epidermal Growth Factor Receptor Mutant Non-Small-Cell Lung Cancer. Drugs 2022, 82, 649–662. [Google Scholar] [CrossRef]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. National Cancer Institute GDC Data Portal: TCGA-GBM. 2022. Available online: https://portal.gdc.cancer.gov/projects/TCGA-GBM (accessed on 9 January 2023).
- Moriyama, T.; Kataoka, H.; Koono, M.; Wakisaka, S. Expression of hepatocyte growth factor/scatter factor and its receptor c-Met in brain tumors: Evidence for a role in progression of astrocytic tumors. Int. J. Mol. Med. 1999, 3, 531–536. [Google Scholar] [CrossRef]
- Li, Y.; Lal, B.; Kwon, S.; Fan, X.; Saldanha, U.; Reznik, T.E.; Kuchner, E.B.; Eberhart, C.; Laterra, J.; Abounader, R. The scatter factor/hepatocyte growth factor: C-met pathway in human embryonal central nervous system tumor malignancy. Cancer Res. 2005, 65, 9355–9362. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.J.; Wang, L.; Su, Y.C.; Gillespie, G.Y.; Salhotra, A.; Lal, B.; Laterra, J. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clin. Cancer Res. 2006, 12, 1292–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol. 2005, 7, 436–451. [Google Scholar] [CrossRef]
- Garcia-Navarrete, R.; Garcia, E.; Arrieta, O.; Sotelo, J. Hepatocyte growth factor in cerebrospinal fluid is associated with mortality and recurrence of glioblastoma, and could be of prognostic value. J. Neurooncol. 2010, 97, 347–351. [Google Scholar] [CrossRef]
- De Bacco, F.; Casanova, E.; Medico, E.; Pellegatta, S.; Orzan, F.; Albano, R.; Luraghi, P.; Reato, G.; D’Ambrosio, A.; Porrati, P.; et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012, 72, 4537–4550. [Google Scholar] [CrossRef]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006, 66, 1721–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, T.; Schmidt, N.O.; Eckerich, C.; Fillbrandt, R.; Merchant, M.; Schwall, R.; Westphal, M.; Lamszus, K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin. Cancer Res. 2006, 12, 6144–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guessous, F.; Zhang, Y.; diPierro, C.; Marcinkiewicz, L.; Sarkaria, J.; Schiff, D.; Buchanan, S.; Abounader, R. An orally bioavailable c-Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anticancer Agents Med. Chem. 2010, 10, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Bradley, R.; Kang, L.; Koeman, J.; Ascierto, M.L.; Worschech, A.; De Giorgi, V.; Wang, E.; Kefene, L.; Su, Y.; et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc. Natl. Acad. Sci. USA 2012, 109, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.T.; Sun, J.; Rex, K.; Radinsky, R.; Kendall, R.; Coxon, A.; Burgess, T.L. AMG 102, a fully human anti-hepatocyte growth factor/scatter factor neutralizing antibody, enhances the efficacy of temozolomide or docetaxel in U-87 MG cells and xenografts. Clin. Cancer Res. 2007, 13, 6735–6742. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.F.; Xie, Q.; Zhang, Y.W.; Su, Y.; Zhao, P.; Cao, B.; Furge, K.; Sun, J.; Rex, K.; Osgood, T.; et al. Therapeutic potential of hepatocyte growth factor/scatter factor neutralizing antibodies: Inhibition of tumor growth in both autocrine and paracrine hepatocyte growth factor/scatter factor: c-Met-driven models of leiomyosarcoma. Mol. Cancer Ther. 2009, 8, 2803–2810. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, I.M.; Scott, T.; Tandle, A.T.; Burgan, W.E.; Burgess, T.L.; Tofilon, P.J.; Camphausen, K. Radiosensitization of glioma cells by modulation of Met signalling with the hepatocyte growth factor neutralizing antibody, AMG102. J. Cell. Mol. Med. 2011, 15, 1999–2006. [Google Scholar] [CrossRef]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, F.; Wright, C.; Bottaro, D.P. Experimental Cancer Therapeutics Targeting the Hepatocyte Growth Factor/Met Signaling Pathway. 2018. Available online: https://ccrod.cancer.gov/confluence/display/CCRHGF/Home (accessed on 9 January 2023).
- Cecchi, F.; Pajalunga, D.; Fowler, C.A.; Uren, A.; Rabe, D.C.; Peruzzi, B.; Macdonald, N.J.; Blackman, D.K.; Stahl, S.J.; Byrd, R.A.; et al. Targeted disruption of heparan sulfate interaction with hepatocyte and vascular endothelial growth factors blocks normal and oncogenic signaling. Cancer Cell 2012, 22, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Boezio, A.A.; Berry, L.; Albrecht, B.K.; Bauer, D.; Bellon, S.F.; Bode, C.; Chen, A.; Choquette, D.; Dussault, I.; Fang, M.; et al. Discovery and optimization of potent and selective triazolopyridazine series of c-Met inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6307–6312. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; DeFrances, M.C.; Zou, C.; Johnson, C.; Ferrell, R.; Zarnegar, R. Somatic mutation and functional polymorphism of a novel regulatory element in the HGF gene promoter causes its aberrant expression in human breast cancer. J. Clin. Investig. 2009, 119, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Cooke, S.L.; Shlien, A.; Marshall, J.; Pipinikas, C.P.; Martincorena, I.; Tubio, J.M.; Li, Y.; Menzies, A.; Mudie, L.; Ramakrishna, M.; et al. Processed pseudogenes acquired somatically during cancer development. Nat. Commun. 2014, 5, 3644. [Google Scholar] [CrossRef] [Green Version]
- Brighenti, M. MicroRNA and MET in lung cancer. Ann. Transl. Med. 2015, 3, 68. [Google Scholar] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, G.M. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989, 49, 1333–1340. [Google Scholar] [PubMed]
- Nielsen, J.L.; Walsh, J.T.; Degen, D.R.; Drabek, S.M.; McGill, J.R.; von Hoff, D.D. Evidence of gene amplification in the form of double minute chromosomes is frequently observed in lung cancer. Cancer Genet. Cytogenet. 1993, 65, 120–124. [Google Scholar] [CrossRef]
- Shimizu, N. Molecular mechanisms of the origin of micronuclei from extrachromosomal elements. Mutagenesis 2011, 26, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Burgess, T.L.; Sun, J.; Meyer, S.; Tsuruda, T.S.; Sun, J.; Elliott, G.; Chen, Q.; Haniu, M.; Barron, W.F.; Juan, T.; et al. Biochemical characterization of AMG 102: A neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol. Cancer Ther. 2010, 9, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Kirchhofer, D.; Yao, X.; Peek, M.; Eigenbrot, C.; Lipari, M.T.; Billeci, K.L.; Maun, H.R.; Moran, P.; Santell, L.; Wiesmann, C.; et al. Structural and functional basis of the serine protease-like hepatocyte growth factor beta-chain in Met binding and signaling. J. Biol. Chem. 2004, 279, 39915–39924. [Google Scholar] [CrossRef] [Green Version]
- Kirchhofer, D.; Lipari, M.T.; Santell, L.; Billeci, K.L.; Maun, H.R.; Sandoval, W.N.; Moran, P.; Ridgway, J.; Eigenbrot, C.; Lazarus, R.A. Utilizing the activation mechanism of serine proteases to engineer hepatocyte growth factor into a Met antagonist. Proc. Natl. Acad. Sci. USA 2007, 104, 5306–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. J. Cell. Physiol. 2020, 235, 5867–5881. [Google Scholar] [CrossRef] [PubMed]
- Low, J.Y.; Laiho, M. Caveolae-associated molecules, tumor stroma, and cancer drug resistance: Current findings and future perspectives. Cancers 2022, 25, 589. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.A.; Klose, C.; Simons, K. Lipid-dependent protein sorting at the trans-Golgi network. Biochim. Biophys. Acta 2012, 1821, 1059–1067. [Google Scholar] [CrossRef]
- Zuidscherwoude, M.; de Winde, C.M.; Cambi, A.; van Spriel, A.B. Microdomains in the membrane landscape shape antigen-presenting cell function. J. Leukoc. Biol. 2014, 95, 251–263. [Google Scholar] [CrossRef]
- Grao-Cruces, E.; Lopez-Enriquez, S.; Martin, M.E.; Montserrat-de la Paz, S. High-density lipoproteins and immune response: A review. Int. J. Biol. Macromol. 2022, 195, 117–123. [Google Scholar] [CrossRef]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergani, E.; Vallacchi, V.; Frigerio, S.; Deho, P.; Mondellini, P.; Perego, P.; Cassinelli, G.; Lanzi, C.; Testi, M.A.; Rivoltini, L.; et al. Identification of MET and SRC activation in melanoma cell lines showing primary resistance to PLX4032. Neoplasia 2011, 13, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.T.; Kim, H.; Liska, D.; Gao, S.; Christensen, J.G.; Weiser, M.R. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol. Cancer Ther. 2012, 11, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.; Mambetsariev, I.; Fricke, J.; Chawla, N.; Nam, A.; Pharaon, R.; Salgia, R. MET receptor in oncology: From biomarker to therapeutic target. Adv. Cancer Res. 2020, 147, 259–301. [Google Scholar]
- Dussault, I.; Bellon, S.F. c-Met inhibitors with different binding modes: Two is better than one. Cell Cycle 2008, 7, 1157–1160. [Google Scholar] [CrossRef]
- Puccini, A.; Marín-Ramos, N.I.; Bergamo, F.; Schirripa, M.; Lonardi, S.; Lenz, H.J.; Loupakis, F.; Battaglin, F. Safety and tolerability of c-MET inhibitors in cancer. Drug Saf. 2019, 42, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Passiglia, F.; Van Der Steen, N.; Raez, L.; Pauwels, P.; Gil-Bazo, I.; Santos, E.; Santini, D.; Tesoriere, G.; Russo, A.; Bronte, G.; et al. The role of cMet in non-small cell lung cancer resistant to EGFR-inhibitors: Did we really find the target? Curr. Drug Targets 2014, 15, 1284–1292. [Google Scholar] [CrossRef]
- Wang, C.; Cao, Y.; Yang, C.; Bernards, R.; Qin, W. Exploring liver cancer biology through functional genetic screens. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 690–704. [Google Scholar] [CrossRef]
- Elshazly, A.M.; Gewirtz, D.A. An overview of resistance to Human epidermal growth factor receptor 2 (Her2) targeted therapies in breast cancer. Cancer Drug Resist. 2022, 5, 472–486. [Google Scholar] [CrossRef]
- Parchment, R.E.; Doroshow, J.H. Pharmacodynamic endpoints as clinical trial objectives to answer important questions in oncology drug development. Semin. Oncol. 2016, 43, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, F.; Giovannetti, E.; Peters, G.J.; Firuzi, O. Combination of HGF/MET-targeting agents and other therapeutic strategies in cancer. Crit. Rev. Oncol. Hematol. 2021, 160, 103234. [Google Scholar] [CrossRef]
- Rodríguez-Camacho, A.; Flores-Vázquez, J.G.; Moscardini-Martelli, J.; Torres-Ríos, J.A.; Olmos-Guzmán, A.; Ortiz-Arce, C.S.; Cid-Sánchez, D.R.; Pérez, S.R.; Macías-González, M.D.S.; Hernández-Sánchez, L.C.; et al. Glioblastoma treatment: State-of-the-art and future perspectives. Int. J. Mol. Sci. 2022, 23, 7207. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Li, S.; Wu, X.; Diao, S.; Zhang, G.; He, H.; Bian, L.; Lu, Y. Cellular origin of glioblastoma and its implication in precision therapy. Cell. Mol. Immunol. 2018, 15, 737–739. [Google Scholar] [CrossRef] [Green Version]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef]
- Metcalfe, C.; de Sauvage, F.J. Hedgehog fights back: Mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011, 71, 5057–5061. [Google Scholar] [CrossRef] [Green Version]
- Bedard, P.L.; Azambuja, E.D.; Cardoso, F. Beyond Trastuzumab: Overcoming resistance to targeted HER-2 therapy in breast cancer. Curr. Cancer Drug Targ. 2009, 9, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, I.V.; Paraiso, K.H.T.; Smalley, K.S.M. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 2011, 82, 201–209. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cecchi, F.; Rex, K.; Schmidt, J.; Vocke, C.D.; Lee, Y.H.; Burkett, S.; Baker, D.; Damore, M.A.; Coxon, A.; Burgess, T.L.; et al. Rilotumumab Resistance Acquired by Intracrine Hepatocyte Growth Factor Signaling. Cancers 2023, 15, 460. https://doi.org/10.3390/cancers15020460
Cecchi F, Rex K, Schmidt J, Vocke CD, Lee YH, Burkett S, Baker D, Damore MA, Coxon A, Burgess TL, et al. Rilotumumab Resistance Acquired by Intracrine Hepatocyte Growth Factor Signaling. Cancers. 2023; 15(2):460. https://doi.org/10.3390/cancers15020460
Chicago/Turabian StyleCecchi, Fabiola, Karen Rex, Joanna Schmidt, Cathy D. Vocke, Young H. Lee, Sandra Burkett, Daniel Baker, Michael A. Damore, Angela Coxon, Teresa L. Burgess, and et al. 2023. "Rilotumumab Resistance Acquired by Intracrine Hepatocyte Growth Factor Signaling" Cancers 15, no. 2: 460. https://doi.org/10.3390/cancers15020460