
Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma

 , , , , , , , ,
, , , , , , , ,  , , ,
, , ,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Characteristics of the Index Patient
2.2. Clonal Immunoglobulin (IG) Quantification
2.3. Fluorescent In Situ Hybridization (FISH)
2.4. Whole Exome Sequencing (WES)
2.5. Exome Sequencing Validation
2.6. Functional Validation of the PSMC2 Y429S Point Mutation
2.7. Meta-Analysis of WGS/WES Cohorts
2.8. Structural Analysis
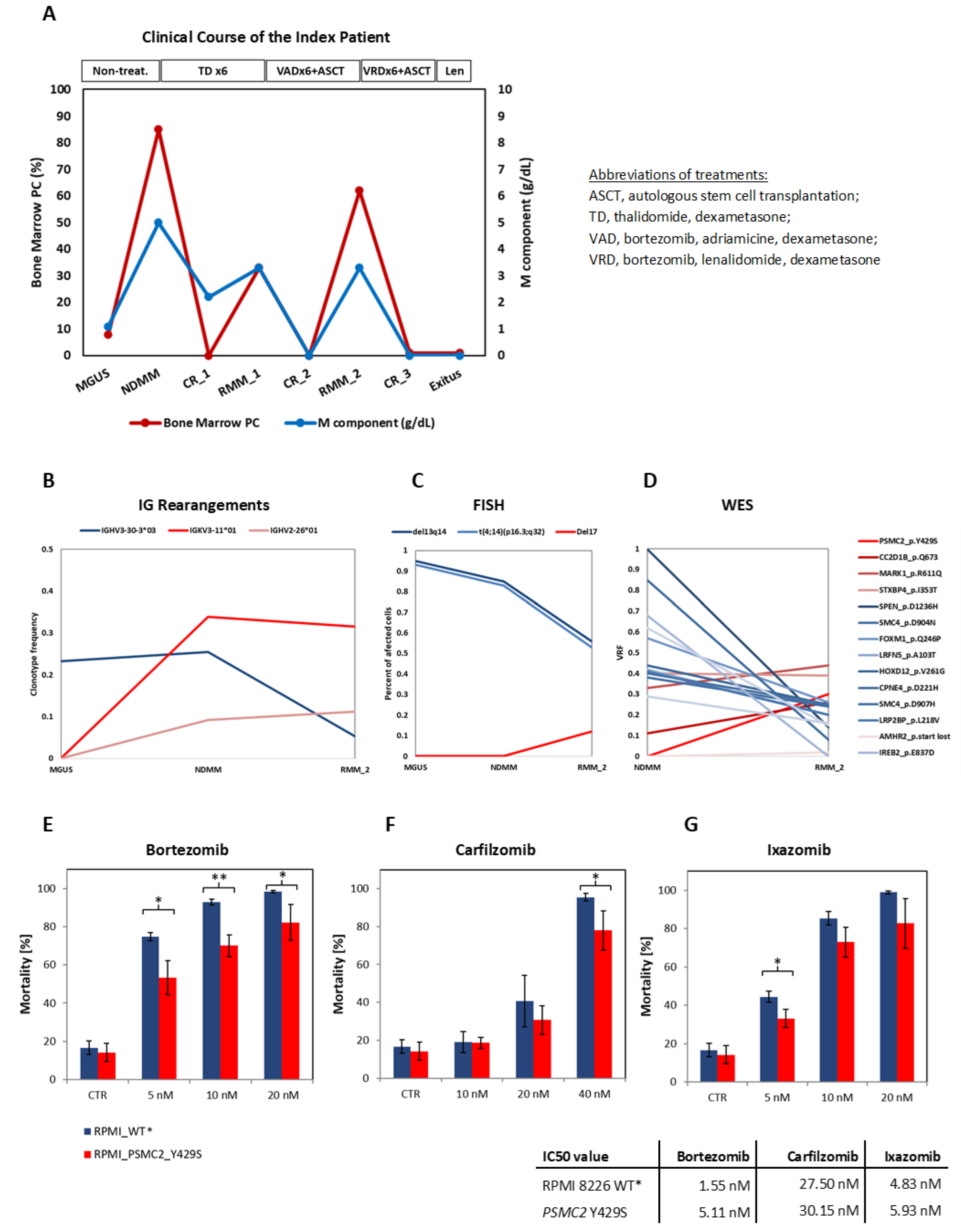
3. Results
3.1. Clinical Course of the Patient
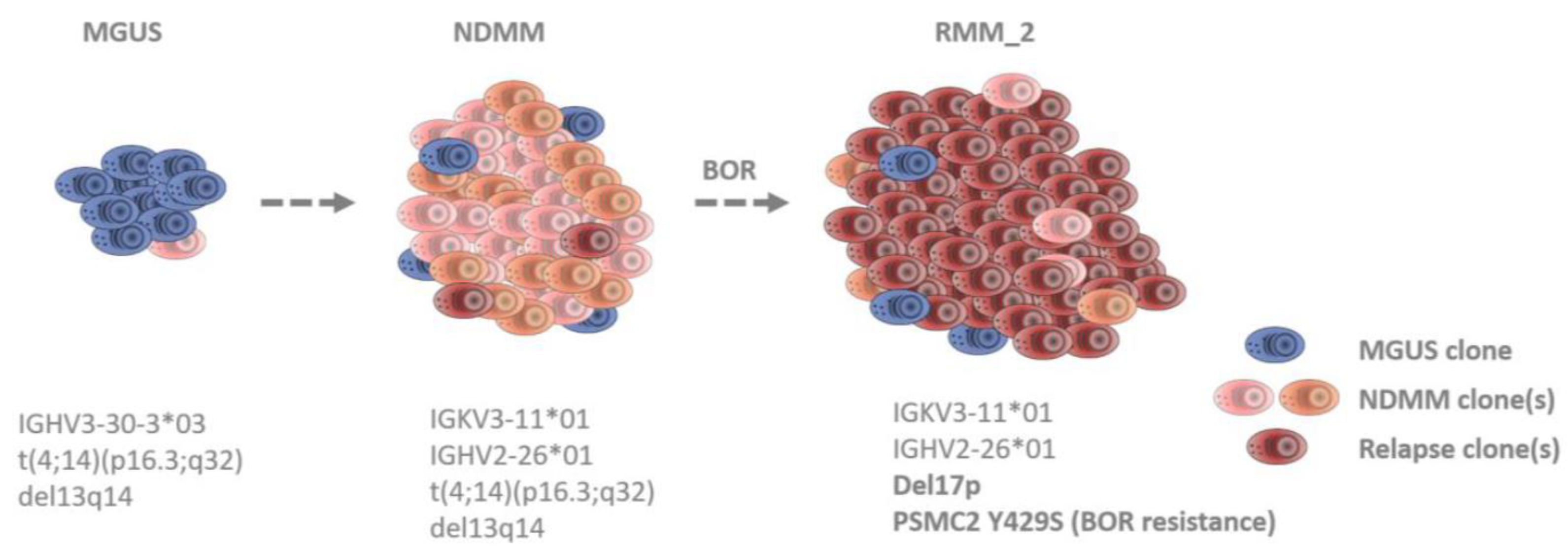
3.2. Identification of Clonal Diversity in the Index Patient Using IG Rearrangement Quantification, FISH, and WES
3.3. The PSMC2 Y429S Single Nucleotide Variant Mediates PI Resistance In Vitro
3.4. Incidence of Genomic Alterations in PSMC Family Member Genes
3.5. Prediction of the Structural Impact of the Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kunjappu, M.J.; Hochstrasser, M. Assembly of the 20S proteasome. Biochim. Biophys. Acta 2014, 1843, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.T.; de Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351.e3. [Google Scholar] [CrossRef] [PubMed]
- VanderLinden, R.T.; Hemmis, C.W.; Yao, T.; Robinson, H.; Hill, C.P. Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem. 2017, 292, 9493–9504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, T.J.; Wu, C.C.; Phan, N.N.; Liu, Y.H.; Ta, H.D.K.; Anuraga, G.; Wu, Y.F.; Lee, K.H.; Chuang, J.Y.; Wang, C.Y. Prognoses and genomic analyses of proteasome 26S subunit, ATPase (PSMC) family genes in clinical breast cancer. Aging 2021, 13, 17970. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nun, S.; Glickman, M.H. Proteasomal AAA-ATPases: Structure and function. Biochim. Biophys. Acta 2012, 1823, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Besche, H.C.; Peth, A.; Goldberg, A.L. Getting to first base in proteasome assembly. Cell 2009, 138, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Laubach, J.; Richardson, P.; Anderson, K. Multiple myeloma. Annu. Rev. Med. 2011, 62, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Garcia, T.A.; Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Mitsiades, C.; Anderson, K.C.; Richardson, P.G. The power of proteasome inhibition in multiple myeloma. Expert Rev. Proteom. 2018, 15, 1033–1052. [Google Scholar] [CrossRef]
- Rajan, A.M.; Rajkumar, S.V. Treatment of newly diagnosed myeloma: Bortezomib-based triplet. Semin. Oncol. 2016, 43, 700–702. [Google Scholar] [CrossRef]
- Barrio, S.; Stuhmer, T.; Da-Via, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Laubach, J.; Garderet, L.; Mahindra, A.; Gahrton, G.; Caers, J.; Sezer, O.; Voorhees, P.; Leleu, X.; Johnsen, H.E.; Streetly, M.; et al. Management of relapsed multiple myeloma: Recommendations of the International Myeloma Working Group. Leukemia 2016, 30, 1005–1017. [Google Scholar] [CrossRef]
- Guang, M.H.Z.; McCann, A.; Bianchi, G.; Zhang, L.; Dowling, P.; Bazou, D.; O’Gorman, P.; Anderson, K.C. Overcoming multiple myeloma drug resistance in the era of cancer ‘omics’. Leuk. Lymphoma 2018, 59, 542–561. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Sokol, E.; Jin, D.; Brune, Z.; Thiru, P.; Ghandi, M.; Garraway, L.A.; Gupta, P.B.; Santagata, S.; Whitesell, L.; et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Haertle, L.; Barrio, S.; Munawar, U.; Han, S.; Zhou, X.; Simicek, M.; Vogt, C.; Truger, M.; Alonso Fernandez, R.; Steinhardt, M.; et al. Single Nucleotide Variants and Epimutations Induce Proteasome Inhibitor Resistance in Multiple Myeloma. Clin. Cancer Res. 2023, 29, 279–288. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Mendillo, M.L.; Zhao, J.; Carette, J.E.; Merrill, P.H.; Cikes, D.; Varadarajan, M.; van Diemen, F.R.; Penninger, J.M.; Goldberg, A.L.; et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. Elife 2015, 4, e08467. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Maric, I.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife 2015, 4, e08153. [Google Scholar] [CrossRef]
- Shi, C.X.; Kortum, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [Green Version]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 Promote Proteasome Inhibitor Resistance in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 1923–1935. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yedidi, R.S.; Wendler, P.; Enenkel, C. AAA-ATPases in Protein Degradation. Front. Mol. Biosci. 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lopez, J.; Sanchez-Vega, B.; Barrio, S.; Cuenca, I.; Ruiz-Heredia, Y.; Alonso, R.; Rapado, I.; Marin, C.; Cedena, M.T.; Paiva, B.; et al. Analytical and clinical validation of a novel in-house deep-sequencing method for minimal residual disease monitoring in a phase II trial for multiple myeloma. Leukemia 2017, 31, 1446–1449. [Google Scholar] [CrossRef]
- van Dongen, J.J.; Langerak, A.W.; Bruggemann, M.; Evans, P.A.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; Garcia-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, C.; Jara-Acevedo, M.; Corchete, L.A.; Castillo, D.; Ordonez, G.R.; Sarasquete, M.E.; Puig, N.; Martinez-Lopez, J.; Prieto-Conde, M.I.; Garcia-Alvarez, M.; et al. A Next-Generation Sequencing Strategy for Evaluating the Most Common Genetic Abnormalities in Multiple Myeloma. J. Mol. Diagn. 2017, 19, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Ross, F.M.; Avet-Loiseau, H.; Ameye, G.; Gutierrez, N.C.; Liebisch, P.; O’Connor, S.; Dalva, K.; Fabris, S.; Testi, A.M.; Jarosova, M.; et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica 2012, 97, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Truger, M.S.; Duell, J.; Zhou, X.; Heimeshoff, L.; Ruckdeschel, A.; John, M.; Riedel, A.; Huper, S.; Peter, J.; Walter, W.; et al. Single- and double-hit events in genes encoding immune targets before and after T cell-engaging antibody therapy in MM. Blood Adv. 2021, 5, 3794–3798. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Giesen, N.; Paramasivam, N.; Toprak, U.H.; Huebschmann, D.; Xu, J.; Uhrig, S.; Samur, M.; Bahr, S.; Frohlich, M.; Mughal, S.S.; et al. Comprehensive genomic analysis of refractory multiple myeloma reveals a complex mutational landscape associated with drug resistance and novel therapeutic vulnerabilities. Haematologica 2022, 107, 1891–1901. [Google Scholar] [CrossRef]
- Ziccheddu, B.; Biancon, G.; Bagnoli, F.; De Philippis, C.; Maura, F.; Rustad, E.H.; Dugo, M.; Devecchi, A.; De Cecco, L.; Sensi, M.; et al. Integrative analysis of the genomic and transcriptomic landscape of double-refractory multiple myeloma. Blood Adv. 2020, 4, 830–844. [Google Scholar] [CrossRef]
- Samur, M.K.; Aktas Samur, A.; Fulciniti, M.; Szalat, R.; Han, T.; Shammas, M.; Richardson, P.; Magrangeas, F.; Minvielle, S.; Corre, J.; et al. Genome-Wide Somatic Alterations in Multiple Myeloma Reveal a Superior Outcome Group. J. Clin. Oncol. 2020, 38, 3107–3118. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Huang, X.; Luan, B.; Wu, J.; Shi, Y. An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 2016, 23, 778–785. [Google Scholar] [CrossRef]
- Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol. 2003, 333, 781–815. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Brioli, A.; Melchor, L.; Cavo, M.; Morgan, G.J. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br. J. Haematol. 2014, 165, 441–454. [Google Scholar] [CrossRef]
- Haertle, L.; Barrio, S.; Munawar, U.; Han, S.; Zhou, X.; Vogt, C.; Fernandez, R.A.; Bittrich, M.; Ruiz-Heredia, Y.; Da Via, M.; et al. Cereblon enhancer methylation and IMiD resistance in multiple myeloma. Blood 2021, 138, 1721–1726. [Google Scholar] [CrossRef]
- Magrangeas, F.; Avet-Loiseau, H.; Gouraud, W.; Lode, L.; Decaux, O.; Godmer, P.; Garderet, L.; Voillat, L.; Facon, T.; Stoppa, A.M.; et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia 2013, 27, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lopez, J.; Fulciniti, M.; Barrio, S.; Carlton, V.; Moorhead, M.; Lahuerta, J.J.; Anderson, K.C.; Magrangeas, F.; Minvielle, S.; Avet-loiseau, H.; et al. Deep Sequencing Reveals Oligoclonality At The Immunoglobulin Locus In Multiple Myeloma Patients. Blood ASH Abstr. 2013, 122, 401. [Google Scholar] [CrossRef]
- Munawar, U.; Rasche, L.; Muller, N.; Vogt, C.; Da-Via, M.; Haertle, L.; Arampatzi, P.; Dietrich, S.; Roth, M.; Garitano-Trojaola, A.; et al. Hierarchy of mono- and bi-allelic TP53 alterations in Multiple Myeloma cell fitness. Blood 2019, 134, 836–840. [Google Scholar] [CrossRef]
- Sheffer, M.; Hu, Y.; Shalem, O.; Sanjana, N.; Dhimolea, E.; Sarkar, S.; Bariteau, M.A.; Aftab, B.T.; Groen, R.W.J.; Zhang, F.; et al. Genome-Scale Crispr-Cas9 Knockout Studies Reveal Mutifactorial and Functionally Overlapping Mechanisms of Myeloma Cell Resistance to Proteasome Inhibition. Blood 2014, 124, 273. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haertle, L.; Buenache, N.; Cuesta Hernández, H.N.; Simicek, M.; Snaurova, R.; Rapado, I.; Martinez, N.; López-Muñoz, N.; Sánchez-Pina, J.M.; Munawar, U.; et al. Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers 2023, 15, 532. https://doi.org/10.3390/cancers15020532
Haertle L, Buenache N, Cuesta Hernández HN, Simicek M, Snaurova R, Rapado I, Martinez N, López-Muñoz N, Sánchez-Pina JM, Munawar U, et al. Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers. 2023; 15(2):532. https://doi.org/10.3390/cancers15020532
Chicago/Turabian StyleHaertle, Larissa, Natalia Buenache, Hipólito Nicolás Cuesta Hernández, Michal Simicek, Renata Snaurova, Inmaculada Rapado, Nerea Martinez, Nieves López-Muñoz, José María Sánchez-Pina, Umair Munawar, and et al. 2023. "Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma" Cancers 15, no. 2: 532. https://doi.org/10.3390/cancers15020532
APA StyleHaertle, L., Buenache, N., Cuesta Hernández, H. N., Simicek, M., Snaurova, R., Rapado, I., Martinez, N., López-Muñoz, N., Sánchez-Pina, J. M., Munawar, U., Han, S., Ruiz-Heredia, Y., Colmenares, R., Gallardo, M., Sanchez-Beato, M., Piris, M. A., Samur, M. K., Munshi, N. C., Ayala, R., ... Martínez-López, J. (2023). Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers, 15(2), 532. https://doi.org/10.3390/cancers15020532