

Intranasal Delivery of Oncolytic Adenovirus XVir-N-31 via Optimized Shuttle Cells Significantly Extends Survival of Glioblastoma-Bearing Mice
 and
and 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viruses
2.2. Immunofluorescence and Microscopy
2.3. Tumor Volumetry
2.4. Animal Experiments
2.5. Statistical Analysis
3. Results
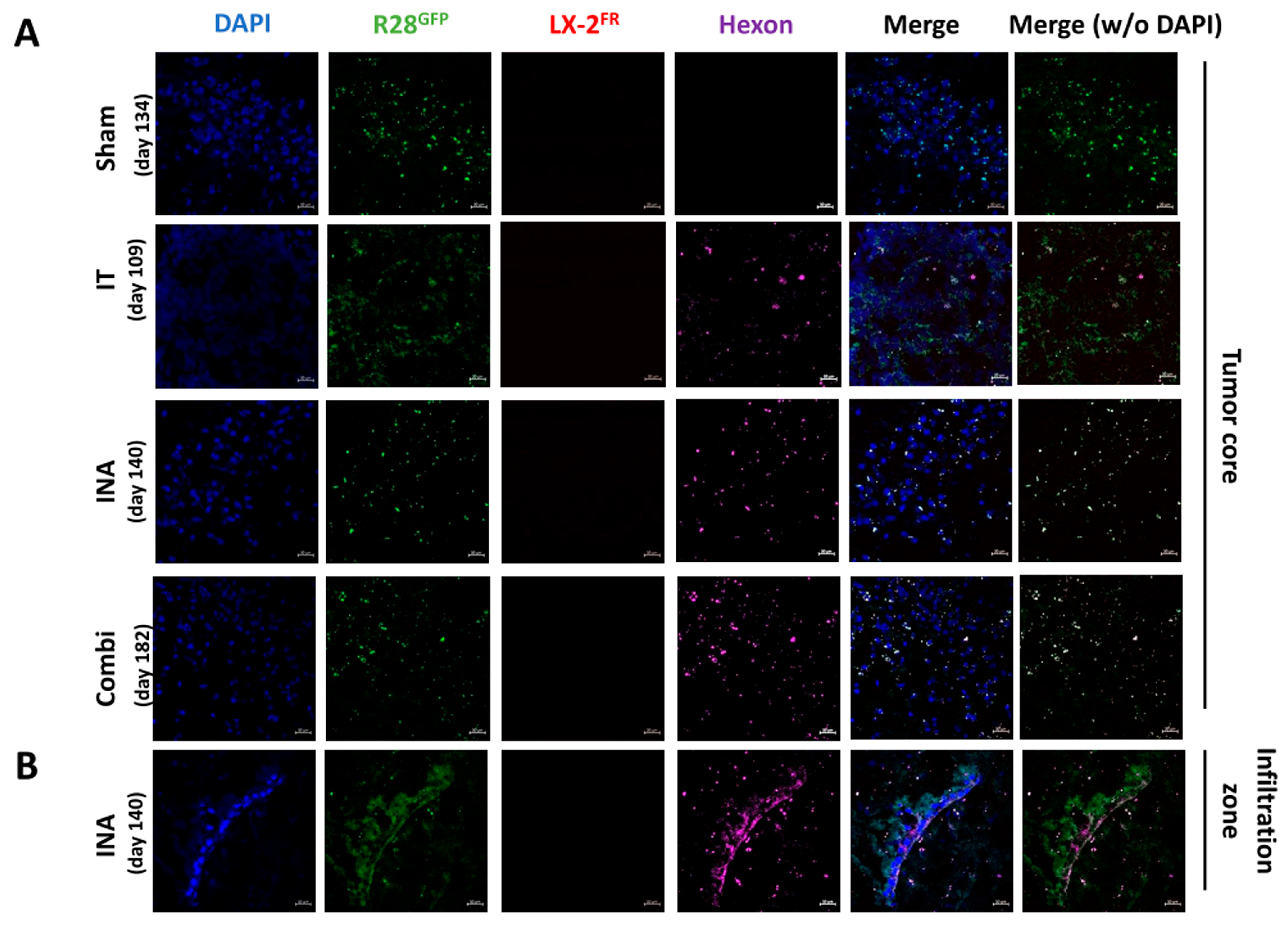
3.1. XVir-N-31 Reaches the GBM after INA of OV-Loaded LX-2FR Cells
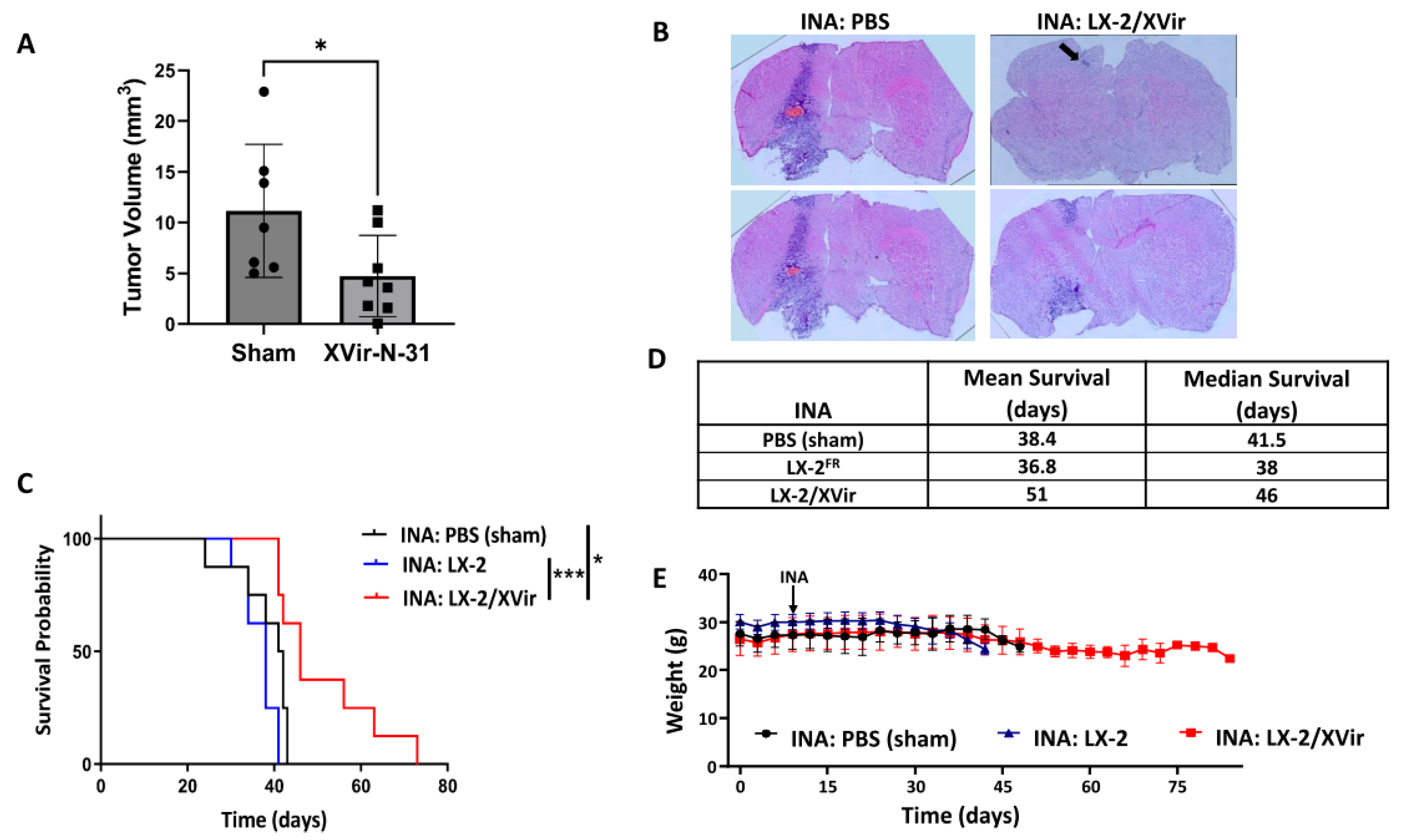
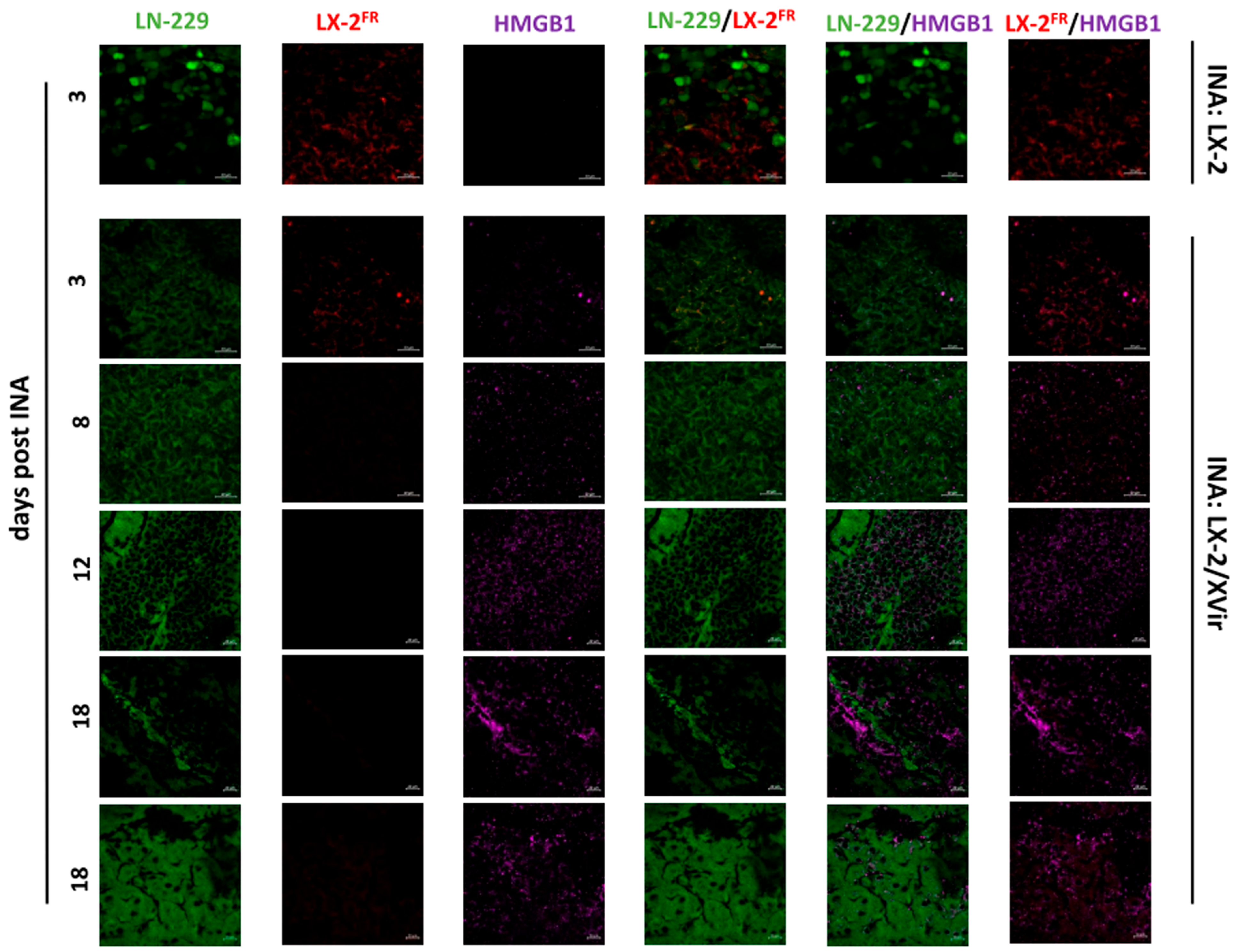
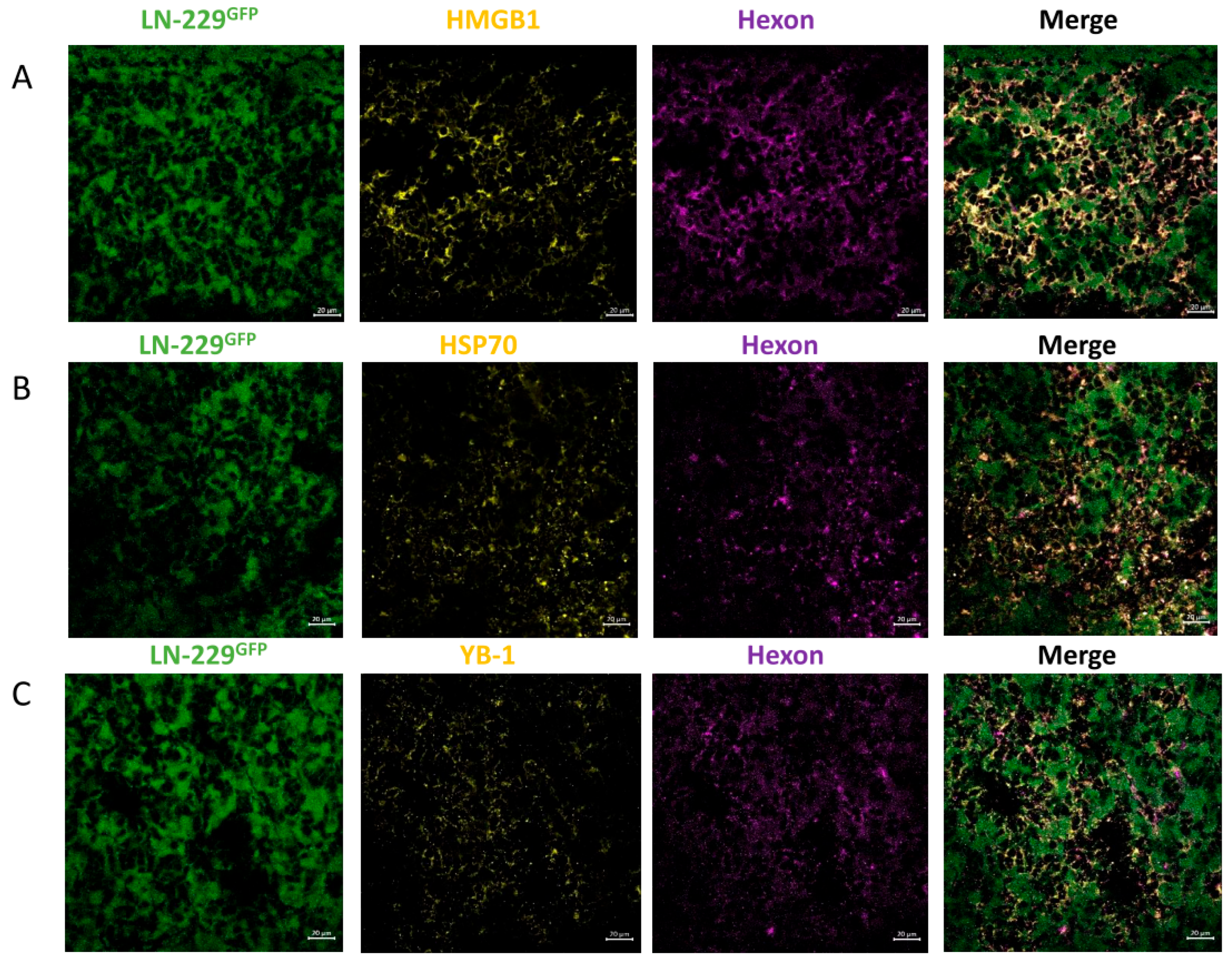
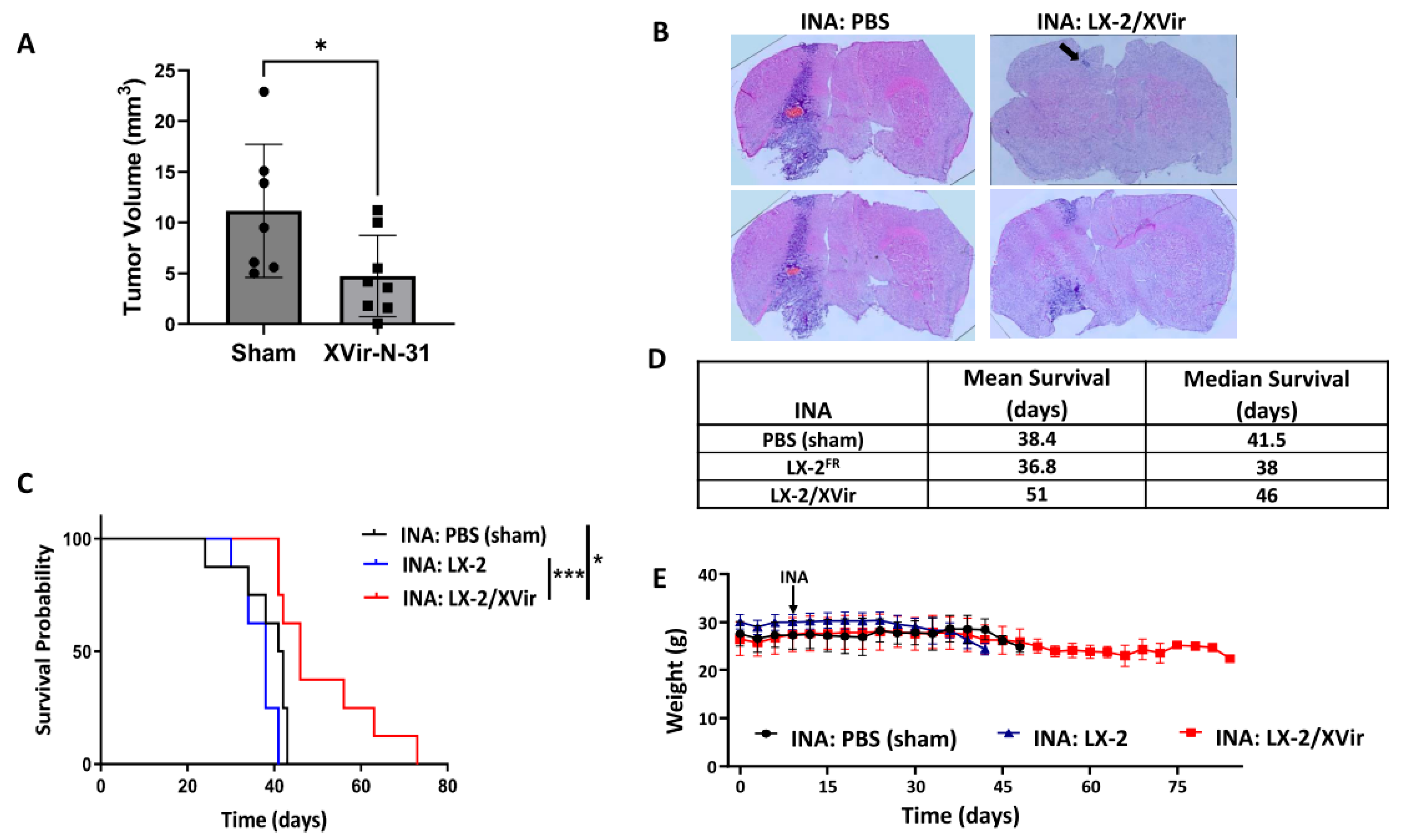
3.2. In LN-229-Derived GBMs, the INA of LX-2/XVir Induced Immunogenic Cell Death, Reduced Tumor Growth and Extended the Survival of Tumor-Bearing Mice
3.3. INA of LX-2/XVir Provides Therapeutical Impact in Mice Bearing Highly Infiltrating, Glioma Stem Cell-Derived GBMs
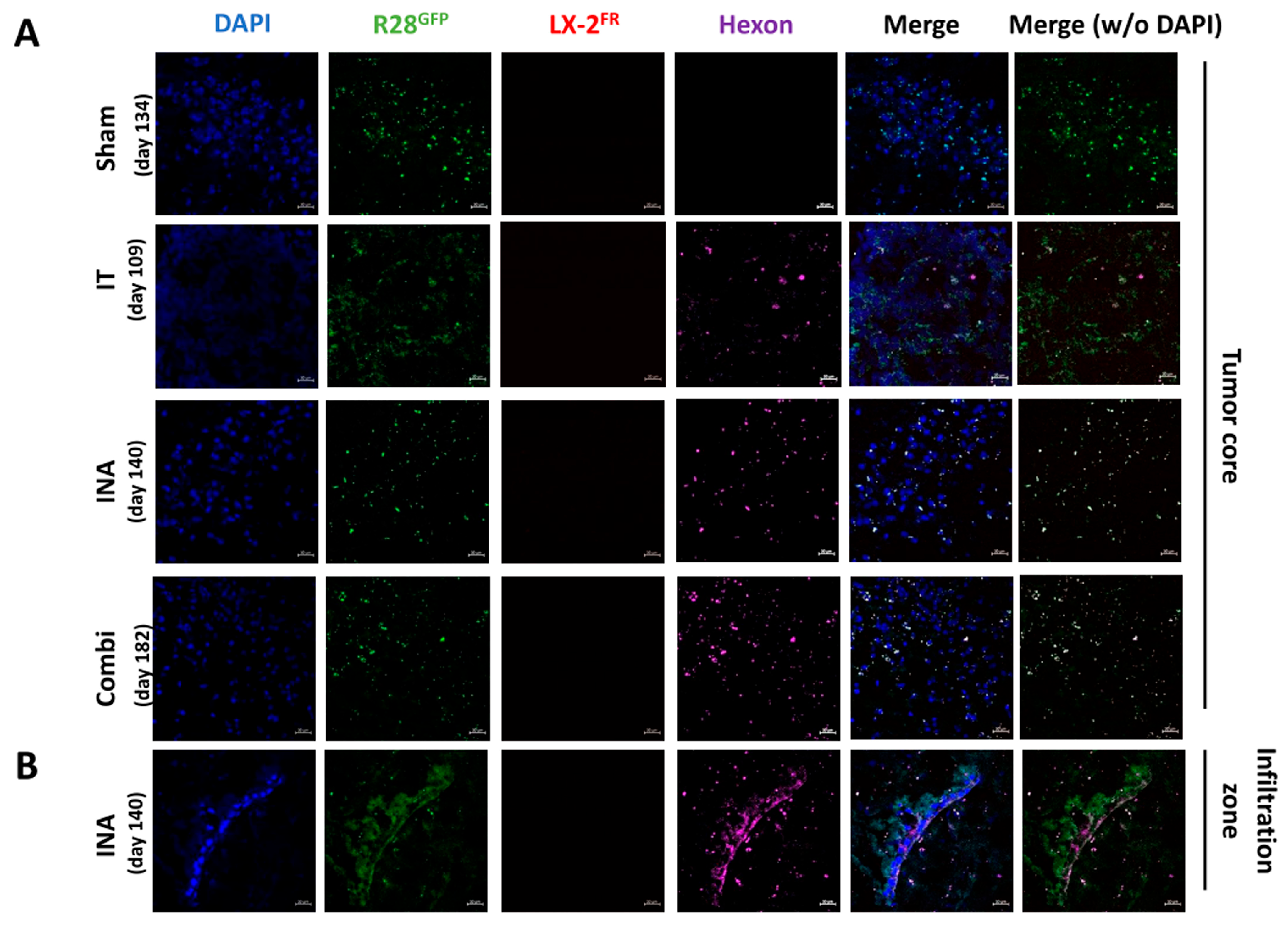
3.4. XVir-N-31 Is Present Long Term after INA of LX-2/XVir
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zreik, J.; Moinuddin, F.M.; Yolcu, Y.U.; Alvi, M.A.; Chaichana, K.L.; Quinones-Hinojosa, A.; Bydon, M. Improved 3-year survival rates for glioblastoma multiforme are associated with trends in treatment: Analysis of the national cancer database from 2004 to 2013. J. Neurooncol. 2020, 148, 69–79. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with oncolytic viruses: Progress and challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef]
- Ma, R.; Li, Z.; Chiocca, E.A.; Caligiuri, M.A.; Yu, J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer 2023, 9, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Lauer, U.M.; Beil, J. Oncolytic viruses: Challenges and considerations in an evolving clinical landscape. Future Oncol. 2022, 18, 2713–2732. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.T.; Hasegawa, K.; Dietz, A.B.; Russell, S.J.; Peng, K.W. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007, 14, 324–333. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes. Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef]
- Liikanen, I.; Koski, A.; Merisalo-Soikkeli, M.; Hemminki, O.; Oksanen, M.; Kairemo, K.; Joensuu, T.; Kanerva, A.; Hemminki, A. Serum HMGB1 is a predictive and prognostic biomarker for oncolytic immunotherapy. Oncoimmunology 2015, 4, e989771. [Google Scholar] [CrossRef]
- Klawitter, M.; El-Ayoubi, A.; Buch, J.; Ruttinger, J.; Ehrenfeld, M.; Lichtenegger, E.; Kruger, M.A.; Mantwill, K.; Koll, F.J.; Kowarik, M.C.; et al. The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model. Int. J. Mol. Sci. 2022, 23, 9965. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Abbasi, L.; Ghanbari Naeini, L.; Kokabian, P.; Nameh Goshay Fard, N.; Givtaj, N. Dendritic cells and natural killer cells: The road to a successful oncolytic virotherapy. Front. Immunol. 2022, 13, 950079. [Google Scholar] [CrossRef]
- Danielyan, L.; Schafer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopfer, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; Ayturan, M.; et al. Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 2009, 88, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Villar-Gomez, N.; Ojeda-Hernandez, D.D.; Lopez-Muguruza, E.; Garcia-Flores, S.; Bonel-Garcia, N.; Benito-Martin, M.S.; Selma-Calvo, B.; Canales-Aguirre, A.A.; Mateos-Diaz, J.C.; Montero-Escribano, P.; et al. Nose-to-Brain: The Next Step for Stem Cell and Biomaterial Therapy in Neurological Disorders. Cells 2022, 11, 3095. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Yu, D.; Kanojia, D.; Li, G.; Sukhanova, M.; Spencer, D.A.; Pituch, K.C.; Zhang, L.; Han, Y.; Ahmed, A.U.; et al. Intranasal Oncolytic Virotherapy with CXCR4-Enhanced Stem Cells Extends Survival in Mouse Model of Glioma. Stem Cell Rep. 2016, 7, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Beer-Hammer, S.; Stolzing, A.; Schafer, R.; Siegel, G.; Fabian, C.; Kahle, P.; Biedermann, T.; Lourhmati, A.; Buadze, M.; et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014, 23 (Suppl. S1), S123–S139. [Google Scholar] [CrossRef]
- Semyachkina-Glushkovskaya, O.; Shirokov, A.; Blokhina, I.; Telnova, V.; Vodovozova, E.; Alekseeva, A.; Boldyrev, I.; Fedosov, I.; Dubrovsky, A.; Khorovodov, A.; et al. Intranasal Delivery of Liposomes to Glioblastoma by Photostimulation of the Lymphatic System. Pharmaceutics 2022, 15, 36. [Google Scholar] [CrossRef]
- Danielyan, L.; Schwab, M.; Siegel, G.; Brawek, B.; Garaschuk, O.; Asavapanumas, N.; Buadze, M.; Lourhmati, A.; Wendel, H.P.; Avci-Adali, M.; et al. Cell motility and migration as determinants of stem cell efficacy. EBioMedicine 2020, 60, 102989. [Google Scholar] [CrossRef]
- Spencer, D.; Yu, D.; Morshed, R.A.; Li, G.; Pituch, K.C.; Gao, D.X.; Bertolino, N.; Procissi, D.; Lesniak, M.S.; Balyasnikova, I.V. Pharmacologic modulation of nasal epithelium augments neural stem cell targeting of glioblastoma. Theranostics 2019, 9, 2071–2083. [Google Scholar] [CrossRef]
- Rognoni, E.; Widmaier, M.; Haczek, C.; Mantwill, K.; Holzmuller, R.; Gansbacher, B.; Kolk, A.; Schuster, T.; Schmid, R.M.; Saur, D.; et al. Adenovirus-based virotherapy enabled by cellular YB-1 expression in vitro and in vivo. Cancer Gene Ther. 2009, 16, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Schober, S.J.; Schoening, C.; Eck, J.; Middendorf, C.; Lutsch, J.; Knoch, P.; von Ofen, A.J.; Gassmann, H.; Thiede, M.; Hauer, J.; et al. The Oncolytic Adenovirus XVir-N-31 Joins Forces with CDK4/6 Inhibition Augmenting Innate and Adaptive Antitumor Immunity in Ewing Sarcoma. Clin. Cancer Res. 2023, 29, 1996–2011. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, E.; Koll, F.; Haas, H.; Mantwill, K.; Janssen, K.P.; Laschinger, M.; Gschwend, J.; Steiger, K.; Black, P.C.; Moskalev, I.; et al. The Oncolytic Adenovirus XVir-N-31 as a Novel Therapy in Muscle-Invasive Bladder Cancer. Hum. Gene Ther. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Czolk, R.; Schwarz, N.; Koch, H.; Schotterl, S.; Wuttke, T.V.; Holm, P.S.; Huber, S.M.; Naumann, U. Irradiation enhances the therapeutic effect of the oncolytic adenovirus XVir-N-31 in brain tumor initiating cells. Int. J. Mol. Med. 2019, 44, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Mantwill, K.; Naumann, U.; Seznec, J.; Girbinger, V.; Lage, H.; Surowiak, P.; Beier, D.; Mittelbronn, M.; Schlegel, J.; Holm, P.S. YB-1 dependent oncolytic adenovirus efficiently inhibits tumor growth of glioma cancer stem like cells. J. Transl. Med. 2013, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Zhu, H.; You, P.; Liu, H.; Wang, W.K.; Fan, X.; Yang, Y.; Xu, K.; Zhu, Y.; Li, Q.; et al. Upregulated YB-1 protein promotes glioblastoma growth through a YB-1/CCT4/mLST8/mTOR pathway. J. Clin. Investig. 2022, 132, e146536. [Google Scholar] [CrossRef] [PubMed]
- El-Ayoubi, A.; Arakelyan, A.; Klawitter, M.; Merk, L.; Hakobyan, S.; Gonzalez-Menendez, I.; Quintanilla-Fend, L.; Holm, P.S.; Mikulits, W.; Schwab, M.; et al. Development of an optimized, non-stem cell line for intranasal delivery of therapeutic cargo to the central nervous system. bioRxiv 2023, 1–21. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Yu-Taeger, L.; Stricker-Shaver, J.; Arnold, K.; Bambynek-Dziuk, P.; Novati, A.; Singer, E.; Lourhmati, A.; Fabian, C.; Magg, J.; Riess, O.; et al. Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells 2019, 8, 595. [Google Scholar] [CrossRef]
- Karunasena, E.; McIver, L.J.; Rood, B.R.; Wu, X.; Zhu, H.; Bavarva, J.H.; Garner, H.R. Somatic intronic microsatellite loci differentiate glioblastoma from lower-grade gliomas. Oncotarget 2014, 5, 6003–6014. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, F.; Sharma, R.; Rasuli, B. Glioblastoma, IDH-Wildtype. Article de Référence. 2021. Available online: radiopaedia.org (accessed on 12 July 2023).
- Naumenko, K.N.; Sukhanova, M.V.; Hamon, L.; Kurgina, T.A.; Alemasova, E.E.; Kutuzov, M.M.; Pastre, D.; Lavrik, O.I. Regulation of Poly(ADP-Ribose) Polymerase 1 Activity by Y-Box-Binding Protein 1. Biomolecules 2020, 10, 1325. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, L.; Ruan, Y.; Zhou, L.; Zhang, D.; Min, Z.; Xie, J.; Yu, M.; Gu, J. An efficient delivery of DAMPs on the cell surface by the unconventional secretion pathway. Biochem. Biophys. Res. Commun. 2011, 404, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C.; et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021, 22, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.M.; Shah, A.H.; Vallejo, F.A.; Eichberg, D.G.; Luther, E.M.; Shah, S.S.; Komotar, R.J.; Ivan, M.E. Clinical trials using oncolytic viral therapy to treat adult glioblastoma: A progress report. Neurosurg. Focus 2021, 50, E3. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- D’Amico, R.S.; Aghi, M.K.; Vogelbaum, M.A.; Bruce, J.N. Convection-enhanced drug delivery for glioblastoma: A review. J. Neurooncol. 2021, 151, 415–427. [Google Scholar] [CrossRef]
- Lu, S.; Niu, N.; Guo, H.; Tang, J.; Guo, W.; Liu, Z.; Shi, L.; Sun, T.; Zhou, F.; Li, H.; et al. ARK5 promotes glioma cell invasion, and its elevated expression is correlated with poor clinical outcome. Eur. J. Cancer 2013, 49, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Chen, Y.L.; Lin, H.W.; Chang, C.F.; Huang, B.S.; Sun, W.Z.; Cheng, W.F. Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and DAMPs. Cancer Lett. 2021, 523, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Baak, L.M.; Wagenaar, N.; van der Aa, N.E.; Groenendaal, F.; Dudink, J.; Tataranno, M.L.; Mahamuud, U.; Verhage, C.H.; Eijsermans, R.; Smit, L.S.; et al. Feasibility and safety of intranasally administered mesenchymal stromal cells after perinatal arterial ischaemic stroke in the Netherlands (PASSIoN): A first-in-human, open-label intervention study. Lancet Neurol. 2022, 21, 528–536. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Ayoubi, A.; Klawitter, M.; Rüttinger, J.; Wellhäusser, G.; Holm, P.S.; Danielyan, L.; Naumann, U. Intranasal Delivery of Oncolytic Adenovirus XVir-N-31 via Optimized Shuttle Cells Significantly Extends Survival of Glioblastoma-Bearing Mice. Cancers 2023, 15, 4912. https://doi.org/10.3390/cancers15204912
El-Ayoubi A, Klawitter M, Rüttinger J, Wellhäusser G, Holm PS, Danielyan L, Naumann U. Intranasal Delivery of Oncolytic Adenovirus XVir-N-31 via Optimized Shuttle Cells Significantly Extends Survival of Glioblastoma-Bearing Mice. Cancers. 2023; 15(20):4912. https://doi.org/10.3390/cancers15204912
Chicago/Turabian StyleEl-Ayoubi, Ali, Moritz Klawitter, Jakob Rüttinger, Giulia Wellhäusser, Per Sonne Holm, Lusine Danielyan, and Ulrike Naumann. 2023. "Intranasal Delivery of Oncolytic Adenovirus XVir-N-31 via Optimized Shuttle Cells Significantly Extends Survival of Glioblastoma-Bearing Mice" Cancers 15, no. 20: 4912. https://doi.org/10.3390/cancers15204912
APA StyleEl-Ayoubi, A., Klawitter, M., Rüttinger, J., Wellhäusser, G., Holm, P. S., Danielyan, L., & Naumann, U. (2023). Intranasal Delivery of Oncolytic Adenovirus XVir-N-31 via Optimized Shuttle Cells Significantly Extends Survival of Glioblastoma-Bearing Mice. Cancers, 15(20), 4912. https://doi.org/10.3390/cancers15204912