
An Increase in HSF1 Expression Directs Human Mammary Epithelial Cells toward a Mesenchymal Phenotype

 , , and
, , and
Abstract
:Simple Summary
Abstract
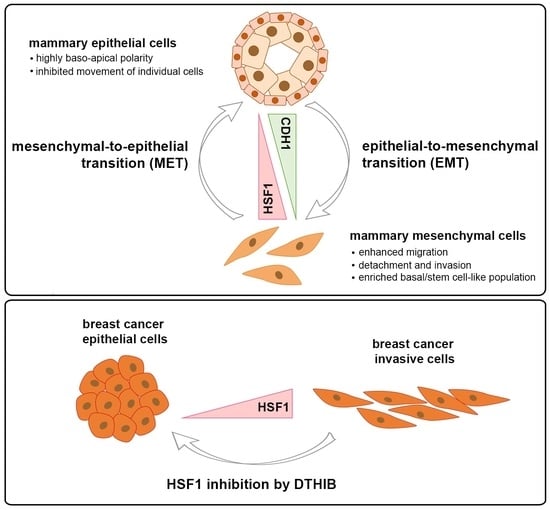
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. HSF1 Down-Regulation Using shRNA
2.3. HSF1 Overexpression
2.4. Cell Proliferation
2.5. Ki-67 Staining
2.6. Senescence-Associated β-Galactosidase (SA-β-Gal) Assay
2.7. Protein Extraction and Western Blotting
2.8. Three-Dimensional Overlay Culture and Spheroid Staining
2.9. Global Gene Expression Profiling
2.10. Boyden Chamber Assay
2.11. Flow Cytometry
2.12. Cell Viability Assay
2.13. Statistical Analyses
3. Results
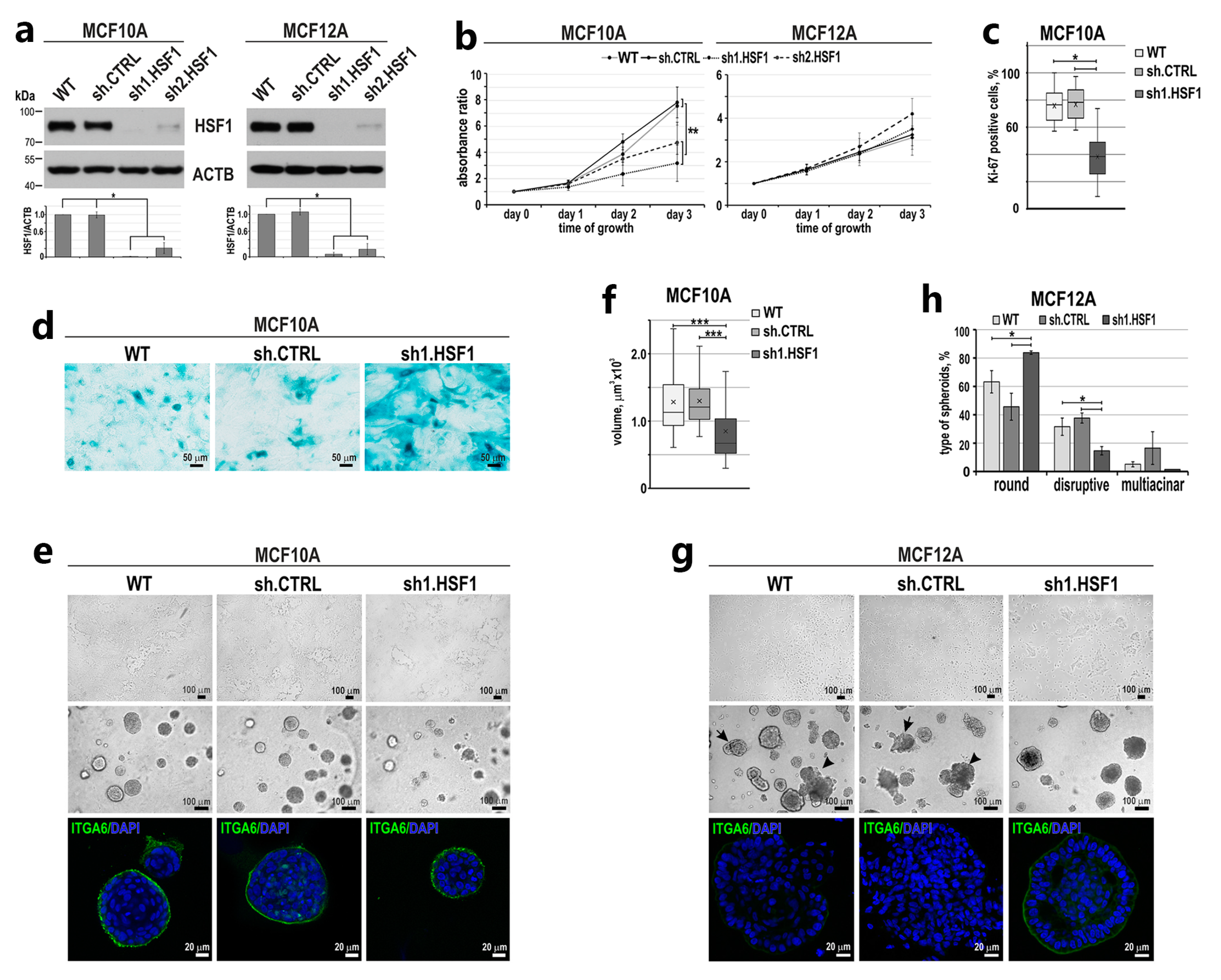
3.1. Down-Regulation of HSF1 Results in Inhibition of MCF10A Cell Proliferation and Alters the Morphology of MCF12A Cells in 2D and 3D Cultures
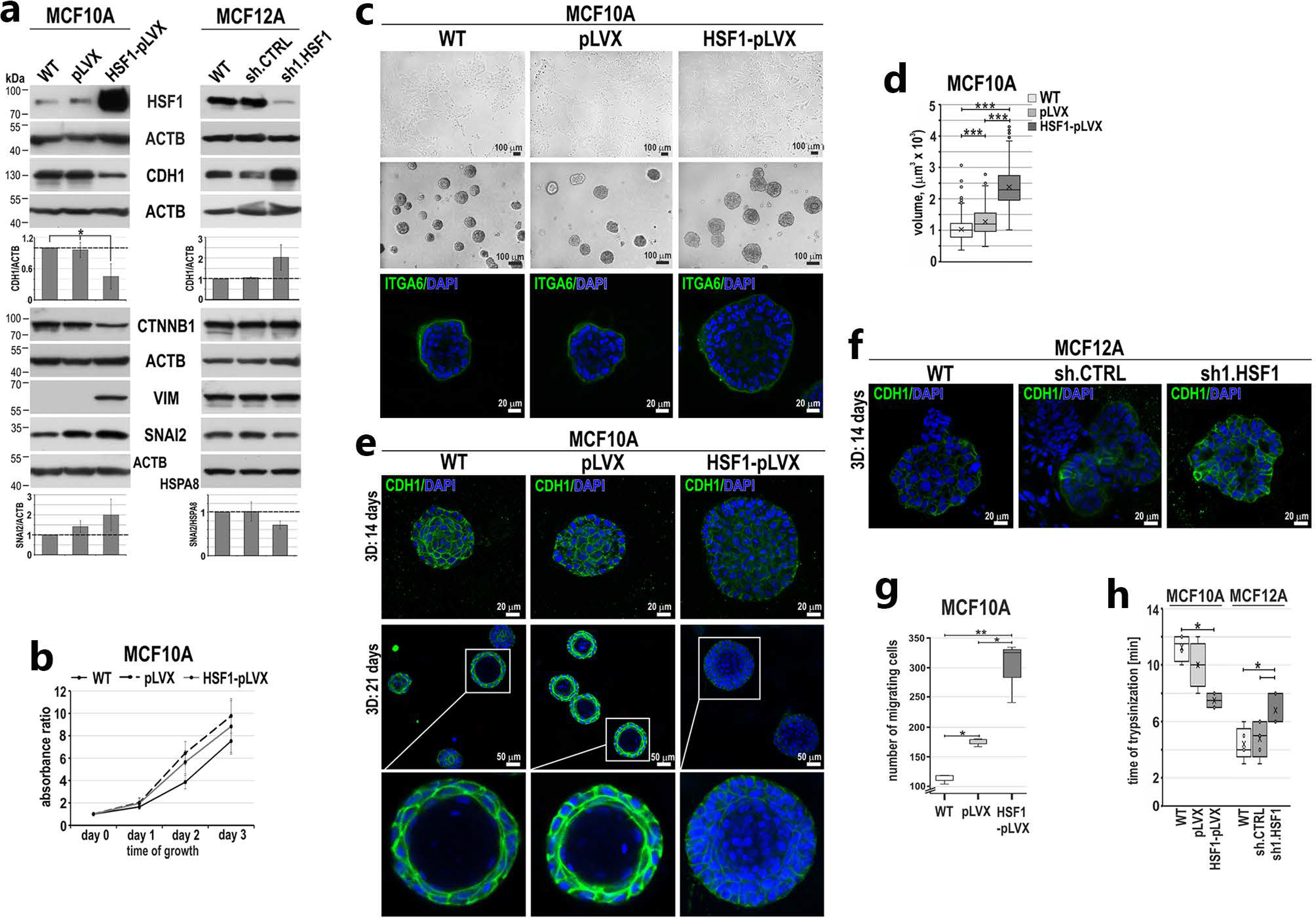
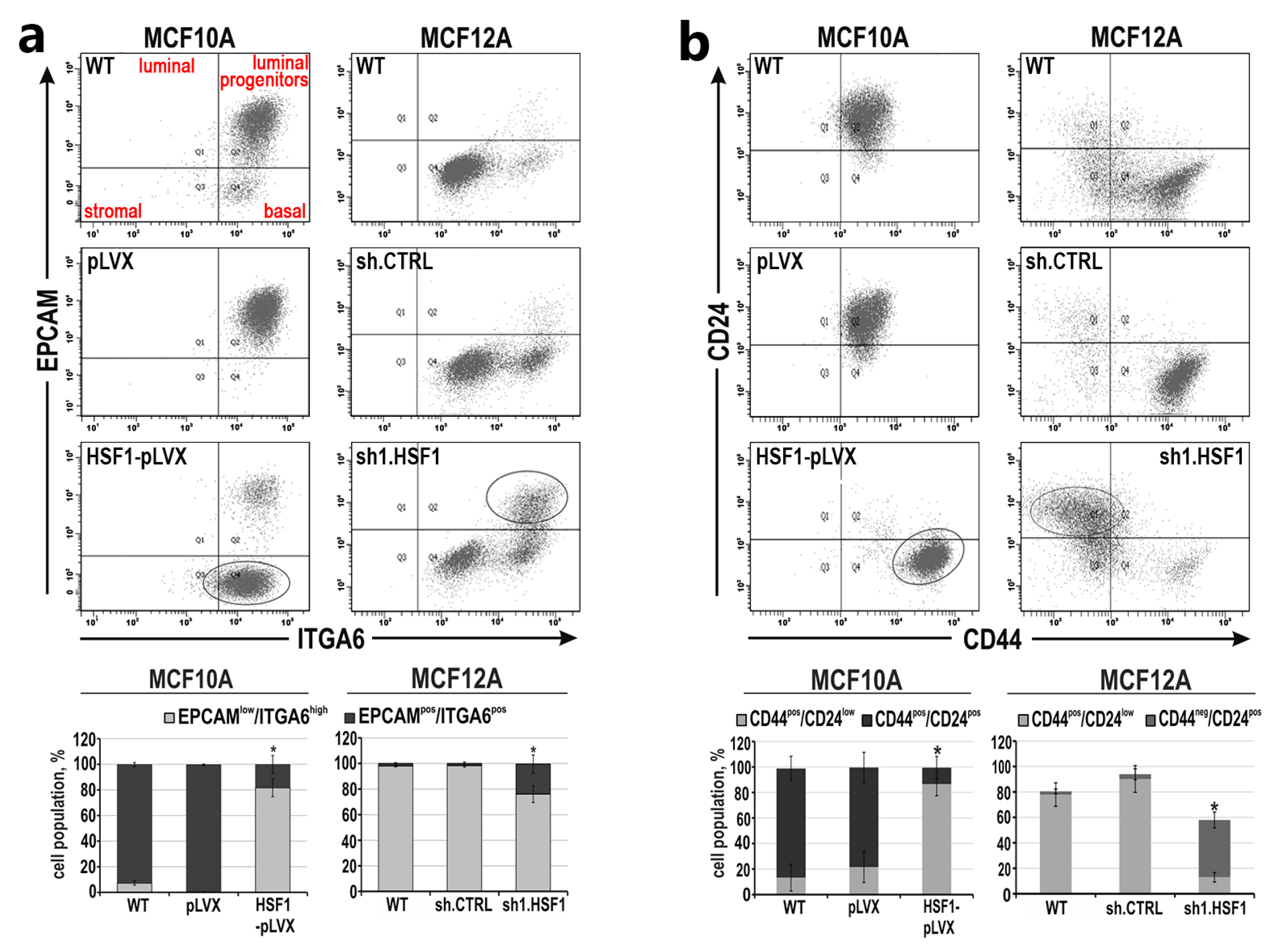
3.2. HSF1 Overexpression May Promote the Epithelial-to-Mesenchymal Transition of MCF10A Cells in a Mechanism Involving CDH1 Down-Regulation
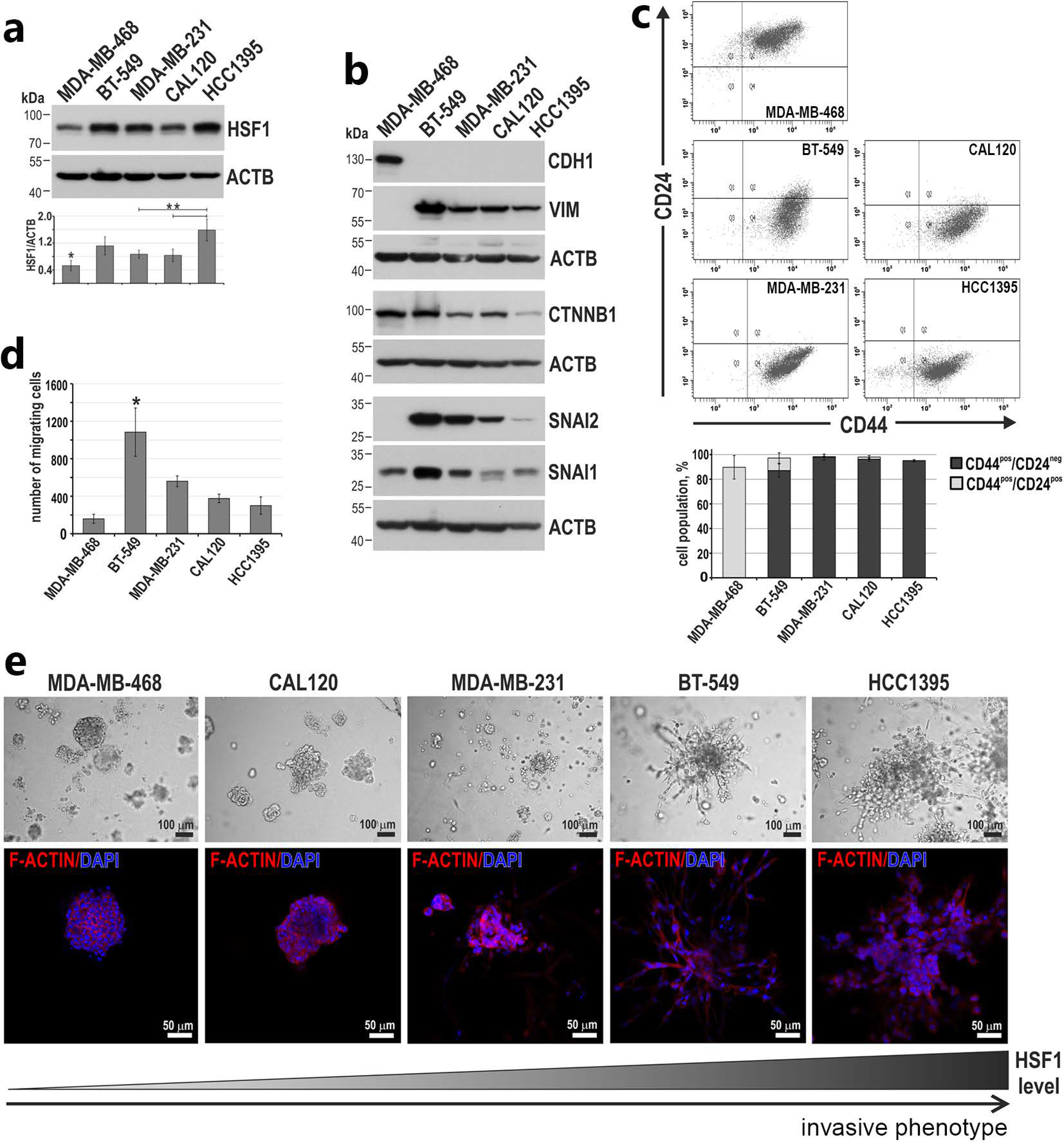
3.3. Higher HSF1 Levels in Triple-Negative Breast Cancer Cells Correlate with a More Invasive Phenotype
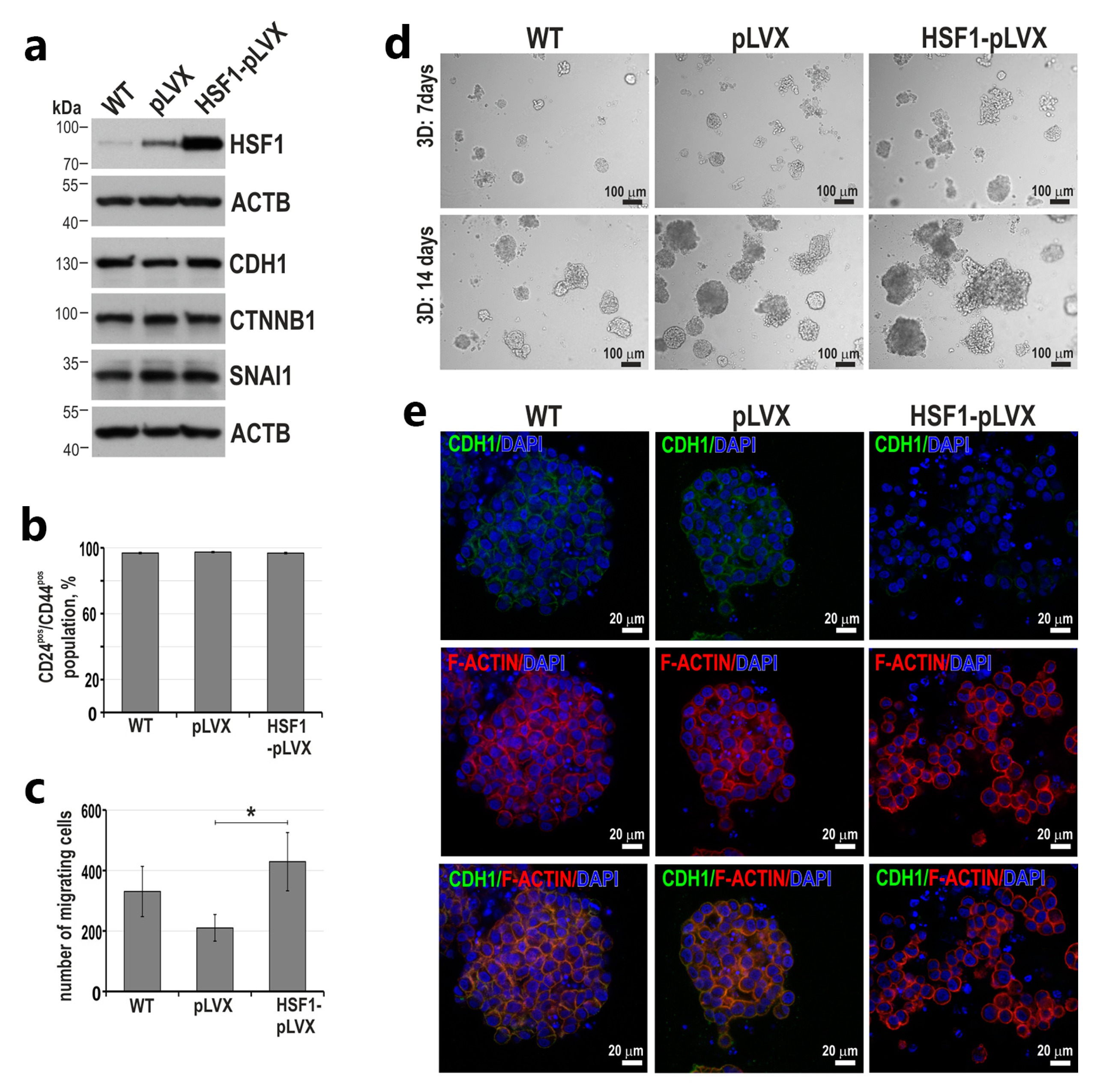
3.4. HSF1 May Affect the Invasive Phenotype of Triple-Negative Breast Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Voulgari, A.; Pintzas, A. Epithelial-Mesenchymal Transition in Cancer Metastasis: Mechanisms, Markers and Strategies to Overcome Drug Resistance in the Clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.; Kuperwasser, C. SLUG: Critical Regulator of Epithelial Cell Identity in Breast Development and Cancer. Cell Adhes. Migr. 2014, 8, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Keeping Abreast of the Mammary Epithelial Hierarchy and Breast Tumorigenesis. Genes Dev. 2009, 23, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
- Githaka, J.M.; Pirayeshfard, L.; Goping, I.S. Cancer Invasion and Metastasis: Insights from Murine Pubertal Mammary Gland Morphogenesis. Biochim. Biophys. Acta Gen. Subj. 2023, 1867, 130375. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Chiappetta, G. The Epithelial-to-Mesenchymal Transition in Breast Cancer: Focus on Basal-Like Carcinomas. Cancers 2017, 9, 134. [Google Scholar] [CrossRef]
- Drasin, D.J.; Robin, T.P.; Ford, H.L. Breast Cancer Epithelial-to-Mesenchymal Transition: Examining the Functional Consequences of Plasticity. Breast Cancer Res. BCR 2011, 13, 226. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef]
- Powell, C.D.; Paullin, T.R.; Aoisa, C.; Menzie, C.J.; Ubaldini, A.; Westerheide, S.D. The Heat Shock Transcription Factor HSF1 Induces Ovarian Cancer Epithelial-Mesenchymal Transition in a 3D Spheroid Growth Model. PLoS ONE 2016, 11, e0168389. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Paw, I.; Dewhirst, M.W.; Lo, H.-W. Akt Phosphorylates and Activates HSF-1 Independent of Heat Shock, Leading to Slug Overexpression and Epithelial-Mesenchymal Transition (EMT) of HER2-Overexpressing Breast Cancer Cells. Oncogene 2015, 34, 546–557. [Google Scholar] [CrossRef]
- Xi, C.; Hu, Y.; Buckhaults, P.; Moskophidis, D.; Mivechi, N.F. Heat Shock Factor Hsf1 Cooperates with ErbB2 (Her2/Neu) Protein to Promote Mammary Tumorigenesis and Metastasis. J. Biol. Chem. 2012, 287, 35646–35657. [Google Scholar] [CrossRef]
- Abane, R.; Mezger, V. Roles of Heat Shock Factors in Gametogenesis and Development. FEBS J. 2010, 277, 4150–4172. [Google Scholar] [CrossRef] [PubMed]
- Barna, J.; Csermely, P.; Vellai, T. Roles of Heat Shock Factor 1 beyond the Heat Shock Response. Cell. Mol. Life Sci. CMLS 2018, 75, 2897–2916. [Google Scholar] [CrossRef]
- Widlak, W.; Vydra, N. The Role of Heat Shock Factors in Mammalian Spermatogenesis. Adv. Anat. Embryol. Cell Biol. 2017, 222, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Dai, C. The Heat-Shock, or HSF1-Mediated Proteotoxic Stress, Response in Cancer: From Proteomic Stability to Oncogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160525. [Google Scholar] [CrossRef] [PubMed]
- Vydra, N.; Toma, A.; Widlak, W. Pleiotropic Role of HSF1 in Neoplastic Transformation. Curr. Cancer Drug Targets 2014, 14, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Vydra, N.; Toma, A.; Glowala-Kosinska, M.; Gogler-Piglowska, A.; Widlak, W. Overexpression of Heat Shock Transcription Factor 1 Enhances the Resistance of Melanoma Cells to Doxorubicin and Paclitaxel. BMC Cancer 2013, 13, 504. [Google Scholar] [CrossRef] [PubMed]
- Vydra, N.; Janus, P.; Kus, P.; Stokowy, T.; Mrowiec, K.; Toma-Jonik, A.; Krzywon, A.; Cortez, A.J.; Wojtas, B.; Gielniewski, B.; et al. Heat Shock Factor 1 (HSF1) Cooperates with Estrogen Receptor α (ERα) in the Regulation of Estrogen Action in Breast Cancer Cells. eLife 2021, 10, e69843. [Google Scholar] [CrossRef]
- Gogler-Pigłowska, A.; Klarzyńska, K.; Sojka, D.R.; Habryka, A.; Głowala-Kosińska, M.; Herok, M.; Kryj, M.; Halczok, M.; Krawczyk, Z.; Scieglinska, D. Novel Role for the Testis-Enriched HSPA2 Protein in Regulating Epidermal Keratinocyte Differentiation. J. Cell. Physiol. 2018, 233, 2629–2644. [Google Scholar] [CrossRef]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and Oncogenesis of MCF-10A Mammary Epithelial Acini Grown in Three-Dimensional Basement Membrane Cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Kurup, A.; Ravindranath, S.; Tran, T.; Keating, M.; Gascard, P.; Valdevit, L.; Tlsty, T.D.; Botvinick, E.L. Novel Insights from 3D Models: The Pivotal Role of Physical Symmetry in Epithelial Organization. Sci. Rep. 2015, 5, 15153. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making Genome-Scale Phylogenetics Accessible to All. Protein Sci. Publ. Protein Soc. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Ince, T.A.; Richardson, A.L.; Bell, G.W.; Saitoh, M.; Godar, S.; Karnoub, A.E.; Iglehart, J.D.; Weinberg, R.A. Transformation of Different Human Breast Epithelial Cell Types Leads to Distinct Tumor Phenotypes. Cancer Cell 2007, 12, 160–170. [Google Scholar] [CrossRef]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 Drives a Transcriptional Program Distinct from Heat Shock to Support Highly Malignant Human Cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef]
- Paine, T.M.; Soule, H.D.; Pauley, R.J.; Dawson, P.J. Characterization of Epithelial Phenotypes in Mortal and Immortal Human Breast Cells. Int. J. Cancer 1992, 50, 463–473. [Google Scholar] [CrossRef]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and Characterization of a Spontaneously Immortalized Human Breast Epithelial Cell Line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Sweeney, M.F.; Sonnenschein, C.; Soto, A.M. Characterization of MCF-12A Cell Phenotype, Response to Estrogens, and Growth in 3D. Cancer Cell Int. 2018, 18, 43. [Google Scholar] [CrossRef]
- Vydra, N.; Janus, P.; Toma-Jonik, A.; Stokowy, T.; Mrowiec, K.; Korfanty, J.; Długajczyk, A.; Wojtaś, B.; Gielniewski, B.; Widłak, W. 17β-Estradiol Activates HSF1 via MAPK Signaling in ERα-Positive Breast Cancer Cells. Cancers 2019, 11, 1533. [Google Scholar] [CrossRef] [PubMed]
- Imbalzano, K.M.; Tatarkova, I.; Imbalzano, A.N.; Nickerson, J.A. Increasingly Transformed MCF-10A Cells Have a Progressively Tumor-like Phenotype in Three-Dimensional Basement Membrane Culture. Cancer Cell Int. 2009, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-Mesenchymal Transition and Cancer Stem Cells: A Dangerously Dynamic Duo in Breast Cancer Progression. Breast Cancer Res. BCR 2011, 13, 202. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant Luminal Progenitors as the Candidate Target Population for Basal Tumor Development in BRCA1 Mutation Carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Sarrio, D.; Franklin, C.K.; Mackay, A.; Reis-Filho, J.S.; Isacke, C.M. Epithelial and Mesenchymal Subpopulations within Normal Basal Breast Cell Lines Exhibit Distinct Stem Cell/Progenitor Properties. Stem Cells Dayt. Ohio 2012, 30, 292–303. [Google Scholar] [CrossRef]
- Chang, C.-C. Recent Translational Research: Stem Cells as the Roots of Breast Cancer. Breast Cancer Res. BCR 2006, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, K.E.; Kendrick, H.; Ashworth, A.; Isacke, C.M.; Smalley, M.J. CD24 Staining of Mouse Mammary Gland Cells Defines Luminal Epithelial, Myoepithelial/Basal and Non-Epithelial Cells. Breast Cancer Res. BCR 2006, 8, R7. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Dong, B.; Jaeger, A.M.; Hughes, P.F.; Loiselle, D.R.; Hauck, J.S.; Fu, Y.; Haystead, T.A.; Huang, J.; Thiele, D.J. Targeting Therapy-Resistant Prostate Cancer via a Direct Inhibitor of the Human Heat Shock Transcription Factor 1. Sci. Transl. Med. 2020, 12, eabb5647. [Google Scholar] [CrossRef]
- Jin, X.; Eroglu, B.; Moskophidis, D.; Mivechi, N.F. Targeted Deletion of Hsf1, 2, and 4 Genes in Mice. In Methods in Molecular Biology; Calderwood, S., Prince, T., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1709, pp. 1–22. [Google Scholar] [CrossRef]
- Xiao, X.; Zuo, X.; Davis, A.A.; McMillan, D.R.; Curry, B.B.; Richardson, J.A.; Benjamin, I.J. HSF1 Is Required for Extra-Embryonic Development, Postnatal Growth and Protection during Inflammatory Responses in Mice. EMBO J. 1999, 18, 5943–5952. [Google Scholar] [CrossRef]
- Christians, E.; Davis, A.A.; Thomas, S.D.; Benjamin, I.J. Maternal Effect of Hsf1 on Reproductive Success. Nature 2000, 407, 693–694. [Google Scholar] [CrossRef] [PubMed]
- Inouye, S.; Izu, H.; Takaki, E.; Suzuki, H.; Shirai, M.; Yokota, Y.; Ichikawa, H.; Fujimoto, M.; Nakai, A. Impaired IgG Production in Mice Deficient for Heat Shock Transcription Factor 1. J. Biol. Chem. 2004, 279, 38701–38709. [Google Scholar] [CrossRef]
- Janus, P.; Kuś, P.; Vydra, N.; Toma-Jonik, A.; Stokowy, T.; Mrowiec, K.; Wojtaś, B.; Gielniewski, B.; Widłak, W. HSF1 Can Prevent Inflammation Following Heat Shock by Inhibiting the Excessive Activation of the ATF3 and JUN&FOS Genes. Cells 2022, 11, 2510. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat Shock Factor 1 Is a Powerful Multifaceted Modifier of Carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Xiang, B.; Muthuswamy, S.K. Controlled Activation of ErbB1/ErbB2 Heterodimers Promote Invasion of Three-Dimensional Organized Epithelia in an ErbB1-Dependent Manner: Implications for Progression of ErbB2-Overexpressing Tumors. Cancer Res. 2006, 66, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, R.; Feng, D.; Huang, W.; Huo, M.; Zhang, J.; Leng, S.; Yang, Y.; Yang, T.; Yin, X.; et al. Snail/PRMT5/NuRD Complex Contributes to DNA Hypermethylation in Cervical Cancer by TET1 Inhibition. Cell Death Differ. 2021, 28, 2818–2836. [Google Scholar] [CrossRef]
- Khaleque, M.A.; Bharti, A.; Gong, J.; Gray, P.J.; Sachdev, V.; Ciocca, D.R.; Stati, A.; Fanelli, M.; Calderwood, S.K. Heat Shock Factor 1 Represses Estrogen-Dependent Transcription through Association with MTA1. Oncogene 2008, 27, 1886–1893. [Google Scholar] [CrossRef]
- Hollier, B.G.; Evans, K.; Mani, S.A. The Epithelial-to-Mesenchymal Transition and Cancer Stem Cells: A Coalition against Cancer Therapies. J. Mammary Gland Biol. Neoplasia 2009, 14, 29–43. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular Definition of Breast Tumor Heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef]
- Wang, B.; Lee, C.-W.; Witt, A.; Thakkar, A.; Ince, T.A. Heat Shock Factor 1 Induces Cancer Stem Cell Phenotype in Breast Cancer Cell Lines. Breast Cancer Res. Treat. 2015, 153, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Hirohashi, Y.; Mariya, T.; Murai, A.; Tabuchi, Y.; Kuroda, T.; Kusumoto, H.; Takaya, A.; Yamamoto, E.; Kubo, T.; et al. Phosphorylation of HSF1 at Serine 326 Residue Is Related to the Maintenance of Gynecologic Cancer Stem Cells through Expression of HSP27. Oncotarget 2017, 8, 31540–31553. [Google Scholar] [CrossRef]
- Dong, Q.; Xiu, Y.; Wang, Y.; Hodgson, C.; Borcherding, N.; Jordan, C.; Buchanan, J.; Taylor, E.; Wagner, B.; Leidinger, M.; et al. HSF1 Is a Driver of Leukemia Stem Cell Self-Renewal in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 6107. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Qian, W.; Li, J.; Jiang, Z.; Cheng, L.; Yan, B.; Cao, J.; Sun, L.; Zhou, C.; Lei, M.; et al. Loss of AMPK Activation Promotes the Invasion and Metastasis of Pancreatic Cancer through an HSF1-Dependent Pathway. Mol. Oncol. 2017, 11, 1475–1492. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sun, L.; Qin, X.; Liu, T.; Zhang, S.; Liu, Y.; Li, S.; Guo, K. HSF1 Promotes the Inhibition of EMT-Associated Migration by Low Glucose via Directly Regulating Snail1 Expression in HCC Cells. Discov. Med. 2016, 22, 87–96. [Google Scholar] [PubMed]
- Fang, F.; Chang, R.; Yang, L. Heat Shock Factor 1 Promotes Invasion and Metastasis of Hepatocellular Carcinoma in Vitro and in Vivo. Cancer 2012, 118, 1782–1794. [Google Scholar] [CrossRef]
- Hoang, A.T.; Huang, J.; Rudra-Ganguly, N.; Zheng, J.; Powell, W.C.; Rabindran, S.K.; Wu, C.; Roy-Burman, P. A Novel Association between the Human Heat Shock Transcription Factor 1 (HSF1) and Prostate Adenocarcinoma. Am. J. Pathol. 2000, 156, 857–864. [Google Scholar] [CrossRef]
- Kourtis, N.; Moubarak, R.S.; Aranda-Orgilles, B.; Lui, K.; Aydin, I.T.; Trimarchi, T.; Darvishian, F.; Salvaggio, C.; Zhong, J.; Bhatt, K.; et al. FBXW7 Modulates Cellular Stress Response and Metastatic Potential through HSF1 Post-Translational Modification. Nat. Cell Biol. 2015, 17, 322–332. [Google Scholar] [CrossRef]
- Fiorenza, M.T.; Farkas, T.; Dissing, M.; Kolding, D.; Zimarino, V. Complex Expression of Murine Heat Shock Transcription Factors. Nucleic Acids Res. 1995, 23, 467–474. [Google Scholar] [CrossRef]
- Rabindran, S.K.; Giorgi, G.; Clos, J.; Wu, C. Molecular Cloning and Expression of a Human Heat Shock Factor, HSF1. Proc. Natl. Acad. Sci. USA 1991, 88, 6906–6910. [Google Scholar] [CrossRef]
- Chen, F.; Fan, Y.; Cao, P.; Liu, B.; Hou, J.; Zhang, B.; Tan, K. Pan-Cancer Analysis of the Prognostic and Immunological Role of HSF1: A Potential Target for Survival and Immunotherapy. Oxid. Med. Cell. Longev. 2021, 2021, 5551036. [Google Scholar] [CrossRef] [PubMed]
- Kourtis, N.; Lazaris, C.; Hockemeyer, K.; Balandrán, J.C.; Jimenez, A.R.; Mullenders, J.; Gong, Y.; Trimarchi, T.; Bhatt, K.; Hu, H.; et al. Oncogenic Hijacking of the Stress Response Machinery in T Cell Acute Lymphoblastic Leukemia. Nat. Med. 2018, 24, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Prince, T.L.; Lang, B.J.; Guerrero-Gimenez, M.E.; Fernandez-Muñoz, J.M.; Ackerman, A.; Calderwood, S.K. HSF1: Primary Factor in Molecular Chaperone Expression and a Major Contributor to Cancer Morbidity. Cells 2020, 9, 1046. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Ghosh, S.; Mandal, S.; Sau, S.; Pal, M. NRF2 Transcriptionally Activates the Heat Shock Factor 1 Promoter under Oxidative Stress and Affects Survival and Migration Potential of MCF7 Cells. J. Biol. Chem. 2018, 293, 19303–19316. [Google Scholar] [CrossRef]
- Cyran, A.M.; Zhitkovich, A. Heat Shock Proteins and HSF1 in Cancer. Front. Oncol. 2022, 12, 860320. [Google Scholar] [CrossRef]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Regulation of the Mammalian Heat Shock Factor 1. FEBS J. 2017, 284, 1606–1627. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PANTHER Pathways | No. of Genes in the Reference Set | No. of Affected Genes | Fold Enrichment | Raw p-Value | FDR | |
|---|---|---|---|---|---|---|
| Observed | Expected | |||||
| Down-regulated (total: 1397) | ||||||
| Cadherin signaling pathway (P00012) | 166 | 33 | 11.14 | 2.96 | 2.98 × 107 | 2.39 × 105 |
| Alzheimer disease–presenilin pathway (P00004) | 131 | 24 | 8.79 | 2.73 | 3.81 × 105 | 2.03 × 103 |
| Wnt signaling pathway (P00057) | 314 | 40 | 21.08 | 1.90 | 3.50 × 104 | 1.40 × 102 |
| Up-regulated (total: 1106) | ||||||
| Serine glycine biosynthesis (P02776) | 6 | 4 | 0.32 | 12.31 | 1.13 × 103 | 3.63 × 102 |
| DNA replication (P00017) | 29 | 13 | 1.57 | 8.28 | 1.03 × 107 | 1.64 × 105 |
| p53 pathway feedback loops 2 (P04398) | 51 | 10 | 2.76 | 3.62 | 1.02 × 103 | 4.06 × 102 |
| p53 pathway (P00059) | 89 | 14 | 4.82 | 2. 90 | 7.89 × 104 | 4.21 × 102 |
| Up- and down-regulated (total: 2503) | ||||||
| DNA replication (P00017) | 29 | 13 | 3.51 | 3.70 | 3.28 × 104 | 1.31 × 102 |
| Alzheimer disease–presenilin pathway (P00004) | 131 | 34 | 15.86 | 2.14 | 2.06 × 104 | 1.65 × 102 |
| Cadherin signaling pathway (P00012) | 166 | 40 | 20.09 | 1.99 | 2.53 × 104 | 1.35 × 102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vydra, N.; Toma-Jonik, A.; Janus, P.; Mrowiec, K.; Stokowy, T.; Głowala-Kosińska, M.; Sojka, D.R.; Olbryt, M.; Widłak, W. An Increase in HSF1 Expression Directs Human Mammary Epithelial Cells toward a Mesenchymal Phenotype. Cancers 2023, 15, 4965. https://doi.org/10.3390/cancers15204965
Vydra N, Toma-Jonik A, Janus P, Mrowiec K, Stokowy T, Głowala-Kosińska M, Sojka DR, Olbryt M, Widłak W. An Increase in HSF1 Expression Directs Human Mammary Epithelial Cells toward a Mesenchymal Phenotype. Cancers. 2023; 15(20):4965. https://doi.org/10.3390/cancers15204965
Chicago/Turabian StyleVydra, Natalia, Agnieszka Toma-Jonik, Patryk Janus, Katarzyna Mrowiec, Tomasz Stokowy, Magdalena Głowala-Kosińska, Damian Robert Sojka, Magdalena Olbryt, and Wiesława Widłak. 2023. "An Increase in HSF1 Expression Directs Human Mammary Epithelial Cells toward a Mesenchymal Phenotype" Cancers 15, no. 20: 4965. https://doi.org/10.3390/cancers15204965
APA StyleVydra, N., Toma-Jonik, A., Janus, P., Mrowiec, K., Stokowy, T., Głowala-Kosińska, M., Sojka, D. R., Olbryt, M., & Widłak, W. (2023). An Increase in HSF1 Expression Directs Human Mammary Epithelial Cells toward a Mesenchymal Phenotype. Cancers, 15(20), 4965. https://doi.org/10.3390/cancers15204965