
Osteosarcoma Multi-Omics Landscape and Subtypes


Abstract
:Simple Summary
Abstract
1. Background
2. Method
2.1. Data Collection
2.2. SCNA Detection
2.3. Gene Expression
2.4. Methylation
2.5. SNF
2.6. MARINa
2.7. Survival Analysis
2.8. Pathway Analysis
2.9. Other: Statistical Analysis
3. Results
3.1. Result 1: Sample and Data Collection
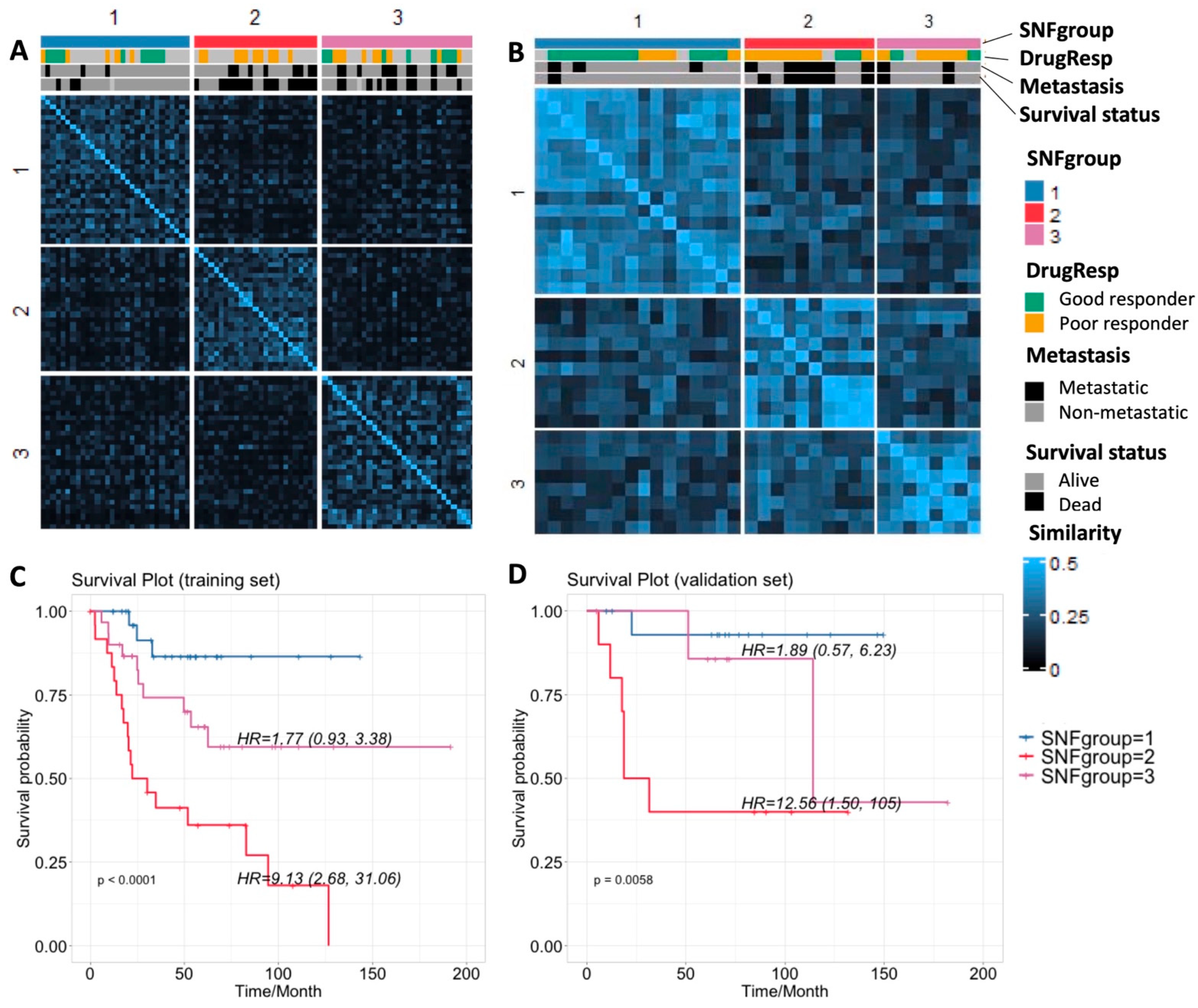
3.2. Result 2: Similarity Network Fusion Analysis Reveals Three Subtypes of Osteosarcoma
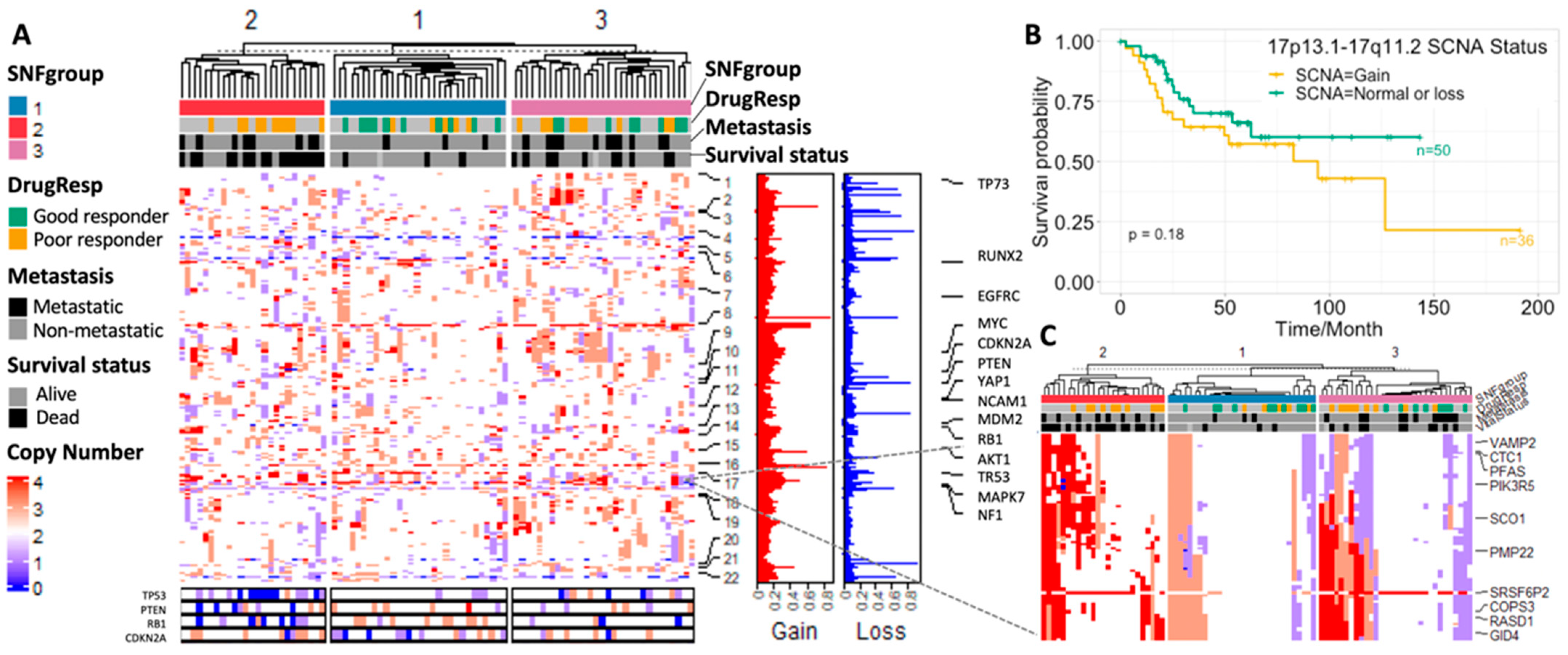
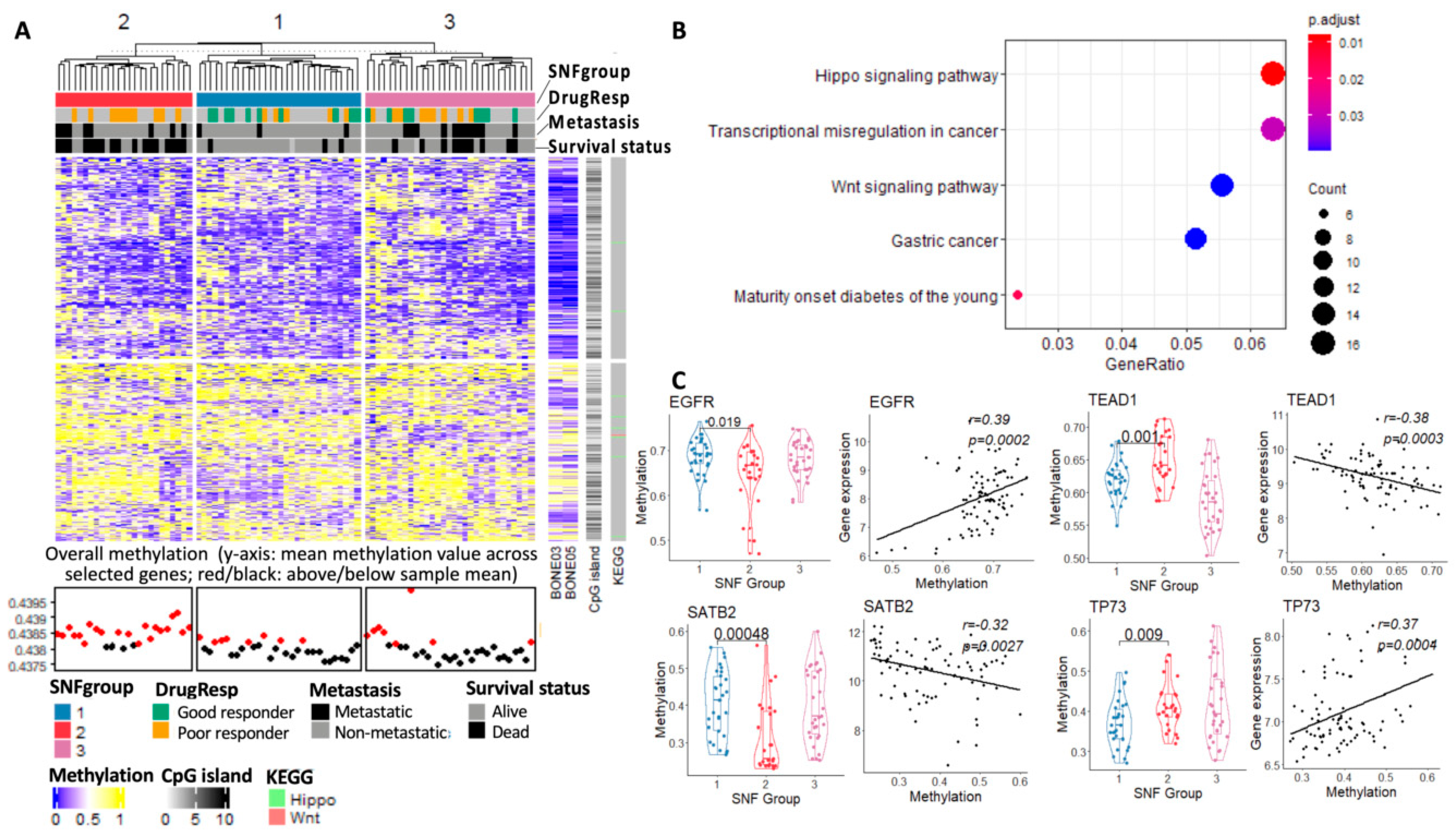
3.3. Result 3: Genomic Landscape of Three Osteosarcoma Subtypes
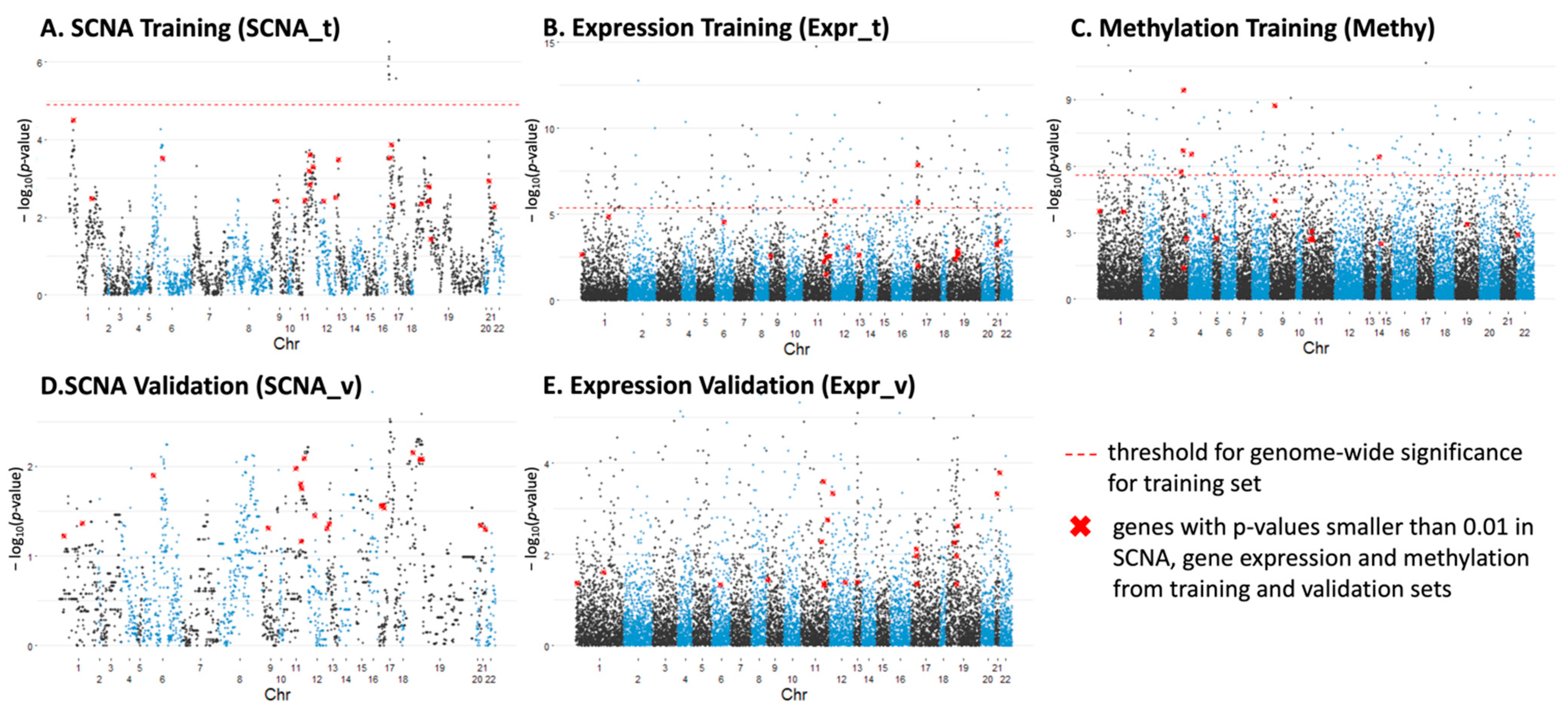
3.3.1. Individual Genes with Significant Differences in SCNA, Expression and Methylation
3.3.2. SCNA Profiles
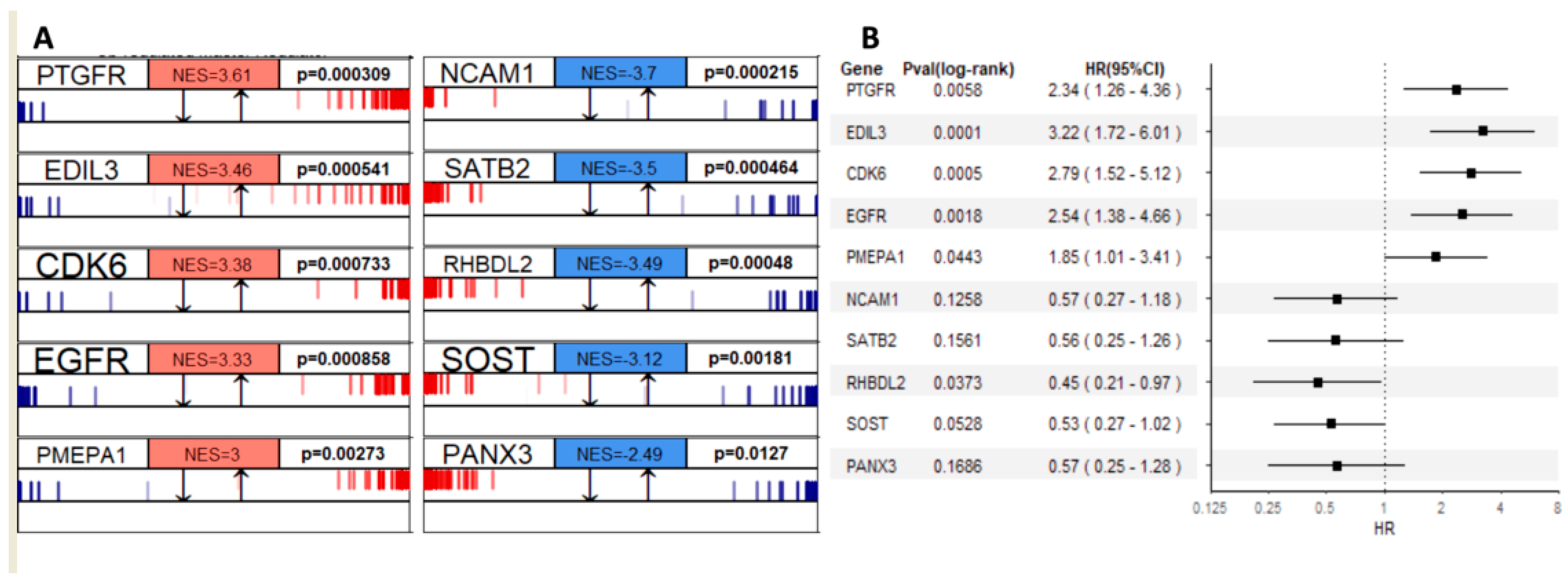
3.3.3. Gene Regulatory Network Hubs Based on the Transcriptome Data
3.3.4. Methylome Landscape of Three Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindsey, B.A.; Markel, J.E.; Kleinerman, E.S. Osteosarcoma overview. Rheumatol. Ther. 2017, 4, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. SICOT-J 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.W.; Squire, J.A.; Zielenska, M. The genetics of osteosarcoma. Sarcoma 2012, 2012, 627254. [Google Scholar] [CrossRef]
- Saraf, A.J.; Fenger, J.M.; Roberts, R.D. Osteosarcoma: Accelerating progress makes for a hopeful future. Front. Oncol. 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Varshney, J.; Scott, M.; Largaespada, D.; Subramanian, S. Understanding the osteosarcoma pathobiology: A comparative oncology approach. Vet. Sci. 2016, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, S.; Xie, X.; Wang, Z.; Lei, Q. Identification of potential crucial genes and key pathways in osteosarcoma. Hereditas 2020, 157, 29. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-H.; Jewell, B.E.; Gingold, J.; Lu, L.; Zhao, R.; Wang, L.L.; Lee, D.-F. Osteosarcoma: Molecular pathogenesis and iPSC modeling. Trends Mol. Med. 2017, 23, 737–755. [Google Scholar] [CrossRef]
- Kovac, M.; Ameline, B.; Ribi, S.; Kovacova, M.; Cross, W.; Barenboim, M.; Witt, O.; Bielack, S.; Krieg, A.; Hartmann, W. The early evolutionary landscape of osteosarcoma provides clues for targeted treatment strategies. J. Pathol. 2021, 254, 556–566. [Google Scholar] [CrossRef]
- Rajan, S.; Zaccaria, S.; Cannon, M.V.; Cam, M.; Gross, A.C.; Raphael, B.J.; Roberts, R.D. Remarkably stable copy-number profiles in osteosarcoma revealed using single-cell DNA sequencing. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wu, C.-C.; Beird, H.C.; Livingston, J.A.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.-L.; Ju, Z. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Southekal, S.; Shakyawar, S.K.; Bajpai, P.; Elkholy, A.; Manne, U.; Mishra, N.K.; Guda, C. Molecular Subtyping and Survival Analysis of Osteosarcoma Reveals Prognostic Biomarkers and Key Canonical Pathways. Cancers 2023, 15, 2134. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Andrade, A.; López-Cortés, A.; Jaramillo-Koupermann, G.; Paz-y-Miño, C.; Pérez-Castillo, Y.; Munteanu, C.R.; González-Díaz, H.; Pazos, A.; Tejera, E. Gene prioritization through consensus strategy, enrichment methodologies analysis, and networking for osteosarcoma pathogenesis. Int. J. Mol. Sci. 2020, 21, 1053. [Google Scholar] [CrossRef] [PubMed]
- Poos, K.; Smida, J.; Maugg, D.; Eckstein, G.; Baumhoer, D.; Nathrath, M.; Korsching, E. Genomic heterogeneity of osteosarcoma-shift from single candidates to functional modules. PLoS ONE 2015, 10, e0123082. [Google Scholar] [CrossRef] [PubMed]
- Pekarek, L.; De la Torre-Escuredo, B.; Fraile-Martinez, O.; García-Montero, C.; Saez, M.A.; Cobo-Prieto, D.; Guijarro, L.G.; Saz, J.V.; De Castro-Martinez, P.; Torres-Carranza, D.; et al. Towards the Search for Potential Biomarkers in Osteosarcoma: State-of-the-Art and Translational Expectations. Int. J. Mol. Sci. 2022, 23, 14939. [Google Scholar] [CrossRef] [PubMed]
- Korn, J.M.; Kuruvilla, F.G.; McCarroll, S.A.; Wysoker, A.; Nemesh, J.; Cawley, S.; Hubbell, E.; Veitch, J.; Collins, P.J.; Darvishi, K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008, 40, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rajbhandari, P.; Alvarez, M.J.; Bandaru, P.; Lim, W.K.; Sato, M.; Wang, K.; Sumazin, P.; Kustagi, M.; Bisikirska, B.C. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010, 6, 377. [Google Scholar] [CrossRef]
- Sun, J.; Ren, P.; Ye, L.; Li, N.; Wang, D. MLLT3 promotes proliferation of osteosarcoma cells by regulating JNK signaling. Int. J. Clin. Exp. Pathol. 2017, 10, 9444. [Google Scholar]
- Baranski, Z.; Booij, T.H.; Cleton-Jansen, A.M.; Price, L.S.; van de Water, B.; Bovée, J.V.; Hogendoorn, P.C.; Danen, E.H. Aven-mediated checkpoint kinase control regulates proliferation and resistance to chemotherapy in conventional osteosarcoma. J. Pathol. 2015, 236, 348–359. [Google Scholar] [CrossRef]
- Li, X.; Dean, D.C.; Cote, G.M.; Zou, L.; Hornicek, F.J.; Yu, S.; Duan, Z. Inhibition of ATR-Chk1 signaling blocks DNA double-strand-break repair and induces cytoplasmic vacuolization in metastatic osteosarcoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920956900. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-H.; Hao, Y.-J. DDX10 overexpression predicts worse prognosis in osteosarcoma and its deletion prohibits cell activities modulated by MAPK pathway. Biochem. Biophys. Res. Commun. 2019, 510, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhou, F.; Zhang, Y.; Zhou, W.; Zhu, L.; Zhang, M.; Luo, J.; Ma, R.; Xu, X.; Zhu, J. Identification of tumorigenicity-associated genes in osteosarcoma cell lines based on bioinformatic analysis and experimental validation. J. Cancer 2020, 11, 3623. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhang, M. A novel risk score model based on eight genes and a nomogram for predicting overall survival of patients with osteosarcoma. BMC Cancer 2020, 20, 456. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, G.; Mu, H.; Ma, X.; Wang, Z.; Lv, Y.; Zhang, T.; Xu, J.; Wang, J.; Li, Y. Bromodomain Inhibition Attenuates the Progression and Sensitizes the Chemosensitivity of Osteosarcoma by Repressing GP130/STAT3 Signaling. Front. Oncol. 2021, 11, 642134. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, Z.; Huang, Z.; Chen, D.-C.; Zhu, X.-X.; Wang, Y.-Z.; Yan, Y.-W.; Tang, S.; Madhavan, S.; Ni, W. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Peng, M.; Yi, L. UBASH3B is a novel prognostic biomarker and correlated with immune infiltrates in prostate cancer. Front. Oncol. 2020, 9, 1517. [Google Scholar] [CrossRef]
- Krupina, K.; Kleiss, C.; Awal, S.; Rodriguez-Hernandez, I.; Sanz-Moreno, V.; Sumara, I. UBASH3B-mediated silencing of the mitotic checkpoint: Therapeutic perspectives in cancer. Mol. Cell. Oncol. 2018, 5, e1271494. [Google Scholar] [CrossRef]
- Hong, W.; Gu, Y.; Guan, R.; Xie, D.; Zhou, H.; Yu, M. Pan-cancer analysis of the CASP gene family in relation to survival, tumor-infiltrating immune cells and therapeutic targets. Genomics 2020, 112, 4304–4315. [Google Scholar] [CrossRef]
- Lin, J.; Liu, H.; Fukumoto, T.; Zundell, J.; Yan, Q.; Tang, C.-H.A.; Wu, S.; Zhou, W.; Guo, D.; Karakashev, S. Targeting the IRE1α/XBP1s pathway suppresses CARM1-expressing ovarian cancer. Nat. Commun. 2021, 12, 5321. [Google Scholar] [CrossRef]
- Wu, D.; He, J.; Zhang, W.; Wang, K.; Jin, S.; Li, J.; Gao, W. CARM1 promotes non-small cell lung cancer progression through upregulating CCNE2 expression. Aging 2020, 12, 10578. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, S.M.; Man, N.; Hamard, P.-J.; Asai, T.; Karl, D.; Martinez, C.; Bilbao, D.; Stathias, V.; Jermakowicz, A.M.; Duffort, S. CARM1 is essential for myeloid leukemogenesis but dispensable for normal hematopoiesis. Cancer Cell 2018, 33, 1111–1127.e1115. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Qin, Y.; Fan, H.; Su, P.; Zhang, X.; Zhang, H.; Zhou, G. Overexpression of CARM1 in breast cancer is correlated with poorly characterized clinicopathologic parameters and molecular subtypes. Diagn. Pathol. 2013, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Di, M.; Wang, M.; Miao, J.; Chen, B.; Huang, H.; Lin, C.; Jian, Y.; Li, Y.; Ouyang, Y.; Chen, X. CHAF1B induces radioresistance by promoting DNA damage repair in nasopharyngeal carcinoma. Biomed. Pharmacother. 2020, 123, 109748. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Liu, T.; Li, S.; Huang, M.; Li, X.; Zhao, H.; Li, J. CHAF1B promotes proliferation and reduces apoptosis in 95-D lung cancer cells and predicts a poor prognosis in non-small cell lung cancer. Oncol. Rep. 2019, 41, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Achinger-Kawecka, J.; Valdes-Mora, F.; Luu, P.-L.; Giles, K.A.; Caldon, C.E.; Qu, W.; Nair, S.; Soto, S.; Locke, W.J.; Yeo-Teh, N.S. Epigenetic reprogramming at estrogen-receptor binding sites alters 3D chromatin landscape in endocrine-resistant breast cancer. Nat. Commun. 2020, 11, 320. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Chen, J.-T.; Lin, M.-W.; Chan, C.-H.; Wen, Y.-F.; Wu, S.-B.; Chung, T.-W.; Lyu, K.W.; Chou, H.-C.; Chan, H.-L. Identification of protein expression alterations in gefitinib-resistant human lung adenocarcinoma: PCNT and mPR play key roles in the development of gefitinib-associated resistance. Toxicol. Appl. Pharmacol. 2015, 288, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Kuijjer, M.L.; Rydbeck, H.; Kresse, S.H.; Buddingh, E.P.; Lid, A.B.; Roelofs, H.; Bürger, H.; Myklebost, O.; Hogendoorn, P.C.; Meza-Zepeda, L.A. Identification of osteosarcoma driver genes by integrative analysis of copy number and gene expression data. Genes Chromosomes Cancer 2012, 51, 696–706. [Google Scholar] [CrossRef]
- Meltzer, P.S.; Davis, S.; Zhu, J.; Wang, Y.; Bilke, S.; Waterfall, J.; Walker, R.; Pineda, M.; Jiang, Y.; Savage, S.; et al. Translational and mechanistic implications of osteosarcoma genomics: A TARGET report. Cancer Res. 2020, 80, LB-307. [Google Scholar] [CrossRef]
- Both, J.; Wu, T.; Bras, J.; Schaap, G.R.; Baas, F.; Hulsebos, T.J. Identification of novel candidate oncogenes in chromosome region 17p11.2-p12 in human osteosarcoma. PLoS ONE 2012, 7, e30907. [Google Scholar] [CrossRef]
- van Dartel, M.; Redeker, S.; Bras, J.; Kool, M.; Hulsebos, T.J. Overexpression through amplification of genes in chromosome region 17p11. 2~p12 in high-grade osteosarcoma. Cancer Genet. Cytogenet. 2004, 152, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, H.J.; Jang, M.H.; Lee, S.; Ahn, S.; Park, S.Y. Centromere 17 copy number gain reflects chromosomal instability in breast cancer. Sci. Rep. 2019, 9, 17968. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.; Aagesen, T.H.; Maelandsmo, G.M.; Lothe, R.A.; Myklebost, O.; Forus, A. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene 2003, 22, 5358–5361. [Google Scholar] [CrossRef] [PubMed]
- van Dartel, M.; Hulsebos, T.J. Amplification and overexpression of genes in 17p11.2~p12 in osteosarcoma. Cancer Genet. Cytogenet. 2004, 153, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Boltz, K.A.; Leehy, K.; Song, X.; Nelson, A.D.; Shippen, D.E. ATR cooperates with CTC1 and STN1 to maintain telomeres and genome integrity in Arabidopsis. Mol. Biol. Cell 2012, 23, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Bergstrand, S.; Böhm, S.; Malmgren, H.; Norberg, A.; Sundin, M.; Nordgren, A.; Farnebo, M. Biallelic mutations in WRAP53 result in dysfunctional telomeres, Cajal bodies and DNA repair, thereby causing Hoyeraal–Hreidarsson syndrome. Cell Death Dis. 2020, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Wang, W.; Hao, Q.; Liu, Y. High RASD1 transcript levels at diagnosis predicted poor survival in adult B-cell acute lymphoblastic leukemia patients. Leuk. Res. 2019, 80, 26–32. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Henriksson, S.; Farnebo, L.; Roberg, K.; Farnebo, M. WRAP53 promotes cancer cell survival and is a potential target for cancer therapy. Cell Death Dis. 2011, 2, e114. [Google Scholar] [CrossRef]
- Martin, J.; Zielenska, M.; Stein, G.; Van Wijnen, A.; Squire, J. The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma 2011, 2011, 282745. [Google Scholar] [CrossRef]
- Sadikovic, B.; Thorner, P.; Chilton-MacNeill, S.; Martin, J.W.; Cervigne, N.K.; Squire, J.; Zielenska, M. Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer 2010, 10, 202. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, R.; Ghanem, N.; El-Makky, N.; Ismail, M. Integrated analysis of SNP, CNV and gene expression data in genetic association studies. Clin. Genet. 2018, 93, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Wu, C.-J.; Siroy, A.E.; Lazar, A.J.; Davies, M.A.; Woodman, S.E.; Kwong, L.N. Somatic copy number alterations at oncogenic loci show diverse correlations with gene expression. Sci. Rep. 2016, 6, 19649. [Google Scholar] [CrossRef] [PubMed]
- Padovan-Merhar, O.; Nair, G.P.; Biaesch, A.G.; Mayer, A.; Scarfone, S.; Foley, S.W.; Wu, A.R.; Churchman, L.S.; Singh, A.; Raj, A. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol. Cell 2015, 58, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, J.; Liu, Z.; Wei, W. MicroRNA-145 inhibits tumour growth and metastasis in osteosarcoma by targeting cyclin-dependent kinase, CDK6. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5117–5125. [Google Scholar]
- Astsaturov, I.; Ratushny, V.; Sukhanova, A.; Einarson, M.B.; Bagnyukova, T.; Zhou, Y.; Devarajan, K.; Silverman, J.S.; Tikhmyanova, N.; Skobeleva, N. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal. 2010, 3, ra67. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Fu, L.; Wang, G.; Wang, J.; Xu, L. MicroRNA-431-5p Inhibits the Tumorigenesis of Osteosarcoma Through Targeting PANX3. Cancer Manag. Res. 2020, 12, 8159. [Google Scholar] [CrossRef] [PubMed]
- Sevelda, F.; Mayr, L.; Kubista, B.; Lötsch, D.; van Schoonhoven, S.; Windhager, R.; Pirker, C.; Micksche, M.; Berger, W. EGFR is not a major driver for osteosarcoma cell growth in vitro but contributes to starvation and chemotherapy resistance. J. Exp. Clin. Cancer Res. 2015, 34, 134. [Google Scholar] [CrossRef]
- Wang, D.; Bao, H. Abemaciclib is synergistic with doxorubicin in osteosarcoma pre-clinical models via inhibition of CDK4/6-Cyclin D-Rb pathway. Cancer Chemother. Pharmacol. 2022, 89, 31–40. [Google Scholar] [CrossRef]
- Yang, G.; Yuan, J.; Li, K. EMT transcription factors: Implication in osteosarcoma. Med. Oncol. 2013, 30, 697. [Google Scholar] [CrossRef]
- Maloney, C.; Kallis, M.P.; Edelman, M.; Tzanavaris, C.; Lesser, M.; Soffer, S.Z.; Symons, M.; Steinberg, B.M. Gefitinib Inhibits Invasion and Metastasis of Osteosarcoma via Inhibition of Macrophage Receptor Interacting Serine-Threonine Kinase 2. Mol. Cancer Ther. 2020, 19, 1340–1350. [Google Scholar] [CrossRef]
- Shen, L.; Kondo, Y.; Ahmed, S.; Boumber, Y.; Konishi, K.; Guo, Y.; Chen, X.; Vilaythong, J.N.; Issa, J.-P.J. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res. 2007, 67, 11335–11343. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. WNT/PCP signaling pathway and human cancer. Oncol. Rep. 2005, 14, 1583–1588. [Google Scholar] [CrossRef]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation patterns between DNA methylation and gene expression in the cancer genome atlas. Cancer Inform. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | n (%) | TARGET (Training, n = 86) | Dr. Korsching (Validation, n = 38) |
|---|---|---|---|
| Median age at diagnosis in year (range) | 15 (3–33) | 16 (8–60) | |
| Gender | Male | 50 (58%) | 25 (66%) |
| Female | 36 (42%) | 13 (34%) | |
| Race | White | 51 (60%) | Not provided |
| Black | 8 (9%) | ||
| Asian | 7 (8%) | ||
| Unknown/not provided | 20 (23%) | ||
| Survival status | Alive | 54 (63%) | 27 (71%) |
| Dead | 30 (35%) | 11 (29%) | |
| Not provided | 2 (2%) | - | |
| Pathologic response to neoadjuvant chemotherapy | Good | 18 (21%) | 14 (37%) |
| Poor | 24 (28%) | 18 (47%) | |
| Not provided | 44 (51%) | 6 (16%) | |
| Metastasis | Metastatic | 22 (26%) | 15 (39%) |
| Non-metastatic | 64 (74%) | 23 (61%) |
| Method and Data Sources | SNF SCNA + Expr + Methy | Clustering SCNA Only | Clustering Expr Only | Clustering Methy Only | |
|---|---|---|---|---|---|
| p-values | Survival (log-rank) | 2.8905 × 10−5 * | 0.011 * | 0.0035 * | 0.0055 * |
| Drug response (Fisher’s exact) | 0.0039 * | 0.1421 | 0.4635 | 0.0388 * | |
| Metastasis (Fisher’s exact) | 0.050 * | 0.0712 | 0.4397 | 0.2712 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.; Roberts, R.D.; Cheng, L.; Li, L. Osteosarcoma Multi-Omics Landscape and Subtypes. Cancers 2023, 15, 4970. https://doi.org/10.3390/cancers15204970
Tang S, Roberts RD, Cheng L, Li L. Osteosarcoma Multi-Omics Landscape and Subtypes. Cancers. 2023; 15(20):4970. https://doi.org/10.3390/cancers15204970
Chicago/Turabian StyleTang, Shan, Ryan D. Roberts, Lijun Cheng, and Lang Li. 2023. "Osteosarcoma Multi-Omics Landscape and Subtypes" Cancers 15, no. 20: 4970. https://doi.org/10.3390/cancers15204970
APA StyleTang, S., Roberts, R. D., Cheng, L., & Li, L. (2023). Osteosarcoma Multi-Omics Landscape and Subtypes. Cancers, 15(20), 4970. https://doi.org/10.3390/cancers15204970