
Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators



Abstract
:Simple Summary
Abstract
1. Introduction
2. Major Classes of Oncogene-Targeted Drugs and Resistance to the Respective Therapeutic Strategies
3. A Primer to Lung Cancer
4. EGFR Mutations in Lung Cancer
5. First and Second Generations of EGFR-Specific Kinase Inhibitors (See Table 1)

{kind=link}
{kind=link}
{kind=link}
| Drug | Inhibitor Type | Drug Target | Status | Relevant Studies |
|---|---|---|---|---|
| Gefitinib | 1st generation TKI Competitive Reversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2003/2009 | NEJ002 [49,50] IPASS [51,52] WJTOG3405 [37,53] |
| Erlotinib | 1st generation TKI Competitive Reversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2004/2005 | OPTIMAL [39,54] ENSURE [55] EUTARC [38] |
| Icotinib | 1st generation TKI Competitive Reversible | Del19/L858R-EGFR | Approved in China in 2011 | CONVINCE [56] EVIDENCE [57] |
| Afatinib | 2nd generation TKI Covalent Irreversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2013 | LUX-Lung3 [58,59] LUX-Lung6 [59,60] |
| Dacomitinib | 2nd generation TKI Covalent Irreversible | Del19/L858R-EGFR | Approved by the FDA/EMA in 2018/2019 | ARCHER1050 [61,62] |
| Osimertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved by the FDA/EMA in 2015/2016 | AURA3 [63] FLAURA [64] |
| Aumolertinib * | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved in China in 2020 | AENEAS [36,65] APOLLO [66] |
| Furmonertinib $ | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved in China in 2021 | FURLONG [67] |
| Lazertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Approved in South Korea in 2021 | LASER201 [68,69] LASER301 (NCT04248829) |
| Befotertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase II/III (active) | NCT03861156 [70] NCT04206072 [71] |
| Abivertinib £ | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase I/II (active) | NCT02274337 [72] AEGIS-1 (NCT02330367) |
| Nazartinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase I/II (active) | NCT02108964 [73] |
| Mavelertinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase I/II (terminated) | NCT02349633 [74] |
| Rociletinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Rejected by the FDA in 2016 | NCT01526928 TIGER-1 (NCT02186301) TIGER-3 (NCT02322281) [75] |
| Olmutinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Terminated ° | NCT01588145 NCT02485652 [76] |
| Naquotinib | 3rd generation TKI Covalent Irreversible | Del19/L858R/T790M-EGFR | Clinical, Phase III (terminated) | NCT02588261 [77] |
| EAI001 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [78,79] |
| EAI045 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [78,79] |
| JBJ-04-125-02 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [80] |
| JBJ-09-063 | 4th generation TKI Allosteric Reversible | L858R/T790M/C797S-EGFR | Preclinical | [81] |
| CH7233163 | 4th generation TKI Non covalent Competitive | Del19/L858R/T790M/C797S-EGFR | Preclinical | [82] |
| BLU-945 | 4th generation TKI Reversible | Del19/L858R/T790M/C797S-EGFR | Phase I/II (Recruiting) | SYMPHONY (NCT04862780) |
| BBT-176 | 4th generation TKI Reversible | Del19/L858R/T790M/C797S-EGFR | Phase I/II (Recruiting) | NCT04820023 [83] |
| TQB3804 | 4th generation TKI | Del19/L858R/T790M/C797S-EGFR | Phase I (Unknown) | NCT04128085 |
| BPI-361175 | 4th generation TKI | Del19/L858R/T790M/C797S-EGFR | Phase I/II (Recruiting) | NCT05329298 |
| HJM-561 | PROTAC | Del19/L858R/T790M/C797S-EGFR | Preclinical | [84] |
| DDC-01-163 | PROTAC | L858R/T790M/C797S-EGFR | Preclinical | [85] |
| Mobocertinib | TKI | Ins20-EGFR | Approved by the FDA in 2021 | EXCLAIM [86] |
| Amivantamab | Bispecific Antibody | Ins20-EGFR MET | Approved by the FDA/EMA in 2021 | CHRYSALIS [87] PAPILLON (NCT04538664) |
6. Third-Generation TKIs (See Table 1)
7. Resistance to Osimertinib and Development of Fourth-Generation EGFR Inhibitors (See Table 1)
8. PROTACs
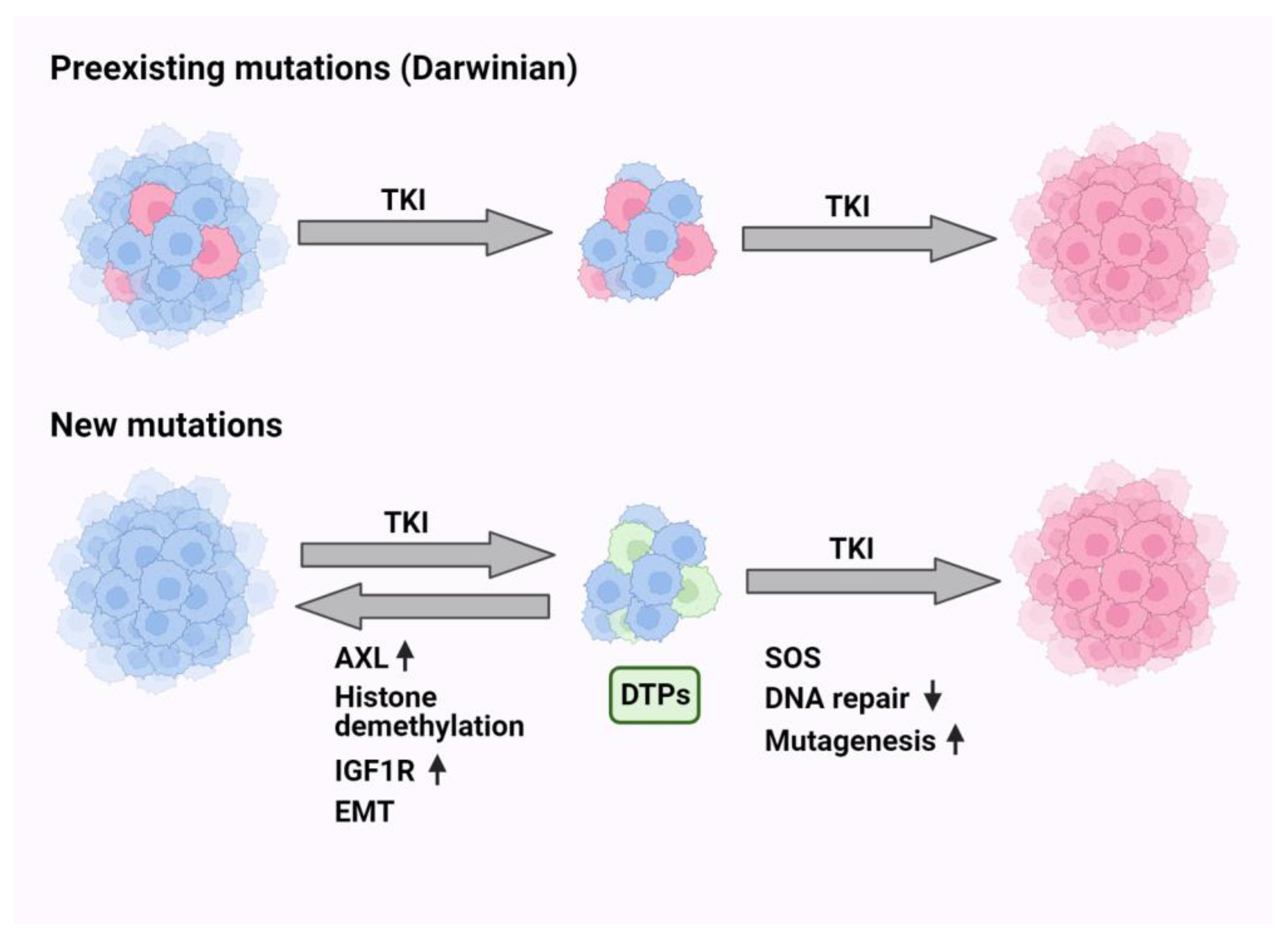
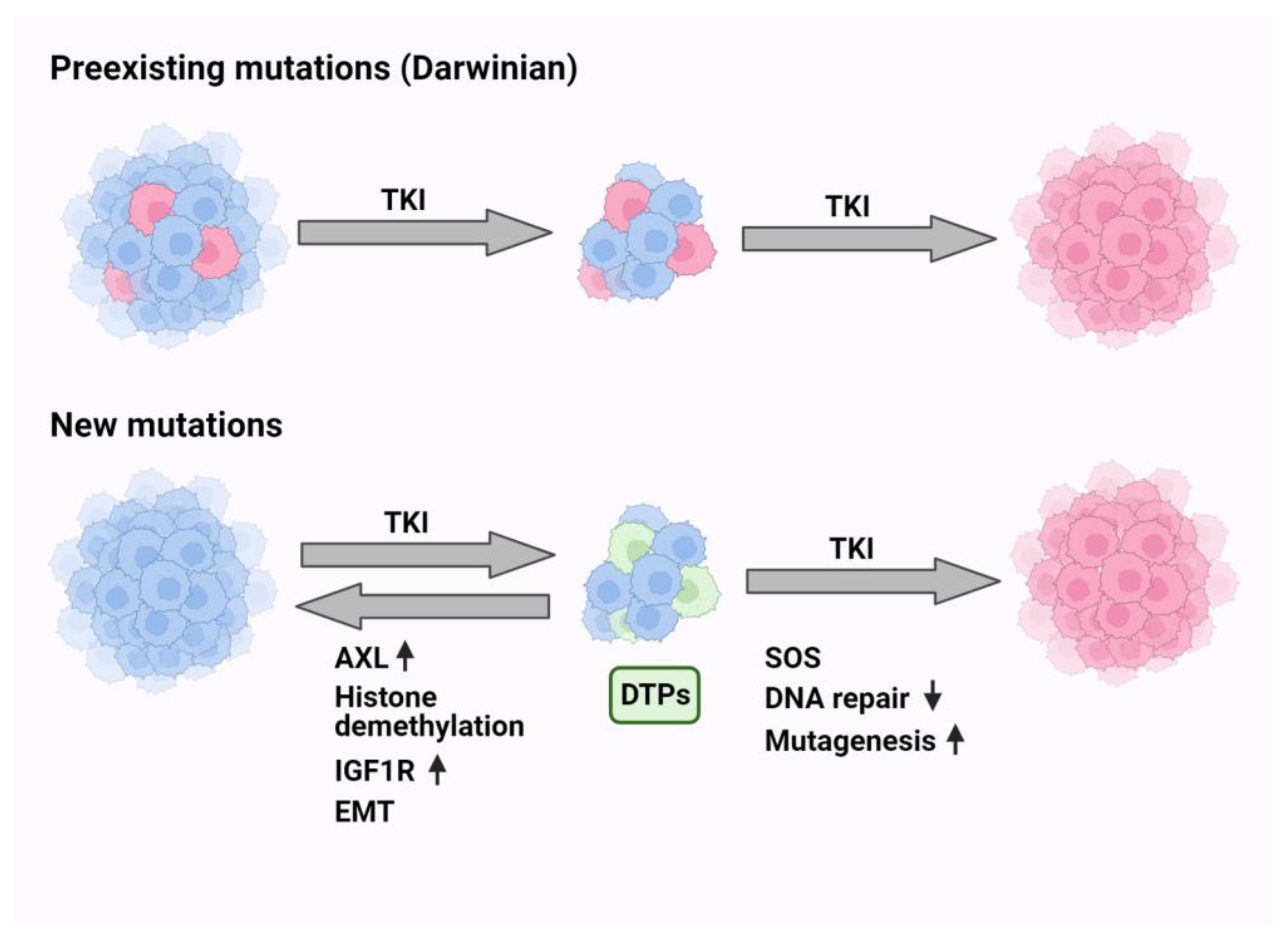
9. Darwinian Mechanisms Underlying Resistance to EGFR-Specific TKIs
10. Non-Darwinian Mechanisms of Resistance and Drug Tolerance Persistence
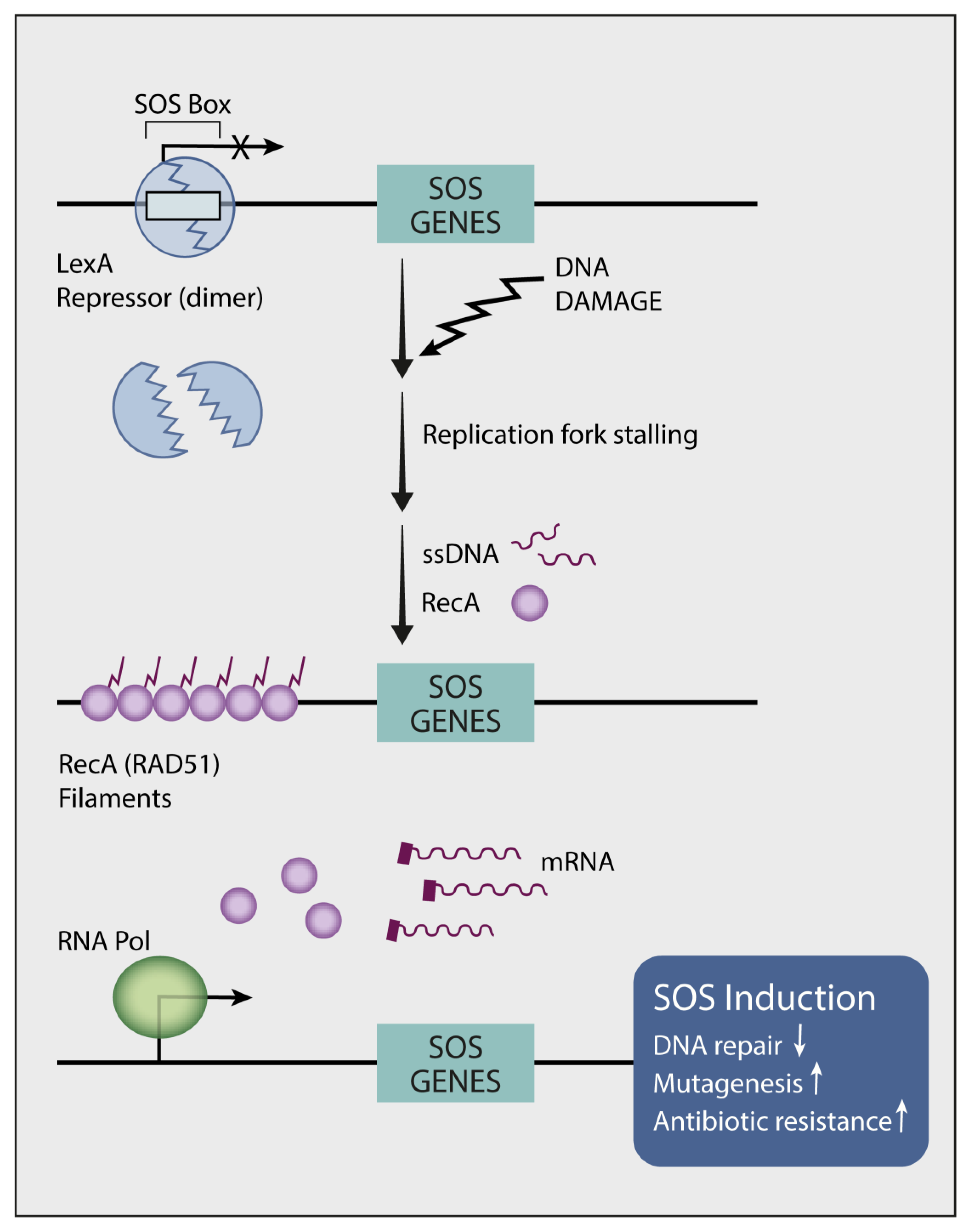
11. Endogenous Mutators Promote the Emergence of New Mutations While under TKI Treatment
12. A Biomarker Predicting Response of EGFR+ Tumors to Antibody Rather Than TKI Treatment
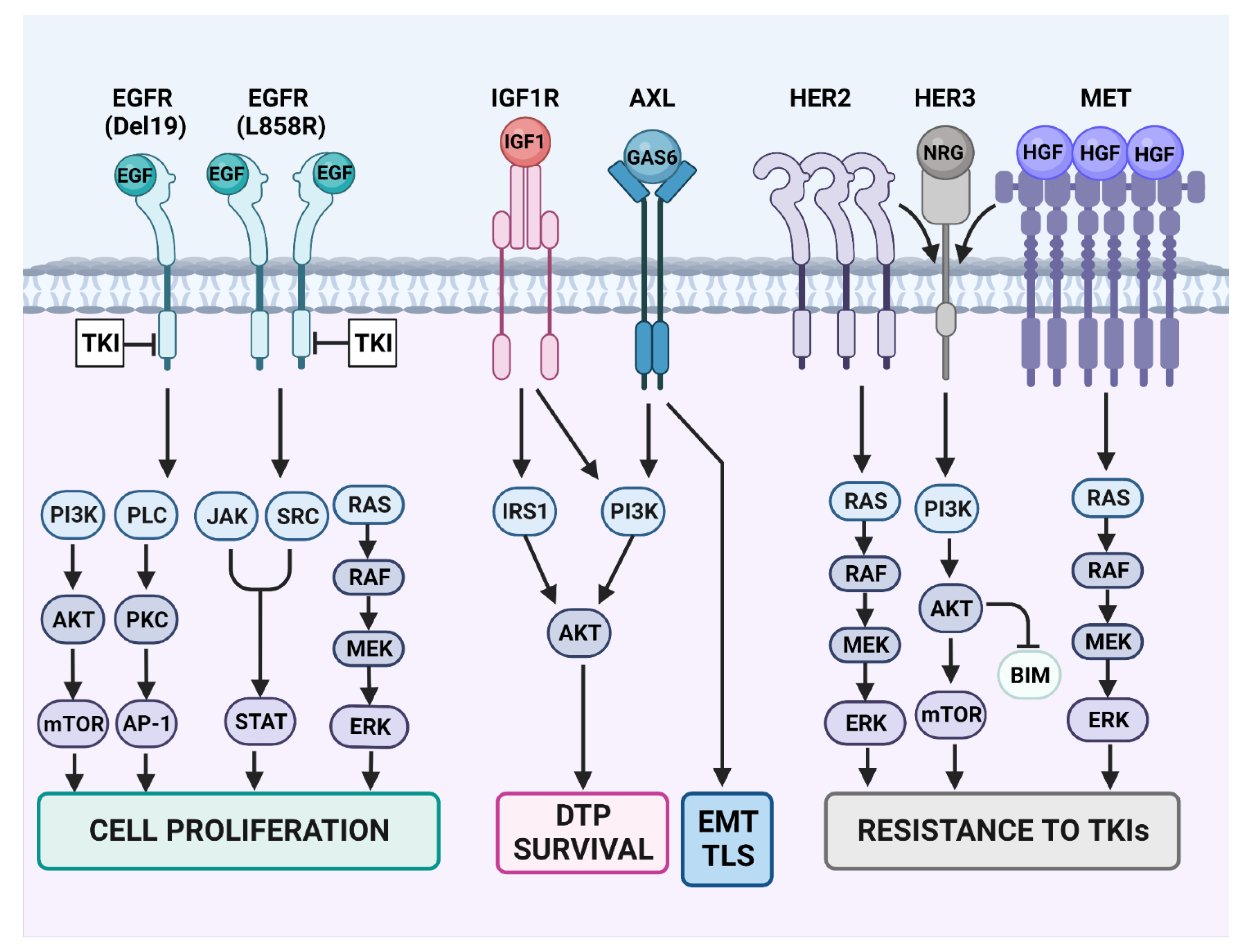
13. Individual Bypass Mechanisms and the Respective Combination Therapies (See Figure 3 and Table 2)
13.1. MET Activation
| Mechanism of Resistance | Strategy to Overcome Resistance | Drugs | Status | Relevant Studies |
|---|---|---|---|---|
| MET alterations | MET TKI | Osimertinib + savolitinib | Phase Ib (active) | NCT02143466 |
| Osimertinib + savolitinib | Phase II (recruiting) | NCT03778229 (SAVANNAH) | ||
| Osimertinib + savolitinib | Phase III (recruiting) | NCT05015608 (SACHI) | ||
| Osimertinib + savolitinib | Phase II (recruiting) | NCT03944772 (ORCHARD) | ||
| Osimertinib + savolitinib | Phase II (not yet recruiting) | NCT05163249 (FLOWERS) | ||
| Osimertinib + savolitinib | Phase III (recruiting) | NCT05261399 (SAFFRON) | ||
| Osimertinib + savolitinib | Phase II (active) | NCT04606771 | ||
| Osimertinib + tepotinib | Phase II (active) | NCT03940703 (INSIGHT) | ||
| Osimertinib + vebreltinib | Phase I/II (recruiting) | NCT04743505 | ||
| Bispecific Antibody (EGFR-MET) | Lazertinib + amivantamab | Phase III (active) | NCT04487080 (MARIPOSA) | |
| Lazertinib ± amivantamab | Phase I (recruiting) | NCT04077463 (CHRYSALIS2) | ||
| Lazertinib + amivantamab | Phase III (recruiting) | NCT05388669 (PALOMA3) | ||
| Lazertinib ± amivantamab ± carboplatin/pemetrexed | Phase I (recruiting) | NCT02609776 (CHRYSALIS) | ||
| Lazertinib + amivantamab + pemetrexed | Phase II (recruiting) | NCT05299125 (AMIGO-1) | ||
| Lazertinib + amivantamab + carboplatin/pemetrexed | Phase III (recruiting) | NCT04988295 (MARIPOSA-2) | ||
| Lazertinib + amivantamab + bevacizumab | Phase II (recruiting) | NCT05601973 (AMAZE-Lung) | ||
| Osimertinib+ EMB-01 | Phase I/II (not yet recruiting) | NCT05498389 | ||
| MET ADC | Osimertinib or erlotinib + telisotuzumab vedotin | Phase I (active) | NCT02099058 | |
| HER2 alterations | HER2 ADC | Trastuzumab deruxtecan | Phase II (active) | NCT03505710 (DESTINY-Lung01) |
| HER2 mAb | Osimertinib + necitumumab + trastuzumab | Phase I/II (recruiting) | NCT04285671 | |
| HER3 alterations | HER3 ADC | Patritumab deruxtecan | Phase II (recruiting) | NCT04619004 (HERTHENA-Lung01) |
| Patritumab deruxtecan | Phase III (recruiting) | NCT05338970 (HERTHENA-Lung02) | ||
| Osimertinib + patritumab deruxtecan | Phase I (recruiting) | NCT04676477 | ||
| Bispecific Antibody (EGFR-HER3) | Osimertinib + izalontamab | Phase II/III (recruiting) | NCT05020769 | |
| EGFR-HER3 ADC (Bispecific Antibody) | Osimertinib + BL-B01D1 | Phase II (not yet recruiting) | NCT05880706 | |
| AXL alterations | AXL TKI | Osimertinib + bemcentinib | Phase I/II (completed) | NCT02424617 |
| Alterations affecting downstream molecules | BRAF inhibitor | Dabrafenib + trametinib | Phase II (recruiting) | NCT04452877 |
| mTOR inhibitor | Osimertinib + sapanisertib | Phase I (recruiting) | NCT02503722 | |
| Osimertinib + sapanisertib | Phase I (not yet recruiting) | NCT04479306 | ||
| JAK inhibitor | Osimertinib + itacitinib | Phase I/II (active) | NCT02917993 | |
| Osimertinib + golidocitinib | Phase I/II (completed) | NCT03450330 (JACKPOT1) | ||
| MEK inhibitor | Osimertinib + selumetinib | Phase I (active) | NCT02143466 (TATTON) | |
| Osimertinib + selumetinib | Phase II (active) | NCT03392246 | ||
| PI3K inhibitor | Osimertinib + TQ-B3525 | Phase I/II (recruiting) | NCT05284994 | |
| RET alterations | RET TKI | Osimertinib + selpercatinib | Phase II (recruiting) | NCT03944772 (ORCHARD) |
| ALK alterations | ALK TKI | Osimertinib + alectinib | Phase II (recruiting) | NCT03944772 (ORCHARD) |
| CDK4/6 amplification | CDK 4/6 inhibitor | Osimertinib + G1738 (lerociclib) | Phase I/II (completed) | NCT03455829 |
| Osimertinib + abemaciclib | Phase II (unknown) | NCT04545710 | ||
| Others | Bcl-2 inhbitor | Osimertinib + navitoclax | Phase I (active) | NCT02520778 |
| Osimertinib + palcitoclax | Phase I (recruiting) | NCT04001777 | ||
| VEGF mAb | Osimertinib + bevacizumab | Phase II (active) | NCT03133546 (BOOSTER) | |
| Erlotinib + bevacizumab | Approved by the EMA in 2016 | BELIEVE JO25567 | ||
| Osimertinib + bevacizumab | Phase I/II (completed) | NCT02803203 | ||
| Osimertinib + bevacizumab | Phase III (recruiting) | NCT04181060 | ||
| Osimertinib + bevacizumab | Phase III (recruiting) | NCT05104281 | ||
| Osimertinib + bevacizumab | Phase II (active) | NCT02971501 | ||
| VEGFR mAb | Osimertinib + ramucirumab | Phase II (recruiting) | NCT03909334 | |
| Osimertinib + ramucirumab or necitumumab | Phase I (completed) | NCT02789345 | ||
| Erlotinib + ramucirumab | Approved by the FDA/EMA in 2020 | NCT02411448 (RELAY) | ||
| VEGFR-PDGFR-FGFR-cKIT TKI | Osimertinib + Anlotinib | Phase I/II (recruiting) | NCT04770688 (AUTOMAN) | |
| Aurora Kinase A inhibitor | Osimertinib + VIC-1911 | Phase I (recruiting) | NCT05489731 | |
| Osimertinib + alisertib | Phase I (not yet recruiting) | NCT04479306 | ||
| Osimertinib + LY3295668 | Phase I/II (active) | NCT05017025 | ||
| MERTK and FLT3 TKI | Osimertinib + MRX-2843 | Phase I (recruiting) | NCT04762199 | |
| ROS17TRK/ALK TKI | Osimertinib + repotrectinib | Phase I (recruiting) | NCT04772235 (TOTEM) |
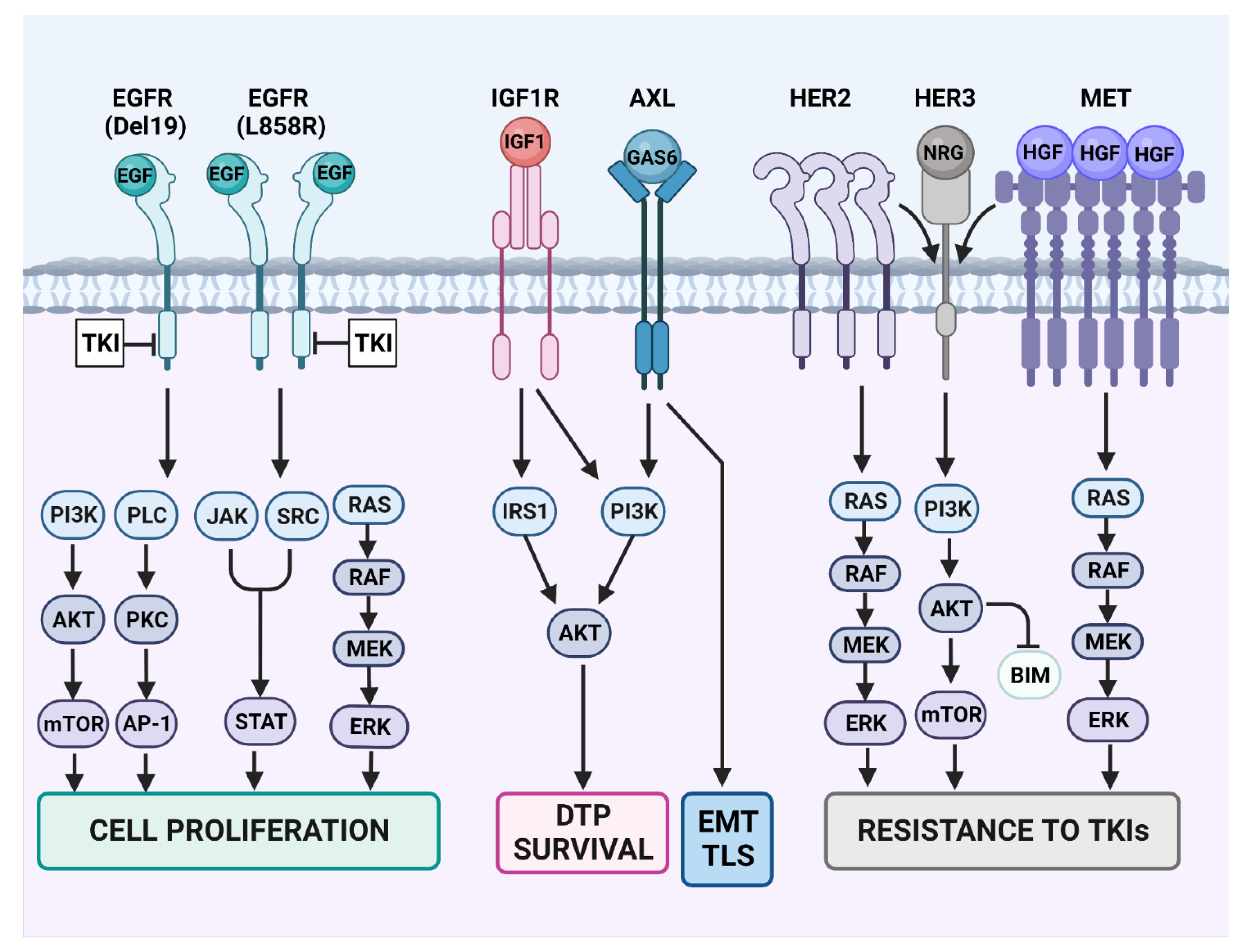
13.2. HER2 and HER3
13.3. Activation of AXL
13.4. IGF1-Receptor (IGF1R)
13.5. Fibroblast Growth Factor Receptors (FGFR)
14. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Wang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Yuan, J.Q.; Wang, K.F.; Fu, X.H.; Han, X.R.; Threapleton, D.; Yang, Z.Y.; Mao, C.; Tang, J.L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Zabransky, D.J.; Yankaskas, C.L.; Cochran, R.L.; Wong, H.Y.; Croessmann, S.; Chu, D.; Kavuri, S.M.; Brewer, M.R.; Rosen, D.M.; Dalton, W.B.; et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc. Natl. Acad. Sci. USA 2015, 112, E6205–E6214. [Google Scholar] [CrossRef]
- Wong, A.J.; Bigner, S.H.; Bigner, D.D.; Kinzler, K.W.; Hamilton, S.R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc. Natl. Acad. Sci. USA 1987, 84, 6899–6903. [Google Scholar] [CrossRef]
- Jeuken, J.; Sijben, A.; Alenda, C.; Rijntjes, J.; Dekkers, M.; Boots-Sprenger, S.; McLendon, R.; Wesseling, P. Robust detection of EGFR copy number changes and EGFR variant III: Technical aspects and relevance for glioma diagnostics. Brain Pathol. 2009, 19, 661–671. [Google Scholar] [CrossRef]
- Stahler, A.; Stintzing, S.; Modest, D.P.; Ricard, I.; Giessen-Jung, C.; Kapaun, C.; Ivanova, B.; Kaiser, F.; Fischer von Weikersthal, L.; Moosmann, N.; et al. Amphiregulin Expression Is a Predictive Biomarker for EGFR Inhibition in Metastatic Colorectal Cancer: Combined Analysis of Three Randomized Trials. Clin. Cancer Res. 2020, 26, 6559–6567. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Levi-Schaffer, F.; Sela, M.; Yarden, Y. Immunotherapy of cancer: From monoclonal to oligoclonal cocktails of anti-cancer antibodies: IUPHAR Review 18. Br. J. Pharmacol. 2016, 173, 1407–1424. [Google Scholar] [CrossRef]
- Pujol, J.L.; Pirker, R.; Lynch, T.J.; Butts, C.A.; Rosell, R.; Shepherd, F.A.; Vansteenkiste, J.; O’Byrne, K.J.; de Blas, B.; Heighway, J.; et al. Meta-analysis of individual patient data from randomized trials of chemotherapy plus cetuximab as first-line treatment for advanced non-small cell lung cancer. Lung Cancer 2014, 83, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Dang, C.V.; Agus, D.B.; Alexander, B.M.; Anderson, K.C.; Ashworth, A.; Barker, A.D.; Bastani, R.; Bhatia, S.; Bluestone, J.A.; et al. Future cancer research priorities in the USA: A Lancet Oncology Commission. Lancet Oncol. 2017, 18, e653–e706. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef]
- Mancini, M.; Yarden, Y. Mutational and network level mechanisms underlying resistance to anti-cancer kinase inhibitors. Semin. Cell Dev. Biol. 2016, 50, 164–176. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.D.; He, X.; Qin, B.D.; Liu, K.; Wu, Y.; Liu, J.; Hou, T.; Zang, Y.S. The prognostic value of tumor mutation burden in EGFR-mutant advanced lung adenocarcinoma, an analysis based on cBioPortal data base. J. Thorac. Dis. 2019, 11, 4507–4515. [Google Scholar] [CrossRef] [PubMed]
- Negrao, M.V.; Skoulidis, F.; Montesion, M.; Schulze, K.; Bara, I.; Shen, V.; Xu, H.; Hu, S.; Sui, D.; Elamin, Y.Y.; et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J. Immunother. Cancer 2021, 9, e002891. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Hill, W.; Lim, E.L.; Weeden, C.E.; Lee, C.; Augustine, M.; Chen, K.; Kuan, F.C.; Marongiu, F.; Evans, E.J., Jr.; Moore, D.A.; et al. Lung adenocarcinoma promotion by air pollutants. Nature 2023, 616, 159–167. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. S1), S24–S31. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Shtiegman, K.; Kochupurakkal, B.S.; Zwang, Y.; Pines, G.; Starr, A.; Vexler, A.; Citri, A.; Katz, M.; Lavi, S.; Ben-Basat, Y.; et al. Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene 2007, 26, 6968–6978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Huang, J.; Meng, L.; Yang, B.; Sun, S.; Luo, Z.; Chen, H. Safety Profile of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors: A Disproportionality Analysis of FDA Adverse Event Reporting System. Sci. Rep. 2020, 10, 4803. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discovery 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C.; et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef]
- Miyauchi, E.; Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Saijo, Y.; Yoshizawa, H.; et al. Efficacy of chemotherapy after first-line gefitinib therapy in EGFR mutation-positive advanced non-small cell lung cancer-data from a randomized Phase III study comparing gefitinib with carboplatin plus paclitaxel (NEJ002). Jpn. J. Clin. Oncol. 2015, 45, 670–676. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Wu, Y.L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.S.; Sriuranpong, V.; Chao, T.Y.; Nakagawa, K.; Chu, D.T.; Saijo, N.; et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Shimokawa, M.; Seto, T.; Morita, S.; Yatabe, Y.; Okamoto, I.; Tsurutani, J.; Satouchi, M.; Hirashima, T.; Atagi, S.; et al. Final overall survival results of WJTOG3405, a randomized phase III trial comparing gefitinib versus cisplatin with docetaxel as the first-line treatment for patients with stage IIIB/IV or postoperative recurrent EGFR mutation-positive non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1978–1984. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.K.; Wang, L.; Han, B.H.; Li, W.; Yu, P.; Liu, Y.P.; Ding, C.M.; Song, X.; Ma, Z.Y.; Ren, X.L.; et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE: A phase 3, open-label, randomized study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 2443–2450. [Google Scholar] [CrossRef]
- He, J.; Su, C.; Liang, W.; Xu, S.; Wu, L.; Fu, X.; Zhang, X.; Ge, D.; Chen, Q.; Mao, W.; et al. Icotinib versus chemotherapy as adjuvant treatment for stage II-IIIA EGFR-mutant non-small-cell lung cancer (EVIDENCE): A randomised, open-label, phase 3 trial. Lancet Respir. Med. 2021, 9, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2023, 41, 2869–2876. [Google Scholar] [CrossRef]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet. Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet. Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet. Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Chawla, A.; Rosell, R.; Corral, J.; Migliorino, M.R.; et al. Updated Overall Survival in a Randomized Study Comparing Dacomitinib with Gefitinib as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. Drugs 2021, 81, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Dong, X.; Jian, H.; Chen, J.; Chen, G.; Sun, Y.; Ji, Y.; Wang, Z.; Shi, J.; Lu, J.; et al. AENEAS: A Randomized Phase III Trial of Aumolertinib Versus Gefitinib as First-Line Therapy for Locally Advanced or MetastaticNon-Small-Cell Lung Cancer with EGFR Exon 19 Deletion or L858R Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 3162–3171. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Q.; Zhang, G.; Dong, X.; Yang, C.T.; Song, Y.; Chang, G.C.; Lu, Y.; Pan, H.; Chiu, C.H.; et al. Efficacy of Aumolertinib (HS-10296) in Patients with Advanced EGFR T790M+ NSCLC: Updated Post-National Medical Products Administration Approval Results From the APOLLO Registrational Trial. J. Thorac. Oncol. 2022, 17, 411–422. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, G.; Wang, X.; Liu, Y.; Wu, L.; Hao, Y.; Liu, C.; Zhu, S.; Zhang, X.; Li, Y.; et al. Furmonertinib (AST2818) versus gefitinib as first-line therapy for Chinese patients with locally advanced or metastatic EGFR mutation-positive non-small-cell lung cancer (FURLONG): A multicentre, double-blind, randomised phase 3 study. Lancet Respir. Med. 2022, 10, 1019–1028. [Google Scholar] [CrossRef]
- Ahn, M.J.; Han, J.Y.; Lee, K.H.; Kim, S.W.; Kim, D.W.; Lee, Y.G.; Cho, E.K.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet. Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef]
- Cho, B.C.; Han, J.Y.; Kim, S.W.; Lee, K.H.; Cho, E.K.; Lee, Y.G.; Kim, D.W.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. A Phase 1/2 Study of Lazertinib 240 mg in Patients with Advanced EGFR T790M-Positive NSCLC after Previous EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2022, 17, 558–567. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Y.; Zhang, G.; Zhou, J.; Cang, S.; Cheng, Y.; Wu, G.; Cao, P.; Lv, D.; Jian, H.; et al. Efficacy and Safety of Befotertinib (D-0316) in Patients with EGFR T790M-Mutated NSCLC That Had Progressed after Prior EGFR Tyrosine Kinase Inhibitor Therapy: A Phase 2, Multicenter, Single-Arm, Open-Label Study. J. Thorac. Oncol. 2022, 17, 1192–1204. [Google Scholar] [CrossRef]
- Lu, S.; Zhou, J.; Jian, H.; Wu, L.; Cheng, Y.; Fan, Y.; Fang, J.; Chen, G.; Zhang, Z.; Lv, D.; et al. Befotertinib (D-0316) versus icotinib as first-line therapy for patients with EGFR-mutated locally advanced or metastatic non-small-cell lung cancer: A multicentre, open-label, randomised phase 3 study. Lancet Respir. Med. 2023, 11, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zheng, X.; Zhao, H.; Fang, W.; Zhang, Y.; Ge, J.; Wang, L.; Wang, W.; Jiang, J.; Chuai, S.; et al. First-in-Human Phase I Study of AC0010, a Mutant-Selective EGFR Inhibitor in Non-Small Cell Lung Cancer: Safety, Efficacy, and Potential Mechanism of Resistance. J. Thorac. Oncol. 2018, 13, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.W.; Kim, S.W.; Ponce Aix, S.; Sequist, L.V.; Smit, E.F.; Yang, J.C.H.; Hida, T.; Toyozawa, R.; Felip, E.; Wolf, J.; et al. Nazartinib for treatment-naive EGFR-mutant non-small cell lung cancer: Results of a phase 2, single-arm, open-label study. Eur. J. Cancer 2022, 172, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.C.; Goldberg, S.B.; Kim, D.W.; Socinski, M.A.; Burns, T.F.; Lwin, Z.; Pathan, N.; Ma, W.D.; Masters, J.C.; Cossons, N.; et al. A phase 1b/2 study of PF-06747775 as monotherapy or in combination with Palbociclib in patients with epidermal growth factor receptor mutant advanced non-small cell lung cancer. Expert. Opin. Investig. Drugs 2022, 31, 747–757. [Google Scholar] [CrossRef]
- Yang, J.C.; Reckamp, K.L.; Kim, Y.C.; Novello, S.; Smit, E.F.; Lee, J.S.; Su, W.C.; Akerley, W.L.; Blakely, C.M.; Groen, H.J.M.; et al. Efficacy and Safety of Rociletinib Versus Chemotherapy in Patients with EGFR-Mutated NSCLC: The Results of TIGER-3, a Phase 3 Randomized Study. JTO Clin. Res. Rep. 2021, 2, 100114. [Google Scholar] [CrossRef]
- Park, K.; Jӓnne, P.A.; Kim, D.W.; Han, J.Y.; Wu, M.F.; Lee, J.S.; Kang, J.H.; Lee, D.H.; Cho, B.C.; Yu, C.J.; et al. Olmutinib in T790M-positive non-small cell lung cancer after failure of first-line epidermal growth factor receptor-tyrosine kinase inhibitor therapy: A global, phase 2 study. Cancer 2021, 127, 1407–1416. [Google Scholar] [CrossRef]
- Kelly, R.J.; Shepherd, F.A.; Krivoshik, A.; Jie, F.; Horn, L. A phase III, randomized, open-label study of ASP8273 versus erlotinib or gefitinib in patients with advanced stage IIIB/IV non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1127–1133. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Zhao, P.; Yao, M.Y.; Zhu, S.J.; Chen, J.Y.; Yun, C.H. Crystal structure of EGFR T790M/C797S/V948R in complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502, 332–337. [Google Scholar] [CrossRef]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef]
- To, C.; Beyett, T.S.; Jang, J.; Feng, W.W.; Bahcall, M.; Haikala, H.M.; Shin, B.H.; Heppner, D.E.; Rana, J.K.; Leeper, B.A.; et al. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat. Cancer 2022, 3, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Fujino, T.; Kim, C.; Lee, G.; Lee, Y.H.; Kim, D.W.; Ahn, J.S.; Mitsudomi, T.; Jin, T.; Lee, S.Y. BBT-176, a Novel Fourth-Generation Tyrosine Kinase Inhibitor for Osimertinib-Resistant Epidermal Growth Factor Receptor Mutations in Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Chen, Y.; Wang, Y.; Chen, J.; Lu, X.; Zhang, L.; Li, Y.; Wang, Z.; Ye, G.; Zhang, G. HJM-561, a Potent, Selective, and Orally Bioavailable EGFR PROTAC that Overcomes Osimertinib-Resistant EGFR Triple Mutations. Mol. Cancer Ther. 2022, 21, 1060–1066. [Google Scholar] [CrossRef]
- Jang, J.; To, C.; De Clercq, D.J.H.; Park, E.; Ponthier, C.M.; Shin, B.H.; Mushajiang, M.; Nowak, R.P.; Fischer, E.S.; Eck, M.J.; et al. Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew. Chem. Int. Ed. Engl. 2020, 59, 14481–14489. [Google Scholar] [CrossRef]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients with EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.Y.; Kim, S.W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results from the CHRYSALIS Phase I Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Sequist, L.V.; Rolfe, L.; Allen, A.R. Rociletinib in EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 578–579. [Google Scholar] [CrossRef]
- Chmielecki, J.; Mok, T.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Shepherd, F.A.; et al. Analysis of acquired resistance mechanisms to osimertinib in patients with EGFR-mutated advanced non-small cell lung cancer from the AURA3 trial. Nat. Commun. 2023, 14, 1071. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Minari, R.; Mazzaschi, G.; Gnetti, L.; La Monica, S.; Alfieri, R.; Campanini, N.; Verzè, M.; Olivani, A.; Ventura, L.; et al. Small Cell Lung Cancer Transformation as a Resistance Mechanism to Osimertinib in Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: Case Report and Literature Review. Front. Oncol. 2021, 11, 642190. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Yang, J.C.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Gray, J.E.; Cheng, Y.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat. Commun. 2023, 14, 1070. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- Eno, M.S.; Brubaker, J.D.; Campbell, J.E.; De Savi, C.; Guzi, T.J.; Williams, B.D.; Wilson, D.; Wilson, K.; Brooijmans, N.; Kim, J.; et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 9662–9677. [Google Scholar] [CrossRef]
- Ferlenghi, F.; Scalvini, L.; Vacondio, F.; Castelli, R.; Bozza, N.; Marseglia, G.; Rivara, S.; Lodola, A.; La Monica, S.; Minari, R.; et al. A sulfonyl fluoride derivative inhibits EGFR(L858R/T790M/C797S) by covalent modification of the catalytic lysine. Eur. J. Med. Chem. 2021, 225, 113786. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Chen, W.; Wang, Y.; Xing, D. Epidermal growth factor receptor PROTACs as an effective strategy for cancer therapy: A review. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188927. [Google Scholar] [CrossRef]
- Cheng, M.; Yu, X.; Lu, K.; Xie, L.; Wang, L.; Meng, F.; Han, X.; Chen, X.; Liu, J.; Xiong, Y.; et al. Discovery of Potent and Selective Epidermal Growth Factor Receptor (EGFR) Bifunctional Small-Molecule Degraders. J. Med. Chem. 2020, 63, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Vartak, R.; Deore, B.; Sanhueza, C.A.; Patel, K. Cetuximab-based PROteolysis targeting chimera for effectual downregulation of NSCLC with varied EGFR mutations. Int. J. Biol. Macromol. 2023, 252, 126413. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Du, Y.; Huang, L.; Cui, J.; Niu, J.; Xu, Y.; Zhu, Q. Discovery of novel potent covalent inhibitor-based EGFR degrader with excellent in vivo efficacy. Bioorg Chem. 2022, 120, 105605. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-E.C.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [PubMed]
- Pfeifer, H.; Lange, T.; Wystub, S.; Wassmann, B.; Maier, J.; Binckebanck, A.; Giagounidis, A.; Stelljes, M.; Schmalzing, M.; Duhrsen, U.; et al. Prevalence and dynamics of bcr-abl kinase domain mutations during imatinib treatment differ in patients with newly diagnosed and recurrent bcr-abl positive acute lymphoblastic leukemia. Leukemia 2012, 26, 1475–1481. [Google Scholar] [CrossRef]
- Kaldalu, N.; Hauryliuk, V.; Tenson, T. Persisters—As elusive as ever. Appl. Microbiol. Biotechnol. 2016, 100, 6545–6553. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef]
- Haga, Y.; Marrocco, I.; Noronha, A.; Uribe, M.L.; Nataraj, N.B.; Sekar, A.; Drago-Garcia, D.; Borgoni, S.; Lindzen, M.; Giri, S.; et al. Host-Dependent Phenotypic Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2021, 81, 3862–3875. [Google Scholar] [CrossRef]
- Moghal, N.; Li, Q.; Stewart, E.L.; Navab, R.; Mikubo, M.; D’Arcangelo, E.; Martins-Filho, S.N.; Raghavan, V.; Pham, N.A.; Li, M.; et al. Single-Cell Analysis Reveals Transcriptomic Features of Drug-Tolerant Persisters and Stromal Adaptation in a Patient-Derived EGFR-Mutated Lung Adenocarcinoma Xenograft Model. J. Thorac. Oncol. 2023, 18, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Woolston, A.; Khan, K.; Spain, G.; Barber, L.J.; Griffiths, B.; Gonzalez-Exposito, R.; Hornsteiner, L.; Punta, M.; Patil, Y.; Newey, A.; et al. Genomic and Transcriptomic Determinants of Therapy Resistance and Immune Landscape Evolution during Anti-EGFR Treatment in Colorectal Cancer. Cancer Cell 2019, 36, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Xu, C.; Yanagita, M.; Monast, C.S.; Pratilas, C.A.; Montero, J.; Butaney, M.; Shimamura, T.; Sholl, L.; Ivanova, E.V.; et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 29–39. [Google Scholar] [CrossRef]
- Kashima, Y.; Shibahara, D.; Suzuki, A.; Muto, K.; Kobayashi, I.S.; Plotnick, D.; Udagawa, H.; Izumi, H.; Shibata, Y.; Tanaka, K.; et al. Single-Cell Analyses Reveal Diverse Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors in Lung Cancer. Cancer Res. 2021, 81, 4835–4848. [Google Scholar] [CrossRef]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-dependent beta-catenin signaling mediates EGFR TKI drug persistence in EGFR mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef]
- Bousquet Mur, E.; Bernardo, S.; Papon, L.; Mancini, M.; Fabbrizio, E.; Goussard, M.; Ferrer, I.; Giry, A.; Quantin, X.; Pujol, J.L.; et al. Notch inhibition overcomes resistance to tyrosine kinase inhibitors in EGFR-driven lung adenocarcinoma. J. Clin. Investig. 2020, 130, 612–624. [Google Scholar] [CrossRef]
- Radman, M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. In Molecular Mechanisms for Repair of DNA: Part A; Basic Life Science Book Series; Springer: Boston, MA, USA, 1975; pp. 355–367. [Google Scholar] [CrossRef]
- wBaharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef]
- Ohmori, H.; Friedberg, E.C.; Fuchs, R.P.; Goodman, M.F.; Hanaoka, F.; Hinkle, D.; Kunkel, T.A.; Lawrence, C.W.; Livneh, Z.; Nohmi, T.; et al. The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [Google Scholar] [CrossRef]
- Noronha, A.; Belugali Nataraj, N.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.T.; Reyes, G.; Gries, K.; Ceylan, C.U.; Sharma, S.; Meurer, M.; Knop, M.; Chabes, A.; Hombauer, H. Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E4442–E4451. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Elledge, S.J. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol. Cell Biol. 1997, 17, 6105–6113. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570.e1522. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Hastings, P.J.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef]
- Garraway, L.A.; Verweij, J.; Ballman, K.V. Precision oncology: An overview. J. Clin. Oncol. 2013, 31, 1803–1805. [Google Scholar] [CrossRef]
- Lee, C.K.; Wu, Y.L.; Ding, P.N.; Lord, S.J.; Inoue, A.; Zhou, C.; Mitsudomi, T.; Rosell, R.; Pavlakis, N.; Links, M.; et al. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes after Treatment with EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFR-Mutant Lung Cancer: A Meta-Analysis. J. Clin. Oncol. 2015, 33, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Papakyriakou, A.; Vourloumis, D.; Tzortzatou-Stathopoulou, F.; Karpusas, M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins Struct. Funct. Bioinform. 2009, 76, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Giri, S.; Simoni-Nieves, A.; Gupta, N.; Rudnitsky, A.; Haga, Y.; Romaniello, D.; Sekar, A.; Zerbib, M.; Oren, R.; et al. L858R emerges as a potential biomarker predicting response of lung cancer models to anti-EGFR antibodies: Comparison of osimertinib vs. cetuximab. Cell Rep. Med. 2023, 4, 101142. [Google Scholar] [CrossRef]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Dhillon, S. Capmatinib: First Approval. Drugs 2020, 80, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Tepotinib: First Approval. Drugs 2020, 80, 829–833. [Google Scholar] [CrossRef]
- Syed, Y.Y. Amivantamab: First Approval. Drugs 2021, 81, 1349–1353. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib after Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients with EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet. Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Shu, C.A.; Goto, K.; Ohe, Y.; Besse, B.; Lee, S.-H.; Wang, Y.; Griesinger, F.; Yang, J.C.-H.; Felip, E.; Sanborn, R.E.; et al. Amivantamab and lazertinib in patients with EGFR-mutant non–small cell lung (NSCLC) after progression on osimertinib and platinum-based chemotherapy: Updated results from CHRYSALIS-2. J. Clin. Oncol. 2022, 40, 9006. [Google Scholar] [CrossRef]
- Cho, B.C.; Felip, E.; Hayashi, H.; Thomas, M.; Lu, S.; Besse, B.; Sun, T.; Martinez, M.; Sethi, S.N.; Shreeve, S.M.; et al. MARIPOSA: Phase 3 study of first-line amivantamab + lazertinib versus osimertinib in EGFR-mutant non-small-cell lung cancer. Future Oncol. 2022, 18, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Gaborit, N.; Lindzen, M.; Salame, T.M.; Dall’Ora, M.; Sevilla-Sharon, M.; Abdul-Hai, A.; Downward, J.; Yarden, Y. Combining three antibodies nullifies feedback-mediated resistance to erlotinib in lung cancer. Sci. Signal 2015, 8, ra53. [Google Scholar] [CrossRef]
- Romaniello, D.; Mazzeo, L.; Mancini, M.; Marrocco, I.; Noronha, A.; Kreitman, M.; Srivastava, S.; Ghosh, S.; Lindzen, M.; Salame, T.M.; et al. A Combination of Approved Antibodies Overcomes Resistance of Lung Cancer to Osimertinib by Blocking Bypass Pathways. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5610–5621. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Romaniello, D.; Vaknin, I.; Drago-Garcia, D.; Oren, R.; Uribe, M.L.; Belugali Nataraj, N.; Ghosh, S.; Eilam, R.; Salame, T.M.; et al. Upfront admixing antibodies and EGFR inhibitors preempts sequential treatments in lung cancer models. EMBO Mol. Med. 2021, 13, e13144. [Google Scholar] [CrossRef] [PubMed]
- La Monica, S.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Digiacomo, G.; Flammini, L.; Barocelli, E.; Minari, R.; Naldi, N.; et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J. Exp. Clin. Cancer Res. 2017, 36, 174. [Google Scholar] [CrossRef] [PubMed]
- Scharpenseel, H.; Hanssen, A.; Loges, S.; Mohme, M.; Bernreuther, C.; Peine, S.; Lamszus, K.; Goy, Y.; Petersen, C.; Westphal, M.; et al. EGFR and HER3 expression in circulating tumor cells and tumor tissue from non-small cell lung cancer patients. Sci. Rep. 2019, 9, 7406. [Google Scholar] [CrossRef]
- Kawano, O.; Sasaki, H.; Endo, K.; Suzuki, E.; Haneda, H.; Yukiue, H.; Kobayashi, Y.; Yano, M.; Fujii, Y. ErbB3 mRNA expression correlated with specific clinicopathologic features of Japanese lung cancers. J. Surg. Res. 2008, 146, 43–48. [Google Scholar] [CrossRef]
- Mancini, M.; Gal, H.; Gaborit, N.; Mazzeo, L.; Romaniello, D.; Salame, T.M.; Lindzen, M.; Mahlknecht, G.; Enuka, Y.; Burton, D.G.; et al. An oligoclonal antibody durably overcomes resistance of lung cancer to third-generation EGFR inhibitors. EMBO Mol. Med. 2018, 10, 294–308. [Google Scholar] [CrossRef]
- Romaniello, D.; Marrocco, I.; Belugali Nataraj, N.; Ferrer, I.; Drago-Garcia, D.; Vaknin, I.; Oren, R.; Lindzen, M.; Ghosh, S.; Kreitman, M.; et al. Targeting HER3, a Catalytically Defective Receptor Tyrosine Kinase, Prevents Resistance of Lung Cancer to a Third-Generation EGFR Kinase Inhibitor. Cancers 2020, 12, 2394. [Google Scholar] [CrossRef]
- Haikala, H.M.; Jänne, P.A. Thirty Years of HER3: From Basic Biology to Therapeutic Interventions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3528–3539. [Google Scholar] [CrossRef]
- Yonesaka, K.; Hirotani, K.; Kawakami, H.; Takeda, M.; Kaneda, H.; Sakai, K.; Okamoto, I.; Nishio, K.; Janne, P.A.; Nakagawa, K. Anti-HER3 monoclonal antibody patritumab sensitizes refractory non-small cell lung cancer to the epidermal growth factor receptor inhibitor erlotinib. Oncogene 2016, 35, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Yonesaka, K.; Hirotani, K.; von Pawel, J.; Dediu, M.; Chen, S.; Copigneaux, C.; Nakagawa, K. Circulating heregulin level is associated with the efficacy of patritumab combined with erlotinib in patients with non-small cell lung cancer. Lung Cancer 2017, 105, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Baik, C.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suda, K.; Shimizu, S.; Sakai, K.; Mizuuchi, H.; Tomizawa, K.; Takemoto, T.; Nishio, K.; Mitsudomi, T. Clinical, Pathological, and Molecular Features of Lung Adenocarcinomas with AXL Expression. PLoS ONE 2016, 11, e0154186. [Google Scholar] [CrossRef]
- Wu, Z.; Bai, F.; Fan, L.; Pang, W.; Han, R.; Wang, J.; Liu, Y.; Yan, X.; Duan, H.; Xing, L. Coexpression of receptor tyrosine kinase AXL and EGFR in human primary lung adenocarcinomas. Hum. Pathol. 2015, 46, 1935–1944. [Google Scholar] [CrossRef]
- Wang, F.; Liu, X.; Bartholdy, B.A.; Cheng, H.; Halmos, B. Blockade of AXL activation overcomes acquired resistance to EGFR tyrosine kinase inhibition in non-small cell lung cancer. Transl. Cancer Res. 2019, 8, 2425–2438. [Google Scholar] [CrossRef]
- Zaman, A.; Bivona, T.G. Targeting AXL in NSCLC. Lung Cancer Targets Ther. 2021, 12, 67–79. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, Y.; James, M.; Jeong, J.H.; You, M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 2016, 55, 991–1001. [Google Scholar] [CrossRef]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Murata, Y.; Kusumoto, S.; Ando, K.; Ishida, H.; Ohnishi, T.; Sasaki, Y. Distinct Afatinib Resistance Mechanisms Identified in Lung Adenocarcinoma Harboring an EGFR Mutation. Mol. Cancer Res. 2017, 15, 915–928. [Google Scholar] [CrossRef]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Jacob Berger, A.; Gigi, E.; Kupershmidt, L.; Meir, Z.; Gavert, N.; Zwang, Y.; Prior, A.; Gilad, S.; Harush, U.; Haviv, I.; et al. IRS1 phosphorylation underlies the non-stochastic probability of cancer cells to persist during EGFR inhibition therapy. Nat. Cancer 2021, 2, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef] [PubMed]
- Varmus, H.; Unni, A.M.; Lockwood, W.W. How Cancer Genomics Drives Cancer Biology: Does Synthetic Lethality Explain Mutually Exclusive Oncogenic Mutations? Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Herbst, R.S.; Boshoff, C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat. Med. 2021, 27, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrocco, I.; Yarden, Y. Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators. Cancers 2023, 15, 5009. https://doi.org/10.3390/cancers15205009
Marrocco I, Yarden Y. Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators. Cancers. 2023; 15(20):5009. https://doi.org/10.3390/cancers15205009
Chicago/Turabian StyleMarrocco, Ilaria, and Yosef Yarden. 2023. "Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators" Cancers 15, no. 20: 5009. https://doi.org/10.3390/cancers15205009
APA StyleMarrocco, I., & Yarden, Y. (2023). Resistance of Lung Cancer to EGFR-Specific Kinase Inhibitors: Activation of Bypass Pathways and Endogenous Mutators. Cancers, 15(20), 5009. https://doi.org/10.3390/cancers15205009