
Molecular Biology and Clinical Management of Esophageal Adenocarcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiological and Biological Characteristics
2.1. Epidemiology and Etiology
2.2. Histology
2.3. Biology and Immunology
2.4. Genetics
2.5. Epigenetics
3. Management of EAC
3.1. Prevention of EAC in Patients with BE
3.2. Treatment of EAC
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Dikken, J.L.; Lemmens, V.E.; Wouters, M.W.; Wijnhoven, B.P.; Siersema, P.D.; Nieuwenhuijzen, G.A.; van Sandick, J.W.; Cats, A.; Verheij, M.; Coebergh, J.W.; et al. Increased incidence and survival for oesophageal cancer but not for gastric cardia cancer in the Netherlands. Eur. J. Cancer 2012, 48, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.F.; Rajput, A.; Chadha, K.S.; Douglas, W.G.; Hill, H.; Nwogu, C.; Nava, H.R.; Sabel, M.S. The changing profile of esophageal cancer presentation and its implication for diagnosis. J. Natl. Med. Assoc. 2007, 99, 620–626. [Google Scholar] [PubMed]
- Coleman, H.G.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.; Bartenhagen, C.; Hoffmann, M.; Will, D.; Fischer, J.C.; Baldus, S.E.; Vay, C.; Fluegen, G.; Dizdar, L.; Vallbohmer, D.; et al. Disseminated tumour cells with highly aberrant genomes are linked to poor prognosis in operable oesophageal adenocarcinoma. Br. J. Cancer 2017, 117, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Bergstrom, R.; Lindgren, A.; Nyren, O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N. Engl. J. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, B.; Yang, Q.; Pan, Y.; Yang, W.; Freedland, S.J.; Ding, L.W.; Freeman, M.R.; Breunig, J.J.; Bhowmick, N.A.; et al. A Transcriptional Regulatory Loop of Master Regulator Transcription Factors, PPARG, and Fatty Acid Synthesis Promotes Esophageal Adenocarcinoma. Cancer Res. 2021, 81, 1216–1229. [Google Scholar] [CrossRef]
- Quante, M.; Graham, T.A.; Jansen, M. Insights Into the Pathophysiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 406–420. [Google Scholar] [CrossRef]
- Sharma, N.; Ho, K.Y. Risk Factors for Barrett’s Oesophagus. Gastrointest. Tumors 2016, 3, 103–108. [Google Scholar] [CrossRef]
- Gregson, E.M.; Bornschein, J.; Fitzgerald, R.C. Genetic progression of Barrett’s oesophagus to oesophageal adenocarcinoma. Br. J. Cancer 2016, 115, 403–410. [Google Scholar] [CrossRef]
- Li, X.; Galipeau, P.C.; Paulson, T.G.; Sanchez, C.A.; Arnaudo, J.; Liu, K.; Sather, C.L.; Kostadinov, R.L.; Odze, R.D.; Kuhner, M.K.; et al. Temporal and spatial evolution of somatic chromosomal alterations: A case-cohort study of Barrett’s esophagus. Cancer Prev. Res. 2014, 7, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Mehtani, K.; Quesenberry, C.; Zhao, W.; de Boer, J.; Weiss, N.S. Impact of endoscopic surveillance on mortality from Barrett’s esophagus-associated esophageal adenocarcinomas. Gastroenterology 2013, 145, 312–319.e311. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Raja, S.; Kamath, S.; Jang, S.; Allende, D.; McNamara, M.; Videtic, G.; Murthy, S.; Bhatt, A. Esophageal adenocarcinoma: A dire need for early detection and treatment. Cleve. Clin. J. Med. 2022, 89, 269–279. [Google Scholar] [CrossRef]
- Eyck, B.M.; van Lanschot, J.J.B.; Hulshof, M.; van der Wilk, B.J.; Shapiro, J.; van Hagen, P.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.L.; van Laarhoven, H.W.M.; Nieuwenhuijzen, G.A.P.; et al. Ten-Year Outcome of Neoadjuvant Chemoradiotherapy Plus Surgery for Esophageal Cancer: The Randomized Controlled CROSS Trial. J. Clin. Oncol. 2021, 39, 1995–2004. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Valkema, M.J.; Mostert, B.; Lagarde, S.M.; Wijnhoven, B.P.L.; van Lanschot, J.J.B. The effectivity of targeted therapy and immunotherapy in patients with advanced metastatic and non-metastatic cancer of the esophagus and esophago-gastric junction. Updates Surg. 2023, 75, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Yoon, H.H. The promise of PD-1 inhibitors in gastro-esophageal cancers: Microsatellite instability vs. PD-L1. J. Gastrointest. Oncol. 2016, 7, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Soerjomataram, I.; Rumgay, H.; Coleman, H.G.; Thrift, A.P.; Vignat, J.; Laversanne, M.; Ferlay, J.; Arnold, M. The Global Landscape of Esophageal Squamous Cell Carcinoma and Esophageal Adenocarcinoma Incidence and Mortality in 2020 and Projections to 2040: New Estimates From GLOBOCAN 2020. Gastroenterology 2022, 163, 649–658.e642. [Google Scholar] [CrossRef]
- Hang, T.P.; Spiritos, Z.; Gamboa, A.M.; Chen, Z.; Force, S.; Patel, V.; Chawla, S.; Keilin, S.; Saba, N.F.; El-Rayes, B.; et al. Epidemiology of early esophageal adenocarcinoma. Clin. Endosc. 2022, 55, 372–380. [Google Scholar] [CrossRef]
- Strauss, A.; Min, E.J.; Long, Q.; Gabriel, P.; Yang, Y.X.; Falk, G.W. Is the age of diagnosis of esophageal adenocarcinoma getting younger? Analysis at a tertiary care center. Dis. Esophagus 2020, 33, doz112. [Google Scholar] [CrossRef]
- Cook, M.B.; Chow, W.H.; Devesa, S.S. Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977–2005. Br. J. Cancer 2009, 101, 855–859. [Google Scholar] [CrossRef] [PubMed]
- To, H.; Clemons, N.J.; Duong, C.P.; Trainer, A.H.; Phillips, W.A. The Genetics of Barrett’s Esophagus: A Familial and Population-Based Perspective. Dig. Dis. Sci. 2016, 61, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, R.E.; Spittuler, L.F.; Peute, A.; van Oijen, M.G.; Ten Kate, F.J.; Vermeijden, J.R.; Oberndorff, A.; van Baal, J.W.; Siersema, P.D. Familial clustering of Barrett’s esophagus and esophageal adenocarcinoma in a European cohort. Clin. Gastroenterol. Hepatol. 2014, 12, 1656–1663.e1651. [Google Scholar] [CrossRef] [PubMed]
- Fecteau, R.E.; Kong, J.; Kresak, A.; Brock, W.; Song, Y.; Fujioka, H.; Elston, R.; Willis, J.E.; Lynch, J.P.; Markowitz, S.D.; et al. Association Between Germline Mutation in VSIG10L and Familial Barrett Neoplasia. JAMA Oncol. 2016, 2, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Buas, M.F.; He, Q.; Johnson, L.G.; Onstad, L.; Levine, D.M.; Thrift, A.P.; Gharahkhani, P.; Palles, C.; Lagergren, J.; Fitzgerald, R.C.; et al. Germline variation in inflammation-related pathways and risk of Barrett’s oesophagus and oesophageal adenocarcinoma. Gut 2017, 66, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.Y.; de Dieu Tapsoba, J.; Buas, M.F.; Onstad, L.E.; Levine, D.M.; Risch, H.A.; Chow, W.H.; Bernstein, L.; Ye, W.; Lagergren, J.; et al. A newly identified susceptibility locus near FOXP1 modifies the association of gastroesophageal reflux with Barrett’s esophagus. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, R.J.; English, D.R.; Hopper, J.L.; Giles, G.G. Body size and composition and the risk of gastric and oesophageal adenocarcinoma. Int. J. Cancer 2006, 118, 2628–2631. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Sadeghi, S.; Pandeya, N.; Smithers, B.M.; Gotley, D.C.; Bain, C.J.; Webb, P.M.; Green, A.C.; Australian Cancer, S. Combined effects of obesity, acid reflux and smoking on the risk of adenocarcinomas of the oesophagus. Gut 2008, 57, 173–180. [Google Scholar] [CrossRef]
- Corley, D.A.; Kubo, A.; Levin, T.R.; Block, G.; Habel, L.; Zhao, W.; Leighton, P.; Quesenberry, C.; Rumore, G.J.; Buffler, P.A. Abdominal obesity and body mass index as risk factors for Barrett’s esophagus. Gastroenterology 2007, 133, 34–41. [Google Scholar] [CrossRef]
- Edelstein, Z.R.; Farrow, D.C.; Bronner, M.P.; Rosen, S.N.; Vaughan, T.L. Central adiposity and risk of Barrett’s esophagus. Gastroenterology 2007, 133, 403–411. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Kvapil, P.; Hacken-Bitar, J.; Kramer, J.R. Abdominal obesity and the risk of Barrett’s esophagus. Am. J. Gastroenterol. 2005, 100, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.C.; Abdalla, S.; Onwuegbusi, B.A.; Sirieix, P.; Saeed, I.T.; Burnham, W.R.; Farthing, M.J. Inflammatory gradient in Barrett’s oesophagus: Implications for disease complications. Gut 2002, 51, 316–322. [Google Scholar] [CrossRef]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Primers 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Munch, N.S.; Fang, H.Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506.e492. [Google Scholar] [CrossRef]
- Reid, B.J.; Li, X.; Galipeau, P.C.; Vaughan, T.L. Barrett’s oesophagus and oesophageal adenocarcinoma: Time for a new synthesis. Nat. Rev. Cancer 2010, 10, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.C.P.; Straub, D.; Read, M.; Hoefnagel, S.J.M.; Romero-Pinedo, S.; Abadia-Molina, A.C.; Clemons, N.J.; Wang, K.; Calpe, S.; Phillips, W.; et al. Inhibition of BMP2 and BMP4 Represses Barrett’s Esophagus While Enhancing the Regeneration of Squamous Epithelium in Preclinical Models. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1199–1217. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.C.; Holub, J.; Lieberman, D.; Hur, C. Low Prevalence of Suspected Barrett’s Esophagus in Patients With Gastroesophageal Reflux Disease Without Alarm Symptoms. Clin. Gastroenterol. Hepatol. 2019, 17, 857–863. [Google Scholar] [CrossRef]
- Rajendra, S. Diagnosis and Management of Barrett’s Esophagus: An Updated ACG Guideline. Am. J. Gastroenterol. 2022, 117, 1880. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef]
- Sawas, T.; Katzka, D.A. Esophageal adenocarcinoma phenotypes and risk factors. Curr. Opin. Gastroenterol. 2022, 38, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Sawas, T.; Killcoyne, S.; Iyer, P.G.; Wang, K.K.; Smyrk, T.C.; Kisiel, J.B.; Qin, Y.; Ahlquist, D.A.; Rustgi, A.K.; Costa, R.J.; et al. Identification of Prognostic Phenotypes of Esophageal Adenocarcinoma in 2 Independent Cohorts. Gastroenterology 2018, 155, 1720–1728.e1724. [Google Scholar] [CrossRef] [PubMed]
- Nowicki-Osuch, K.; Zhuang, L.; Jammula, S.; Bleaney, C.W.; Mahbubani, K.T.; Devonshire, G.; Katz-Summercorn, A.; Eling, N.; Wilbrey-Clark, A.; Madissoon, E.; et al. Molecular phenotyping reveals the identity of Barrett’s esophagus and its malignant transition. Science 2021, 373, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Straub, D.; Elferink, R.P.J.O.; Jansen, P.L.M.; Bergman, J.J.G.H.M.; Parikh, K.; Krishnadath, K.K. Glyco-conjugated bile acids drive the initial metaplastic gland formation from multi-layered glands through crypt-fission in a murine model. PLoS ONE 2019, 14, e0220050. [Google Scholar] [CrossRef] [PubMed]
- Minacapelli, C.D.; Bajpai, M.; Geng, X.; Cheng, C.L.; Chouthai, A.A.; Souza, R.; Spechler, S.J.; Das, K.M. Barrett’s metaplasia develops from cellular reprograming of esophageal squamous epithelium due to gastroesophageal reflux. Am. J. Physiol-Gastr. Liver Physiol. 2017, 312, G615–G622. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, H.Y.; Zhang, Y.C.; Yang, Y.; Lu, R.; Liu, K.C.; Lin, S.J.; Lan, X.P.; Wang, H.K.; Wu, H.; et al. Transitional basal cells at the squamous-columnar junction generate Barrett’s oesophagus. Nature 2017, 550, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ouyang, H.; Yamamoto, Y.; Kumar, P.A.; Wei, T.S.; Dagher, R.; Vincent, M.; Lu, X.; Bellizzi, A.M.; Ho, K.Y.; et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 2011, 145, 1023–1035. [Google Scholar] [CrossRef]
- Leedham, S.J.; Preston, S.L.; McDonald, S.A.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef]
- Desai, T.K.; Krishnan, K.; Samala, N.; Singh, J.; Cluley, J.; Perla, S.; Howden, C.W. The incidence of oesophageal adenocarcinoma in non-dysplastic Barrett’s oesophagus: A meta-analysis. Gut 2012, 61, 970–976. [Google Scholar] [CrossRef]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sorensen, H.T.; Funch-Jensen, P. Incidence of Adenocarcinoma among Patients with Barrett’s Esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef]
- Vennalaganti, P.R.; Kanakadandi, V.N.; Gross, S.A.; Parasa, S.; Wang, K.K.; Gupta, N.; Sharma, P. Inter-Observer Agreement among Pathologists Using Wide-Area Transepithelial Sampling With Computer-Assisted Analysis in Patients With Barrett’s Esophagus. Am. J. Gastroenterol. 2015, 110, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Curvers, W.L.; ten Kate, F.J.; Krishnadath, K.K.; Visser, M.; Elzer, B.; Baak, L.C.; Bohmer, C.; Mallant-Hent, R.C.; van Oijen, A.; Naber, A.H.; et al. Low-grade dysplasia in Barrett’s esophagus: Overdiagnosed and underestimated. Am. J. Gastroenterol. 2010, 105, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Wani, S.; Falk, G.W.; Post, J.; Yerian, L.; Hall, M.; Wang, A.; Gupta, N.; Gaddam, S.; Singh, M.; Singh, V.; et al. Risk factors for progression of low-grade dysplasia in patients with Barrett’s esophagus. Gastroenterology 2011, 141, 1179–1186.e1171. [Google Scholar] [CrossRef] [PubMed]
- Falk, G.W. Current Management of Low-Grade Dysplasia in Barrett Esophagus. Gastroenterol. Hepatol. 2017, 13, 221–225. [Google Scholar]
- Hoefnagel, S.J.M.; Mostafavi, N.; Timmer, M.R.; Lau, C.T.; Meijer, S.L.; Wang, K.K.; Krishnadath, K.K. A genomic biomarker-based model for cancer risk stratification of non-dysplastic Barrett’s esophagus patients after extended follow up; results from Dutch surveillance cohorts. PLoS ONE 2020, 15, e0231419. [Google Scholar] [CrossRef]
- Falk, G.W. Barrett’s oesophagus: Frequency and prediction of dysplasia and cancer. Best. Pract. Res. Clin. Gastroenterol. 2015, 29, 125–138. [Google Scholar] [CrossRef]
- Chandrasekar, V.T.; Hamade, N.; Desai, M.; Rai, T.; Gorrepati, V.S.; Jegadeesan, R.; Sathyamurthy, A.; Sharma, P. Significantly lower annual rates of neoplastic progression in short- compared to long-segment non-dysplastic Barrett’s esophagus: A systematic review and meta-analysis. Endoscopy 2019, 51, 665–672. [Google Scholar] [CrossRef]
- Lam, A.K. Updates on World Health Organization classification and staging of esophageal tumors: Implications for future clinical practice. Hum. Pathol. 2021, 108, 100–112. [Google Scholar] [CrossRef]
- Fiocca, R.; Mastracci, L.; Lugaresi, M.; Grillo, F.; D’Errico, A.; Malvi, D.; Spaggiari, P.; Tomezzoli, A.; Albarello, L.; Ristimaki, A.; et al. The Prognostic Impact of Histology in Esophageal and Esophago-Gastric Junction Adenocarcinoma. Cancers 2021, 13, 5211. [Google Scholar] [CrossRef]
- Jimenez Fonseca, P.; Carmona-Bayonas, A.; Hernandez, R.; Custodio, A.; Cano, J.M.; Lacalle, A.; Echavarria, I.; Macias, I.; Mangas, M.; Visa, L.; et al. Lauren subtypes of advanced gastric cancer influence survival and response to chemotherapy: Real-world data from the AGAMENON National Cancer Registry. Br. J. Cancer 2017, 117, 775–782. [Google Scholar] [CrossRef]
- van der Kaaij, R.T.; Snaebjornsson, P.; Voncken, F.E.; van Dieren, J.M.; Jansen, E.P.; Sikorska, K.; Cats, A.; van Sandick, J.W. The prognostic and potentially predictive value of the Lauren classification in oesophageal adenocarcinoma. Eur. J. Cancer 2017, 76, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.; Kumarasinghe, M. Adenocarcinoma of the oesophagus and oesophagogastric junction NOS. WHO Classif. Tumours 2019, 38–43. [Google Scholar]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; the WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Zhang, X.; Yu, C.; Zhang, Q.; Cheng, E.; Wang, D.H.; Pham, T.H.; Spechler, S.J.; Souza, R.F. In oesophageal squamous cells exposed to acidic bile salt medium, omeprazole inhibits IL-8 expression through effects on nuclear factor-kappaB and activator protein-1. Gut 2014, 63, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.F.; Zhang, H.Y.; Zhang, X.I.; Lynch, J.P.; Strauch, E.D.; Wang, J.Y.; Melton, S.D.; Genta, R.M.; Wang, D.H.; Spechler, S.J.; et al. Acid and Bile Salt-Induced CDX2 Expression Differs in Esophageal Squamous Cells From Patients With and Without Barrett’s Esophagus. Gastroenterology 2010, 139, 194–203. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, J.M.; Abdel-latif, M.M.; Ravi, N.; McNamara, D.; Byrne, P.J.; McDonald, G.S.A.; Keeling, P.W.N.; Kelleher, D.; Reynolds, J.V. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am. J. Gastroenterol. 2005, 100, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFkappaB-signaling pathway in cancer. Onco. Targets. Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, M.M.; O’Riordan, J.; Windle, H.J.; Carton, E.; Ravi, N.; Kelleher, D.; Reynolds, J.V. NF-kappaB activation in esophageal adenocarcinoma: Relationship to Barrett’s metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann. Surg. 2004, 239, 491–500. [Google Scholar] [CrossRef]
- Eda, A.; Osawa, H.; Satoh, K.; Yanaka, I.; Kihira, K.; Ishino, Y.; Mutoh, H.; Sugano, K. Aberrant expression of CDX2 in Barrett’s epithelium and inflammatory esophageal mucosa. J. Gastroenterol. 2003, 38, 14–22. [Google Scholar] [CrossRef]
- Groisman, G.M.; Amar, M.; Meir, A. Expression of the intestinal marker Cdx2 in the columnar-lined esophagus with and without intestinal (Barrett’s) metaplasia. Mod. Pathol. 2004, 17, 1282–1288. [Google Scholar] [CrossRef]
- Phillips, R.W.; Frierson, H.F., Jr.; Moskaluk, C.A. Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am. J. Surg. Pathol. 2003, 27, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Vallbohmer, D.; DeMeester, S.R.; Peters, J.H.; Oh, D.S.; Kuramochi, H.; Shimizu, D.; Hagen, J.A.; Danenberg, K.D.; Danenberg, P.V.; DeMeester, T.R.; et al. Cdx-2 expression in squamous and metaplastic columnar epithelia of the esophagus. Dis. Esophagus 2006, 19, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Kazumori, H.; Ishihara, S.; Rumi, M.A.; Kadowaki, Y.; Kinoshita, Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett’s epithelium. Gut 2006, 55, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa, Y.; Ishimura, N.; Uno, G.; Aimi, M.; Oshima, N.; Yuki, T.; Sato, S.; Ishihara, S.; Kinoshita, Y. Bile acids induce Delta-like 1 expression via Cdx2-dependent pathway in the development of Barrett’s esophagus. Lab. Investig. 2016, 96, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mari, L.; Milano, F.; Parikh, K.; Straub, D.; Everts, V.; Hoeben, K.K.; Fockens, P.; Buttar, N.S.; Krishnadath, K.K. A pSMAD/CDX2 Complex Is Essential for the Intestinalization of Epithelial Metaplasia. Cell Rep. 2014, 7, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Milano, F.; van Baal, J.W.; Buttar, N.S.; Rygiel, A.M.; de Kort, F.; DeMars, C.J.; Rosmolen, W.D.; Bergman, J.J.; van Marle, J.; Wang, K.K.; et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology 2007, 132, 2412–2421. [Google Scholar] [CrossRef]
- Li, S.; Hoefnagel, S.J.M.; Read, M.; Meijer, S.; van Berge Henegouwen, M.I.; Gisbertz, S.S.; Bonora, E.; Liu, D.S.H.; Phillips, W.A.; Calpe, S.; et al. Selective targeting BMP2 and 4 in SMAD4 negative esophageal adenocarcinoma inhibits tumor growth and aggressiveness in preclinical models. Cell. Oncol. 2022, 45, 639–658. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, M.M.; Kelleher, D.; Reynolds, J.V. Potential role of NF-kappaB in esophageal adenocarcinoma: As an emerging molecular target. J. Surg. Res. 2009, 153, 172–180. [Google Scholar] [CrossRef]
- Shirvani, V.N.; Ouatu-Lascar, R.; Kaur, B.S.; Omary, M.B.; Triadafilopoulos, G. Cyclooxygenase 2 expression in Barrett’s esophagus and adenocarcinoma: Ex vivo induction by bile salts and acid exposure. Gastroenterology 2000, 118, 487–496. [Google Scholar] [CrossRef]
- Souza, R.F.; Shewmake, K.; Beer, D.G.; Cryer, B.; Spechler, S.J. Selective inhibition of cyclooxygenase-2 suppresses growth and induces apoptosis in human esophageal adenocarcinoma cells. Cancer Res. 2000, 60, 5767–5772. [Google Scholar]
- Ferguson, H.R.; Wild, C.P.; Anderson, L.A.; Murphy, S.J.; Johnston, B.T.; Murray, L.J.; Watson, R.G.; McGuigan, J.; Reynolds, J.V.; Hardie, L.J. Cyclooxygenase-2 and inducible nitric oxide synthase gene polymorphisms and risk of reflux esophagitis, Barrett’s esophagus, and esophageal adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Moons, L.M.G.; Kuipers, E.J.; Rygiel, A.M.; Groothuismink, A.Z.M.; Geldof, H.; Bode, W.A.; Krishnadath, K.K.; Bergman, J.J.G.H.M.; van Vliet, A.H.M.; Siersema, P.D.; et al. COX-2 CA-haplotype is a risk factor for the development of esophageal adenocarcinoma. Am. J. Gastroenterol. 2007, 102, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Buttar, N.S.; Wang, K.K.; Leontovich, O.; Westcott, J.Y.; Pacifico, R.J.; Anderson, M.A.; Krishnadath, K.K.; Lutzke, L.S.; Burgart, L.J. Chemoprevention of esophageal adenocarcinoma by COX-2 inhibitors in an animal model of Barrett’s esophagus. Gastroenterology 2002, 122, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Mayr, C.; Winkelmann, P.; Neumayer, B.; Klieser, E.; Wagner, A.; Hufnagl, C.; Emmanuel, K.; Holzinger, J.; Koch, O.; et al. Expression of the microRNA-200 Family, microRNA-205, and Markers of Epithelial-Mesenchymal Transition as Predictors for Endoscopic Submucosal Dissection over Esophagectomy in Esophageal Adenocarcinoma: A Single-Center Experience. Cells 2020, 9, 486. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Munch, N.S.; Schottelius, M.; Ingermann, J.; Liu, H.B.; Schauer, M.; Stangl, S.; Multhoff, G.; Steiger, K.; Gerngross, C.; et al. CXCR4 Is a Potential Target for Diagnostic PET/CT Imaging in Barrett’s Dysplasia and Esophageal Adenocarcinoma. Clin. Cancer Res. 2018, 24, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Milano, F.; Jorritsma, T.; Rygiel, A.M.; Bergman, J.J.; Sondermeijer, C.; Ten Brinke, A.; vanHam, S.M.; Krishnadath, K.K. Expression pattern of immune suppressive cytokines and growth factors in oesophageal adenocarcinoma reveal a tumour immune escape-promoting microenvironment. Scand. J. Immunol. 2008, 68, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.S.; DeMeester, S.R.; Vallbohmer, D.; Mori, R.; Kuramochi, H.; Hagen, J.A.; Lipham, J.; Danenberg, K.D.; Danenberg, P.V.; Chandrasoma, P.; et al. Reduction of interleukin 8 gene expression in reflux esophagitis and Barrett’s esophagus with antireflux surgery. Arch. Surg. 2007, 142, 554–559; discussion 559–560. [Google Scholar] [CrossRef]
- Karstens, K.F.; Kempski, J.; Giannou, A.D.; Pelczar, P.; Steglich, B.; Steurer, S.; Freiwald, E.; Woestemeier, A.; Konczalla, L.; Tachezy, M.; et al. Anti-inflammatory microenvironment of esophageal adenocarcinomas negatively impacts survival. Cancer Immunol. Immunother. 2020, 69, 1043–1056. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef]
- Gockel, I.; Schimanski, C.C.; Heinrich, C.; Wehler, T.; Frerichs, K.; Drescher, D.; von Langsdorff, C.; Domeyer, M.; Biesterfeld, S.; Galle, P.R.; et al. Expression of chemokine receptor CXCR4 in esophageal squamous cell and adenocarcinoma. BMC Cancer 2006, 6, 290. [Google Scholar] [CrossRef]
- Goto, M.; Shibahara, Y.; Baciu, C.; Allison, F.; Yeung, J.C.; Darling, G.E.; Liu, M. Prognostic Impact of CXCR7 and CXCL12 Expression in Patients with Esophageal Adenocarcinoma. Ann. Surg. Oncol. 2021, 28, 4943–4951. [Google Scholar] [CrossRef] [PubMed]
- Lagisetty, K.H.; McEwen, D.P.; Nancarrow, D.J.; Schiebel, J.G.; Ferrer-Torres, D.; Ray, D.; Frankel, T.L.; Lin, J.; Chang, A.C.; Kresty, L.A.; et al. Immune determinants of Barrett’s progression to esophageal adenocarcinoma. JCI Insight 2021, 6, e143888. [Google Scholar] [CrossRef]
- Lind, A.; Siersema, P.D.; Kusters, J.G.; Konijn, T.; Mebius, R.E.; Koenderman, L. The Microenvironment in Barrett’s Esophagus Tissue Is Characterized by High FOXP3 and RALDH2 Levels. Front. Immunol. 2018, 9, 1375. [Google Scholar] [CrossRef] [PubMed]
- Rubinkiewicz, M.; Migaczewski, M.; Hankus, J.; Major, P.; Pedziwiatr, M.; Budzynski, P.; Okon, K.; Budzynski, A. Foxp3+ lymphocyte count in Barrett’s esophagus tissue is higher than in in amed esophageal tissue. Folia. Med. Cracov. 2016, 56, 51–59. [Google Scholar] [PubMed]
- Gokon, Y.; Fujishima, F.; Taniyama, Y.; Ishida, H.; Yamagata, T.; Sawai, T.; Uzuki, M.; Ichikawa, H.; Itakura, Y.; Takahashi, K.; et al. Immune microenvironment in Barrett’s esophagus adjacent to esophageal adenocarcinoma: Possible influence of adjacent mucosa on cancer development and progression. Virchows Arch. 2020, 477, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Volkweis, B.S.; Gurski, R.R.; Meurer, L.; Pretto, G.G.; Mazzini Gda, S.; Edelweiss, M.I. Ki-67 Antigen Overexpression Is Associated with the Metaplasia-Adenocarcinoma Sequence in Barrett’s Esophagus. Gastroenterol. Res. Pract. 2012, 2012, 639748. [Google Scholar] [CrossRef] [PubMed]
- Derks, S.; Nason, K.S.; Liao, X.; Stachler, M.D.; Liu, K.X.; Liu, J.B.; Sicinska, E.; Goldberg, M.S.; Freeman, G.J.; Rodig, S.J.; et al. Epithelial PD-L2 Expression Marks Barrett’s Esophagus and Esophageal Adenocarcinoma. Cancer Immunol. Res. 2015, 3, 1123–1129. [Google Scholar] [CrossRef]
- Moons, L.M.G.; Kusters, J.G.; Bultman, E.; Kuipers, E.J.; van Dekken, H.; Tra, W.M.W.; Kleinjan, A.; Kwekkeboom, J.; van Vliet, A.H.M.; Siersema, P.D. Barrett’s oesophagus is characterized by a predominantly humoral inflammatory response. J. Pathol. 2005, 207, 269–276. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Onwuegbusi, B.A.; Bajaj-Elliott, M.; Saeed, I.T.; Burnham, W.R.; Farthing, M.J. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: Immunological determinants. Gut 2002, 50, 451–459. [Google Scholar] [CrossRef]
- Liu, J.Y.; Geng, X.F.; Hou, J.X.; Wu, G.S. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef]
- Kotsafti, A.; Fassan, M.; Cavallin, F.; Angerilli, V.; Saadeh, L.; Cagol, M.; Alfieri, R.; Pilati, P.; Castoro, C.; Castagliuolo, I.; et al. Tumor immune microenvironment in therapy-naive esophageal adenocarcinoma could predict the nodal status. Cancer Med. 2023, 12, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.P.; Rothenberg, M.E. Eosinophils and cancer. Cancer Immunol. Res. 2014, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Croft, W.; Evans, R.P.T.; Pearce, H.; Elshafie, M.; Griffiths, E.A.; Moss, P. The single cell transcriptional landscape of esophageal adenocarcinoma and its modulation by neoadjuvant chemotherapy. Mol. Cancer 2022, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Caspa Gokulan, R.; Garcia-Buitrago, M.T.; Zaika, A.I. From genetics to signaling pathways: Molecular pathogenesis of esophageal adenocarcinoma. Biochim. Biophys. Acta. Rev. Cancer 2019, 1872, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.M.J.; Ross-Innes, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.J.; Lao-Sirieix, P.; Dunning, M.J.; et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat. Genet. 2014, 46, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Cai, H.; Jing, C.; Chang, X.; Ding, D.; Han, T.; Yang, J.; Lu, Z.; Hu, X.; Liu, Z.; Wang, J.; et al. Mutational landscape of gastric cancer and clinical application of genomic profiling based on target next-generation sequencing. J. Transl. Med. 2019, 17, 189. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.A.; Rabinovitch, P.S.; et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006, 38, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.J.; Barrett, M.T.; Galipeau, P.C.; Sanchez, C.A.; Neshat, K.; Cowan, D.S.; Levine, D.S. Barrett’s esophagus: Ordering the events that lead to cancer. Eur. J. Cancer Prev. 1996, 5, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Timmer, M.R.; Lau, C.T.; Calpe, S.; Sancho-Serra Mdel, C.; Straub, D.; Baker, A.M.; Meijer, S.L.; Kate, F.J.; Mallant-Hent, R.C.; et al. Dynamic clonal equilibrium and predetermined cancer risk in Barrett’s oesophagus. Nat. Commun. 2016, 7, 12158. [Google Scholar] [CrossRef]
- Gharahkhani, P.; Fitzgerald, R.C.; Vaughan, T.L.; Palles, C.; Gockel, I.; Tomlinson, I.; Buas, M.F.; May, A.; Gerges, C.; Anders, M.; et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: A large-scale meta-analysis. Lancet Oncol. 2016, 17, 1363–1373. [Google Scholar] [CrossRef]
- Su, Z.; Gay, L.J.; Strange, A.; Palles, C.; Band, G.; Whiteman, D.C.; Lescai, F.; Langford, C.; Nanji, M.; Edkins, S.; et al. Common variants at the MHC locus and at chromosome 16q24.1 predispose to Barrett’s esophagus. Nat. Genet. 2012, 44, 1131–1136. [Google Scholar] [CrossRef]
- Frankell, A.M.; Jammula, S.; Li, X.D.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Bower, L.; Devonshire, G.; Cocks, E.; et al. The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef]
- Lin, D.C.; Dinh, H.Q.; Xie, J.J.; Mayakonda, A.; Silva, T.C.; Jiang, Y.Y.; Ding, L.W.; He, J.Z.; Xu, X.E.; Hao, J.J.; et al. Identification of distinct mutational patterns and new driver genes in oesophageal squamous cell carcinomas and adenocarcinomas. Gut 2018, 67, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Orsini, A.; Mastracci, L.; Bozzarelli, I.; Ferrari, A.; Isidori, F.; Fiocca, R.; Lugaresi, M.; D’Errico, A.; Malvi, D.; Cataldi-Stagetti, E.; et al. Correlations between Molecular Alterations, Histopathological Characteristics, and Poor Prognosis in Esophageal Adenocarcinoma. Cancers 2023, 15, 1408. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Madani, K.; Zhao, R.; Lim, H.J.; Casson, A.G. Prognostic value of p53 mutations in oesophageal adenocarcinoma: Final results of a 15-year prospective study. Eur. J. Cardiothorac. Surg. 2010, 37, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Foxwell, T.J.; Nason, K.; Cressman, K.L.; McGrath, K.M.; Sun, W.J.; Bahary, N.; Zeh, H.J.; Levy, R.M.; Luketich, J.D.; et al. Smad4 Loss in Esophageal Adenocarcinoma Is Associated With an Increased Propensity for Disease Recurrence and Poor Survival. Am. J. Surg. Pathol. 2015, 39, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Pasello, G.; Agata, S.; Bonaldi, L.; Corradin, A.; Montagna, M.; Zamarchi, R.; Parenti, A.; Cagol, M.; Zaninotto, G.; Ruol, A.; et al. DNA copy number alterations correlate with survival of esophageal adenocarcinoma patients. Mod. Pathol. 2009, 22, 58–65. [Google Scholar] [CrossRef]
- Sihag, S.; Nussenzweig, S.C.; Walch, H.S.; Hsu, M.; Tan, K.S.; Sanchez-Vega, F.; Chatila, W.K.; De La Torre, S.A.; Patel, A.; Janjigian, Y.Y.; et al. Next-Generation Sequencing of 487 Esophageal Adenocarcinomas Reveals Independently Prognostic Genomic Driver Alterations and Pathways. Clin. Cancer Res. 2021, 27, 3491–3498. [Google Scholar] [CrossRef]
- Paulson, T.G.; Maley, C.C.; Li, X.; Li, H.; Sanchez, C.A.; Chao, D.L.; Odze, R.D.; Vaughan, T.L.; Blount, P.L.; Reid, B.J. Chromosomal instability and copy number alterations in Barrett’s esophagus and esophageal adenocarcinoma. Clin. Cancer Res. 2009, 15, 3305–3314. [Google Scholar] [CrossRef]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta. Rev. Cancer 2017, 1867, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.J.; Prevo, L.J.; Galipeau, P.C.; Sanchez, C.A.; Longton, G.; Levine, D.S.; Blount, P.L.; Rabinovitch, P.S. Predictors of progression in Barrett’s esophagus II: Baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am. J. Gastroenterol. 2001, 96, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Timmer, M.R.; Martinez, P.; Lau, C.T.; Westra, W.M.; Calpe, S.; Rygiel, A.M.; Rosmolen, W.D.; Meijer, S.L.; Ten Kate, F.J.; Dijkgraaf, M.G.; et al. Derivation of genetic biomarkers for cancer risk stratification in Barrett’s oesophagus: A prospective cohort study. Gut 2016, 65, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, P.C.; Li, X.; Blount, P.L.; Maley, C.C.; Sanchez, C.A.; Odze, R.D.; Ayub, K.; Rabinovitch, P.S.; Vaughan, T.L.; Reid, B.J. NSAIDs modulate CDKN2A, TP53, and DNA content risk for progression to esophageal adenocarcinoma. PLoS Med. 2007, 4, e67. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Schumacher, S.E.; van Lieshout, J.; Imamura, Y.; Fox, C.; Shim, B.; Ramos, A.H.; Saksena, G.; Baca, S.C.; Baselga, J.; et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012, 72, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.J.; Ruurda, J.P.; van Diest, P.J.; van Hillegersberg, R.; Ten Kate, F.J. The significance of the HER-2 status in esophageal adenocarcinoma for survival: An immunohistochemical and an in situ hybridization study. Ann. Oncol. 2013, 24, 1290–1297. [Google Scholar] [CrossRef]
- Finley, J.C.; Reid, B.J.; Odze, R.D.; Sanchez, C.A.; Galipeau, P.; Li, X.H.; Self, S.G.; Gollahon, K.A.; Blount, P.L.; Rabinovitch, P.S. Chromosomal instability in Barrett’s esophagus is related to telomere shortening. Cancer Epidem. Biomar. 2006, 15, 1451–1457. [Google Scholar] [CrossRef]
- Risques, R.A.; Vaughan, T.L.; Li, X.; Odze, R.D.; Blount, P.L.; Ayub, K.; Gallaher, J.L.; Reid, B.J.; Rabinovitch, P.S. Leukocyte telomere length predicts cancer risk in Barrett’s esophagus. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2649–2655. [Google Scholar] [CrossRef]
- Xing, J.; Ajani, J.A.; Chen, M.; Izzo, J.; Lin, J.; Chen, Z.; Gu, J.; Wu, X. Constitutive short telomere length of chromosome 17p and 12q but not 11q and 2p is associated with an increased risk for esophageal cancer. Cancer Prev. Res. 2009, 2, 459–465. [Google Scholar] [CrossRef]
- Demissie, S.; Levy, D.; Benjamin, E.J.; Cupples, L.A.; Gardner, J.P.; Herbert, A.; Kimura, M.; Larson, M.G.; Meigs, J.B.; Keaney, J.F.; et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell 2006, 5, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Andrew, T.; Gardner, J.P.; Kimura, M.; Oelsner, E.; Cherkas, L.F.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Hoefnagel, S.J.M.; Koemans, W.J.; Khan, H.N.; Koster, J.; Meijer, S.L.; van Dieren, J.M.; Kodach, L.L.; van Sandick, J.W.; Calpe, S.; Del Sancho-Serra, C.M.; et al. Identification of Novel Molecular Subgroups in Esophageal Adenocarcinoma to Predict Response to Neo-Adjuvant Therapies. Cancers 2022, 14, 4498. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.P. Genome-wide methylation analysis shows similar patterns in Barrett’s esophagus and esophageal adenocarcinoma (vol 34, pg 2750, 2013). Carcinogenesis 2014, 35, 738. [Google Scholar]
- Smith, E.; De Young, N.J.; Pavey, S.J.; Hayward, N.K.; Nancarrow, D.J.; Whiteman, D.C.; Smithers, B.M.; Ruszkiewicz, A.R.; Clouston, A.D.; Gotley, D.C.; et al. Similarity of aberrant DNA methylation in Barrett’s esophagus and esophageal adenocarcinoma. Mol. Cancer 2008, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Klump, B.; Hsieh, C.J.; Holzmann, K.; Gregor, M.; Porschen, R. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 1998, 115, 1381–1386. [Google Scholar] [CrossRef]
- Wong, D.J.; Barrett, M.T.; Stoger, R.; Emond, M.J.; Reid, B.J. p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res. 1997, 57, 2619–2622. [Google Scholar]
- Peng, D.; Hu, T.L.; Jiang, A.; Washington, M.K.; Moskaluk, C.A.; Schneider-Stock, R.; El-Rifai, W. Location-specific epigenetic regulation of the metallothionein 3 gene in esophageal adenocarcinomas. PLoS ONE 2011, 6, e22009. [Google Scholar] [CrossRef]
- Alvi, M.A.; Liu, X.X.; O’Donovan, M.; Newton, R.; Wernisch, L.; Shannon, N.B.; Shariff, K.; di Pietro, M.; Bergman, J.J.G.H.M.; Ragunath, K.; et al. DNA Methylation as an Adjunct to Histopathology to Detect Prevalent, Inconspicuous Dysplasia and Early-Stage Neoplasia in Barrett’s Esophagus. Clin. Cancer Res. 2013, 19, 878–888. [Google Scholar] [CrossRef]
- Fassan, M.; Volinia, S.; Palatini, J.; Pizzi, M.; Baffa, R.; De Bernard, M.; Battaglia, G.; Parente, P.; Croce, C.M.; Zaninotto, G.; et al. MicroRNA expression profiling in human Barrett’s carcinogenesis. Int. J. Cancer 2011, 129, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, S.Y.; Gao, Y.M.; Liu, Y.F.; Liu, Y.B.; Zhao, Z.G.; Yang, K. MicroRNAs as oncogenes or tumour suppressors in oesophageal cancer: Potential biomarkers and therapeutic targets. Cell Proliferat. 2014, 47, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Kailasam, A.; Mittal, S.K.; Agrawal, D.K. Epigenetics in the Pathogenesis of Esophageal Adenocarcinoma. Clin. Transl. Sci. 2015, 8, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Saad, R.; Chen, Z.; Zhu, S.; Jia, P.; Zhao, Z.; Washington, M.K.; Belkhiri, A.; El-Rifai, W. Deciphering the unique microRNA signature in human esophageal adenocarcinoma. PLoS ONE 2013, 8, e64463. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Saad, R.; Jia, P.; Peng, D.; Zhu, S.; Washington, M.K.; Zhao, Z.; Xu, Z.; El-Rifai, W. Gastric adenocarcinoma has a unique microRNA signature not present in esophageal adenocarcinoma. Cancer 2013, 119, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Chen, D.; Li, X.; Yang, K.; Wang, H.; Wu, W. miR-133b regulates the MET proto-oncogene and inhibits the growth of colorectal cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Crawford, M.; Yu, B.; Mao, Y.; Nana-Sinkam, S.P.; Lee, L.J. MicroRNA delivery by cationic lipoplexes for lung cancer therapy. Mol. Pharm. 2011, 8, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef]
- Mari, L.; Hoefnagel, S.J.M.; Zito, D.; van de Meent, M.; van Endert, P.; Calpe, S.; Sancho Serra, M.D.C.; Heemskerk, M.H.M.; van Laarhoven, H.W.M.; Hulshof, M.; et al. microRNA 125a Regulates MHC-I Expression on Esophageal Adenocarcinoma Cells, Associated With Suppression of Antitumor Immune Response and Poor Outcomes of Patients. Gastroenterology 2018, 155, 784–798. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e315. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R. Screening for Barrett Esophagus With a Nonendoscopic Sponge Capsule. Gastroenterol. Hepatol. 2015, 11, 623–625. [Google Scholar]
- Peters, Y.; Schrauwen, R.W.M.; Tan, A.C.; Bogers, S.K.; de Jong, B.; Siersema, P.D. Detection of Barrett’s oesophagus through exhaled breath using an electronic nose device. Gut 2020, 69, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Codipilly, D.C.; Chandar, A.K.; Singh, S.; Wani, S.; Shaheen, N.J.; Inadomi, J.M.; Chak, A.; Iyer, P.G. The Effect of Endoscopic Surveillance in Patients With Barrett’s Esophagus: A Systematic Review and Meta-analysis. Gastroenterology 2018, 154, 2068–2086.e2065. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Kou, T.D.; Chak, A. Receipt of previous diagnoses and endoscopy and outcome from esophageal adenocarcinoma: A population-based study with temporal trends. Am. J. Gastroenterol. 2009, 104, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, A.; Zafirellis, K.D.; Dolan, K.; Dexter, S.P.; Martin, I.G.; Sue-Ling, H.M. Effect of surveillance of Barrett’s oesophagus on the clinical outcome of oesophageal cancer. Br. J. Surg. 2004, 91, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Sonnenberg, A.; Davis, J.; McMahon, L.; Inadomi, J.M. Effect of a prior endoscopy on outcomes of esophageal adenocarcinoma among United States veterans. Gastrointest. Endosc. 2008, 68, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Hillman, L.C.; Chiragakis, L.; Shadbolt, B.; Kaye, G.L.; Clarke, A.C. Proton-pump inhibitor therapy and the development of dysplasia in patients with Barrett’s oesophagus. Med. J. Aust. 2004, 180, 387–391. [Google Scholar] [CrossRef]
- Kastelein, F.; Spaander, M.C.; Steyerberg, E.W.; Biermann, K.; Valkhoff, V.E.; Kuipers, E.J.; Bruno, M.J.; ProBar Study, G. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett’s esophagus. Clin. Gastroenterol. Hepatol. 2013, 11, 382–388. [Google Scholar] [CrossRef]
- Nguyen, D.M.; El-Serag, H.B.; Henderson, L.; Stein, D.; Bhattacharyya, A.; Sampliner, R.E. Medication usage and the risk of neoplasia in patients with Barrett’s esophagus. Clin. Gastroenterol. Hepatol. 2009, 7, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Kerlikowske, K.; Verma, R.; Buffler, P. Protective association of aspirin/NSAIDs and esophageal cancer: A systematic review and meta-analysis. Gastroenterology 2003, 124, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.M.; Vaughan, T.L.; Corley, D.A.; Cook, M.B.; Casson, A.G.; Kamangar, F.; Abnet, C.C.; Risch, H.A.; Giffen, C.; Freedman, N.D.; et al. Nonsteroidal anti-inflammatory drug use reduces risk of adenocarcinomas of the esophagus and esophagogastric junction in a pooled analysis. Gastroenterology 2012, 142, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.M.; Richardson, P.; El-Serag, H.B. Medications (NSAIDs, Statins, Proton Pump Inhibitors) and the Risk of Esophageal Adenocarcinoma in Patients With Barrett’s Esophagus. Gastroenterology 2010, 138, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.Q.; Ding, X.W.; Yang, R.K.; Huang, S.L.; Kastelein, F.; Bruno, M.; Yu, X.J.; Zhou, D.; Zou, X.P. Cyclooxygenase inhibitors use is associated with reduced risk of esophageal adenocarcinoma in patients with Barrett’s esophagus: A meta-analysis. Br. J. Cancer 2014, 110, 2378–2388. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.A.; Sitja-Arnau, M.; Lemoine, M.G.; Frazier, M.L.; Sinicrope, F.A. Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: Growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 1999, 59, 4356–4362. [Google Scholar] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Overholt, B.F.; Wang, K.K.; Burdick, J.S.; Lightdale, C.J.; Kimmey, M.; Nava, H.R.; Sivak, M.V., Jr.; Nishioka, N.; Barr, H.; Marcon, N.; et al. Five-year efficacy and safety of photodynamic therapy with Photofrin in Barrett’s high-grade dysplasia. Gastrointest. Endosc. 2007, 66, 460–468. [Google Scholar] [CrossRef]
- Genere, J.R.; Visrodia, K.; Zakko, L.; Hoefnagel, S.J.M.; Wang, K.K. Spray cryotherapy versus continued radiofrequency ablation in persistent Barrett’s esophagus. Dis. Esophagus 2022, 35, doab084. [Google Scholar] [CrossRef]
- Mansour, N.M.; Groth, S.S.; Anandasabapathy, S. Esophageal Adenocarcinoma: Screening, Surveillance, and Management. Annu. Rev. Med. 2017, 68, 213–227. [Google Scholar] [CrossRef]
- Shaheen, N.J.; Sharma, P.; Overholt, B.F.; Wolfsen, H.C.; Sampliner, R.E.; Wang, K.K.; Galanko, J.A.; Bronner, M.P.; Goldblum, J.R.; Bennett, A.E.; et al. Radiofrequency ablation in Barrett’s esophagus with dysplasia. N. Engl. J. Med. 2009, 360, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Aadam, A.A. T1b esophageal cancer: Is it time for endoscopic submucosal dissection to enter the stage? Gastrointest. Endosc. 2022, 96, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Obermannova, R.; Alsina, M.; Cervantes, A.; Leong, T.; Lordick, F.; Nilsson, M.; van Grieken, N.C.T.; Vogel, A.; Smyth, E.C. Oesophageal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- van Hagen, P.; Hulshof, M.C.; van Lanschot, J.J.; Steyerberg, E.W.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.; Richel, D.J.; Nieuwenhuijzen, G.A.; Hospers, G.A.; Bonenkamp, J.J.; et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N. Engl. J. Med. 2012, 366, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.E.; Homann, N.; Pauligk, C.; Goetze, T.O.; Meiler, J.; Kasper, S.; Kopp, H.G.; Mayer, F.; Haag, G.M.; Luley, K.; et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): A randomised, phase 2/3 trial. Lancet 2019, 393, 1948–1957. [Google Scholar] [PubMed]
- Moussa, O.; Bhogal, R.H.; Malietzis, G.; Fribbens, C.; Starling, N.; Gerlinger, M.; Watkins, D.; Chau, I.; Rao, S.; Cunningham, D.; et al. Effect of perioperative FLOT versus ECF/ECX on short-term outcomes after surgery for resectable oesophagogastric adenocarcinoma: Propensity score-matched study. BJS Open 2022, 6, zrac003. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.V.; Preston, S.R.; O’Neill, B.; Lowery, M.A.; Baeksgaard, L.; Crosby, T.; Cunningham, M.; Cuffe, S.; Griffiths, G.O.; Roy, R.; et al. Neo-AEGIS (Neoadjuvant trial in Adenocarcinoma of the Esophagus and Esophago-Gastric Junction International Study): Preliminary results of phase III RCT of CROSS versus perioperative chemotherapy (Modified MAGIC or FLOT protocol). (NCT01726452). J. Clin. Oncol. 2021, 39, 4004. [Google Scholar] [CrossRef]
- Hoeppner, J.; Lordick, F.; Brunner, T.; Glatz, T.; Bronsert, P.; Rothling, N.; Schmoor, C.; Lorenz, D.; Ell, C.; Hopt, U.T.; et al. ESOPEC: Prospective randomized controlled multicenter phase III trial comparing perioperative chemotherapy (FLOT protocol) to neoadjuvant chemoradiation (CROSS protocol) in patients with adenocarcinoma of the esophagus (NCT02509286). BMC Cancer 2016, 16, 503. [Google Scholar] [CrossRef]
- Mariette, C.; Markar, S.R.; Dabakuyo-Yonli, T.S.; Meunier, B.; Pezet, D.; Collet, D.; D’Journo, X.B.; Brigand, C.; Perniceni, T.; Carrere, N.; et al. Hybrid Minimally Invasive Esophagectomy for Esophageal Cancer. N. Engl. J. Med. 2019, 380, 152–162. [Google Scholar] [CrossRef]
- van der Sluis, P.C.; van der Horst, S.; May, A.M.; Schippers, C.; Brosens, L.A.A.; Joore, H.C.A.; Kroese, C.C.; Haj Mohammad, N.; Mook, S.; Vleggaar, F.P.; et al. Robot-assisted Minimally Invasive Thoracolaparoscopic Esophagectomy Versus Open Transthoracic Esophagectomy for Resectable Esophageal Cancer: A Randomized Controlled Trial. Ann. Surg. 2019, 269, 621–630. [Google Scholar] [CrossRef]
- Crosby, T.; Hurt, C.N.; Falk, S.; Gollins, S.; Staffurth, J.; Ray, R.; Bridgewater, J.A.; Geh, J.I.; Cunningham, D.; Blazeby, J.; et al. Long-term results and recurrence patterns from SCOPE-1: A phase II/III randomised trial of definitive chemoradiotherapy +/− cetuximab in oesophageal cancer. Br. J. Cancer 2017, 116, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Galais, M.P.; Raoul, J.L.; Bouche, O.; Gourgou-Bourgade, S.; Douillard, J.Y.; Etienne, P.L.; Boige, V.; Martel-Lafay, I.; Michel, P.; et al. Definitive chemoradiotherapy with FOLFOX versus fluorouracil and cisplatin in patients with oesophageal cancer (PRODIGE5/ACCORD17): Final results of a randomised, phase 2/3 trial. Lancet Oncol. 2014, 15, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; D’Amico, T.A.; Bentrem, D.J.; Chao, J.; Corvera, C.; Das, P.; Denlinger, C.S.; Enzinger, P.C.; Fanta, P.; Farjah, F.; et al. Esophageal and Esophagogastric Junction Cancers, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019, 17, 855–883. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.P.; Li, Z.; Kim, S.B.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): A randomised, placebo-controlled, phase 3 study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Doi, T.; Dvorkin, M.; Mansoor, W.; Arkenau, H.T.; Prokharau, A.; Alsina, M.; Ghidini, M.; Faustino, C.; Gorbunova, V.; et al. Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.E.; Hartmann, J.T.; Probst, S.; Schmalenberg, H.; Hollerbach, S.; Hofheinz, R.; Rethwisch, V.; Seipelt, G.; Homann, N.; Wilhelm, G.; et al. Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: A study of the Arbeitsgemeinschaft Internistische Onkologie. J. Clin. Oncol. 2008, 26, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Bang, Y.J.; Iwasa, S.; Sugimoto, N.; Ryu, M.H.; Sakai, D.; Chung, H.C.C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab deruxtecan (T-DXd; DS-8201) in patients with HER2-positive advanced gastric or gastroesophageal junction (GEJ) adenocarcinoma: Final overall survival (OS) results from a randomized, multicenter, open-label, phase 2 study (DESTINY-Gastric01). J. Clin. Oncol. 2021, 40, 4048. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Kawazoe, A.; Yanez, P.E.; Luo, S.X.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.X.; Shen, L.; Tang, Y.; et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2+metastatic gastric or gastroesophageal junction (G/GEJ) cancer: Initial findings of the global phase 3 KEYNOTE-811 study. J. Clin. Oncol. 2021, 39, iv25. [Google Scholar] [CrossRef]
- Li, S.; Hoefnagel, S.J.M.; Krishnadath, K.K. Single domain Camelid antibody fragments for molecular imaging and therapy of cancer. Front. Oncol. 2023, 13, 1257175. [Google Scholar] [CrossRef] [PubMed]
- Hazelton, W.D.; Curtius, K.; Inadomi, J.M.; Vaughan, T.L.; Meza, R.; Rubenstein, J.H.; Hur, C.; Luebeck, E.G. The Role of Gastroesophageal Reflux and Other Factors during Progression to Esophageal Adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Miyashita, T.; Lay, F.; Ahmed, A.K.; Matsangos, A.E.; Born, L.; Ng, C.; Cohen, R.M.; Stricker-Krongrad, A.H.; Salimian, K.; et al. Targeting the hedgehog pathway in esophageal adenocarcinoma (EAC) using Itraconazole. J. Clin. Oncol. 2018, 36, e13552. [Google Scholar] [CrossRef]
- Vaninetti, N.; Williams, L.; Geldenhuys, L.; Porter, G.A.; Guernsey, D.L.; Casson, A.G. Regulation of CDX2 expression in esophageal adenocarcinoma. Mol. Carcinog. 2009, 48, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Revenko, A.; Carnevalli, L.S.; Sinclair, C.; Johnson, B.; Peter, A.; Taylor, M.; Hettrick, L.; Chapman, M.; Klein, S.; Solanki, A.; et al. Direct targeting of FOXP3 in Tregs with AZD8701, a novel antisense oligonucleotide to relieve immunosuppression in cancer. J. Immunother. Cancer 2022, 10, e003892. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Geboes, K.; Hoorens, A. The cell of origin for Barrett’s esophagus. Science 2021, 373, 737–738. [Google Scholar] [CrossRef]
- Correia, A.C.P.; Straub, D.; Calpe, S.; Krishnadath, K.K. Novel In Vivo Mouse Cryoablation Model to Explore Unique Therapeutic Approaches for Premalignant Columnar Lesions. Methods Protoc. 2021, 4, 6. [Google Scholar] [CrossRef]
- Sihag, S.; Nussenzweig, S.C.; Walch, H.S.; Hsu, M.; Tan, K.S.; De La Torre, S.; Janjigian, Y.Y.; Maron, S.B.; Ku, G.Y.; Tang, L.H.; et al. The Role of the TP53 Pathway in Predicting Response to Neoadjuvant Therapy in Esophageal Adenocarcinoma. Clin. Cancer Res. 2022, 28, 2669–2678. [Google Scholar] [CrossRef]
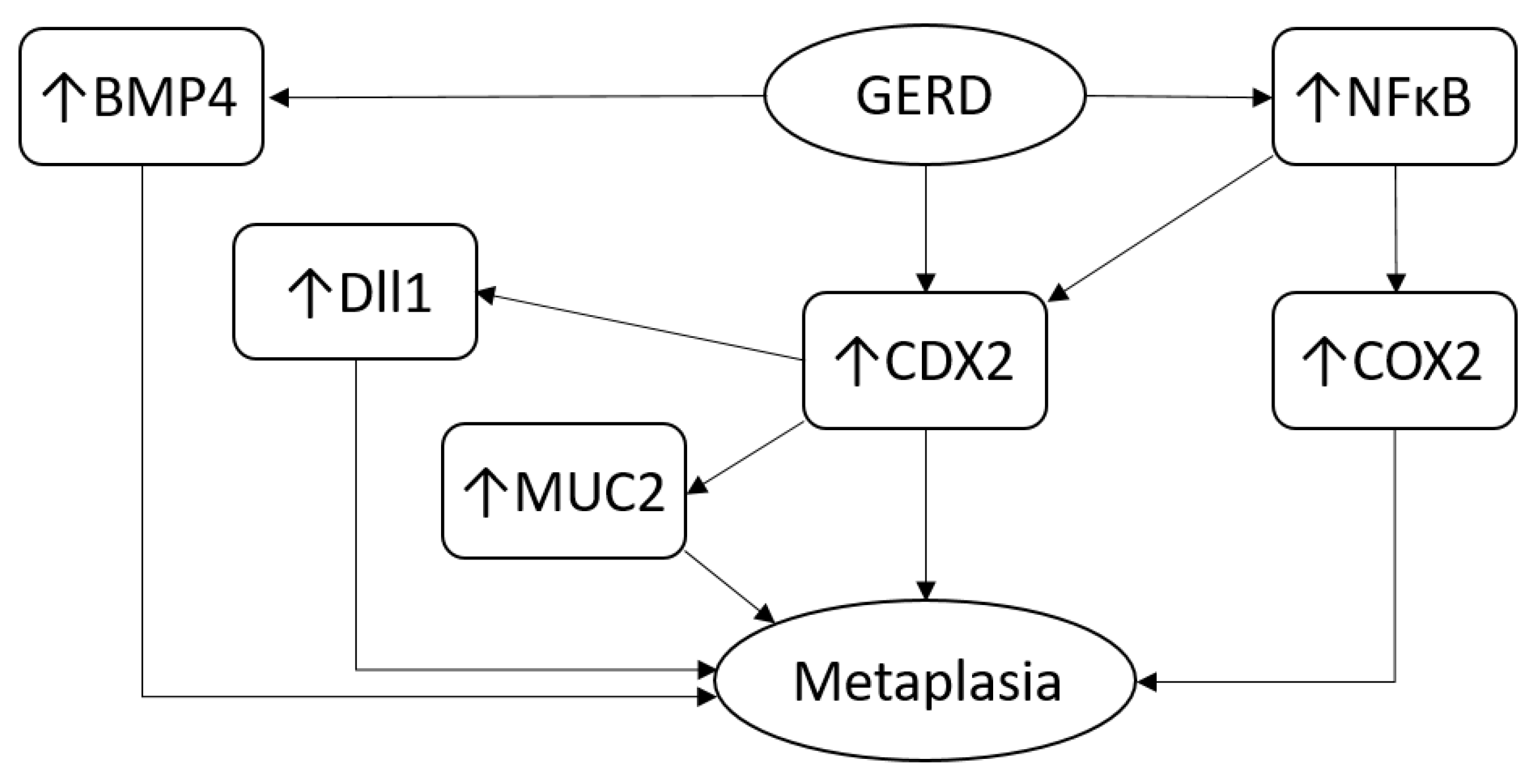

{kind=link}
| Reference | Authors | Year | Origin | Drivers |
|---|---|---|---|---|
| [43] | K. Nowicki-osuch et al. | 2021 | Gastric cardia | c-MYC- and HNF4A-driven transcriptional programs |
| [44] | D. Straub et al. | 2019 | Non-squamous cells residing in submucosal gland ducts | Glycine-conjugated bile acids |
| [45] | C. Minacapelli et al. | 2018 | Normal esophageal squamous epithelial cells | Acid and bile |
| [46] | M. Jiang | 2017 | p63+KRT5+KRT7+ basal cells in the upper gastrointestinal tract | Ectopic expression of CDX2 |
| [40] | M. Quante | 2012 | Gastric cardia stem cells | Bile acids and/or nitrosamines |
| [47] | X. Wang et al. | 2011 | Embryonic cells at the squamocolumnar junction | Competitive interactions between cell lineages |
| [48] | S. Leedham | 2008 | Squamous gland ducts situated throughout the esophagus | Gene mutations |
| Reference | Authors | Year | Mutated Genes | Detecting Methods |
|---|---|---|---|---|
| [122] | A. Orsini et al. | 2023 | TP53, ATM, MSH6, APC, PIK3CA, SMAD4, CDKN2A, SMARCA4, ERBB2, HNF1A, CHEK2, FLT3, PTEN, IDH2, CTNNB1, MET, STK11, ALK, KRAS, RET, EGFR, ARID2, CDK6, TSSC1, MAP2K1, SRC | Targeted sequencing |
| [120] | A. Frankell et al. | 2019 | TP53, CDKN2A, KRAS, MYC, ERBB2, GATA4, CCND1, GATA6, SMAD4, CDK6, ARID1A, EGFR, CCNE1, CCND3, MUC6, MDM2, KCNQ3, APC, SMARCA4, PIK3CA, ABCB1, PTEN, MET, RNF43, DNAH7, TSHZ3, LRRK2, TRPA1, NAV3, ARID2, SLIT2, EPHA3, SCN3A, CRISPLD1, AXIN1, FBXW7, PPM1D, ACVR2A, RASA1, CD1A, CCDC102B, CHL1, LIN7A, COIL, MAP2K7, EPHA2, PBRM1, POLQ, ARID1B, CTNNB1, SIN3A, RPL22, PIK3R1, MAP3K1, NIPBL, B2M, FAM196B, HIST1H3B, TGFBR2, MBD6, BRAF, MSH3, CHD4, CDH1, GATAD1, KDM6A, CDKN1B, ACVR1B, STK11, NOTCH1, ZFHX3, JAK1, PCDH17, ELF3, GPATCH8, C3orf62 | Whole-genome sequencing |
| [121] | D. Lin et al. | 2018 | PIK3CA, PBRM1, SMARCA4, CTNNB1, PCDH18, C6orf114, CHRNB1, EPHA2, SEMA5A, PGCP, DOCK2, CDKN2A, ARID1A, SMAD4, FBXW7, KRAS, TP53 | Whole-genome sequencing |
| [1] | TCGA Research Network | 2017 | TP53, CDKN2A, ARID1A, SMAD4, ERBB2, VEGFA, GATA6, CCNE1, KRAS, EGFR, IGF1R, VEGFA, GATA4, ARID1A, SMARCA4, PBRM1 | Whole-exome sequencing |
| [123] | M. Secrier et al. | 2016 | SMYD3, RUNX1, CTNNA3, RBFOX1, AGBL4, INK4/ARF, SAMD5, CDK14, KIF26B, THADA, SASH1, MECOM, JUP, IKZF3, FHIT, WWOX, MACROD2, IMMP2L, CCSER1, PDE4D, NAALADL2, PARK2, PARD3B, PRKG1, TP53, SMAD4, ARID1A, CDKN2A, KCNQ3, CCDC102B, CYP7B1 | Whole-genome sequencing |
| [106] | J. Weaver et al. | 2014 | ABCB1, ARID1A, CCDC102B, CCDC153, CDKN2A, CNTNAP5, FGF10, MMP16, MYD88, MYF6, MYO18B, PCDH9, PNLIPRP3, SEMA5A, SMAD4, SMARCA4, SSTR4, TLR1, TLR4, TLR7, TLR9, TP53, TRAF3, TRAF6, TRIM58, UNC13C | Whole-genome sequencing |
| [105] | A. Dulak et al. | 2013 | TP53, CDKN2A, EYS, ARID1A, SMAD4, PIK3CA, SLC39A12, SPG20, DOCK2, AKAP6, TLL1, TLR4, ARID2, HECW1, ELMO1, SYNE1, SMARCA4, AJAP1, C6orf118, ACTL7B, F5, KCNU1, NUAK1, MYST3, SCN10A, CNTNAP5 | Whole-exome sequencing |
| Reference | Authors | Years | Genes with CNVs | Detected Method |
|---|---|---|---|---|
| [129] | Sihag et al. | 2021 | ERBB2, KRAS, CCNE1, MYC, CCND1, MDM2, VEGFA, EGFR, CDK6, CCND3, CDKN2A/B, SMAD4, ARID1A, PIK3CA, APC, TP53 NGS | Next-generation sequencing |
| [120] | Frankell et al. | 2019 | ERBB2, KRAS, SMAD4 | Whole-genome sequencing |
| [1] | TCGA Research Network | 2017 | VEGFA, ERBB2, SMAD4, GATA6, CCNE1 | Whole-genome sequencing |
| [123] | Secrier et al. | 2016 | 50 genes: CCND1, EGFR, ERBB2, VEGFA, KRAS etc. | Whole-genome sequencing |
| [107] | Ross-Innes et al. | 2015 | 35 genes: GATA4, KLF5, MYB, PRKCI, CCND1, FGF3, FGF4, FGF19, VEGFA, A2BP1, CDKN2A, PDE4D, PTPRD, PARK2 etc. | Whole-genome sequencing |
| [108] | Nones et al. | 2014 | 210 genes: CCNE1, ERBB2, FRS2, GATA4, KRAS, MTMR9, MDM2, CDKN2A, FHIT, RUNX1 etc. | Whole-genome sequencing |
| [130] | Paulson et al. | 2009 | 47 genes: EGFR, MYC, EGFR, MTAP, CDKN2A, CDKN2B, SMAD2, SMAD4, SMAD7 etc. | BAC array CGH |
| [128] | Pasello et al. | 2009 | 97 genes: VEGF, PTK2, ING1, SCYA3, ABCG2, DCC etc. | MLPA |
| Reference | Authors | Year | Genes | Status | Type of Lesion |
|---|---|---|---|---|---|
| [145] | Xu et al. | 2013 | 20 genes: SH3GL3, GBX2, SLC18A3, SLC6A2 etc. | hypermethylated | BE (vs. NE) |
| 20 genes: ZNF625, PTPRT, ST6GAL2, SLC18A3 etc. | hypermethylated | EAC (vs. NE) | |||
| [150] | Alvi et al. | 2012 | SLC22A18, PIGR, GJA12, RIN2, RGN, TCEAL7 | hypermethylated | EAC (vs. BE) |
| [149] | Peng et al. | 2011 | MT3 | hypermethylated | EAC (vs. NE) |
| [146] | Smith et al. | 2008 | APC, CDKN2A, ID4, MGMT, RBP1, RUNX3, SFRP1, TIMP3, TMEFF2 | hypermethylated | EAC (vs. NE) |
| APC, ID4, MGMT, RUNX3, SFRP1, TIMP3, TMEFF2 | hypermethylated | BE (vs. NE) | |||
| [147] | Klump et al. | 1998 | p16 | hypermethylated | BE (vs. NE) |
| [148] | Wong et al. | 1997 | p16 | hypermethylated | EAC/BE (vs. NE) |
| Reference | Authors | Year | MicroRNA | Status | Type of Lesion |
|---|---|---|---|---|---|
| [84] | Neureiter et al. | 2020 | miR-205 | upregulated | LE-EAC (vs. RI-EAC) |
| [161] | Mari et al. | 2018 | miR125a-5p | downregulated | EAC (vs. NE) |
| [153] | Kailasam et al. | 2015 | miR-28, miR-30a-5p, miR-126, miR-143, miR-145, miR-181a/b, miR-199a | upregulated | EAC (vs. NE) |
| miR-27b, miR-99a, miR-149, miR-193a/b, miR-210, miR-345, miR-494, miR-513, miR-617, let-7a/b/c | downregulated | EAC (vs. NE) | |||
| [152] | Huang et al. | 2014 | miR-126, miR-143, miR-145, miR-181a, miR-181b, miR-199a, miR-28, miR-30a-5p | upregulated | EAC (vs. HGD) |
| miR-149, miR-203, miR-210, miR-27b, miR-513, miR-617, miR-99a let-7a/b/c, miR-193a, miR-345, miR-494 | downregulated | EAC (vs. HGD) | |||
| miR-25, miR-93, miR-106b, miR-192 | upregulated | EAC (vs. BE) | |||
| miR-203, let-7 | downregulated | EAC (vs. BE) | |||
| miR-200a, miR-513, miR-125b, miR-101, miR-197 | upregulated | HGD (vs. LGD) | |||
| miR-23b, miR-20b, miR-181b, miR-203, miR-193b, miR-636 | downregulated | HGD (vs. LGD) | |||
| [154] | Saad et al. | 2013 | miR-194, miR-31, miR-192, miR-200a | upregulated | EAC (vs. BE) |
| miR-203, miR-205 | downregulated | EAC (vs. BE) | |||
| miR-194, miR-192, miR-21, miR-31 | upregulated | HGD (vs. BE) | |||
| [155] | Chen et al. | 2013 | miRNA-21, miRNA-133b, miR-200a | upregulated | EAC (vs. NE) |
| [151] | Fassan et al. | 2011 | miR-215, miR-560, miR-615-3p, miR-192, miR-326, miR-147 | upregulated | BE (vs. NE) |
| miR-100, miR-23a, miR-605, miR-99a, miR-205, let-7c, miR-203 | downregulated | BE (vs. NE) |
| Therapy | Target | Patients | Treatment | Approval | Clinical Trials | Current Status |
|---|---|---|---|---|---|---|
| Targeted therapy | HER2 | HER2 positive metastatic EAC | Trastuzumab plus chemotherapy | FDA | Phase III (NCT01041404) [15] | In clinic |
| Targeted therapy | HER2 | HER2 positive metastatic EAC | Fam-Trastuzumab Deruxtecan-nxki | FDA | Phase II (NCT03329690) [197] | In clinic |
| Monotherapy/ Combination therapy | VEGFR2 | advanced or metastatic EAC | Ramucirumab (plus paclitaxel) | FDA | Phase III (NCT00917384; NCT01170663) [198,199] | In clinic |
| Immunotherapy | PD-1 | resectable EAC | Nivolumab following nCRT plus radical resection | FDA; EMA | Phase III (NCT02743494) [200] | In clinic |
| Immunotherapy | PD-1 | unresectable HER2 negative metastatic EAC | Nivolumab plus chemotherapy | FDA; EMA | Phase III (NCT02872116) [201] | In clinic |
| Immunotherapy | PD-1 | unresectable HER2 positive metastatic EAC | Pembrolizumab plus Trastuzumab plus chemotherapy | FDA | Phase III (NCT03615326) [202] | In clinic |
| Immunotherapy | PD-1 | locally advanced or metastatic EAC | Pembrolizumab plus chemotherapy | FDA | Phase III (NCT03189719) [194] | In clinic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Hoefnagel, S.J.M.; Krishnadath, K.K. Molecular Biology and Clinical Management of Esophageal Adenocarcinoma. Cancers 2023, 15, 5410. https://doi.org/10.3390/cancers15225410
Li S, Hoefnagel SJM, Krishnadath KK. Molecular Biology and Clinical Management of Esophageal Adenocarcinoma. Cancers. 2023; 15(22):5410. https://doi.org/10.3390/cancers15225410
Chicago/Turabian StyleLi, Shulin, Sanne Johanna Maria Hoefnagel, and Kausilia Krishnawatie Krishnadath. 2023. "Molecular Biology and Clinical Management of Esophageal Adenocarcinoma" Cancers 15, no. 22: 5410. https://doi.org/10.3390/cancers15225410
APA StyleLi, S., Hoefnagel, S. J. M., & Krishnadath, K. K. (2023). Molecular Biology and Clinical Management of Esophageal Adenocarcinoma. Cancers, 15(22), 5410. https://doi.org/10.3390/cancers15225410