


Targeting Metabolic Vulnerabilities to Overcome Prostate Cancer Resistance: Dual Therapy with Apalutamide and Complex I Inhibition



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Pharmacological Compounds
2.2. Cell Culture
2.3. Galactose Medium Preparation
2.4. Cell Proliferation Assay
2.5. Mitochondrial Substrate Utilization
2.6. Extracellular Flux Analysis by Seahorse Assay
2.7. Monitoring of Glucose and Lactate Levels
2.8. Protein Analysis
2.9. Immunostaining of Mitochondrial Network
2.10. Statistical Analysis
3. Results
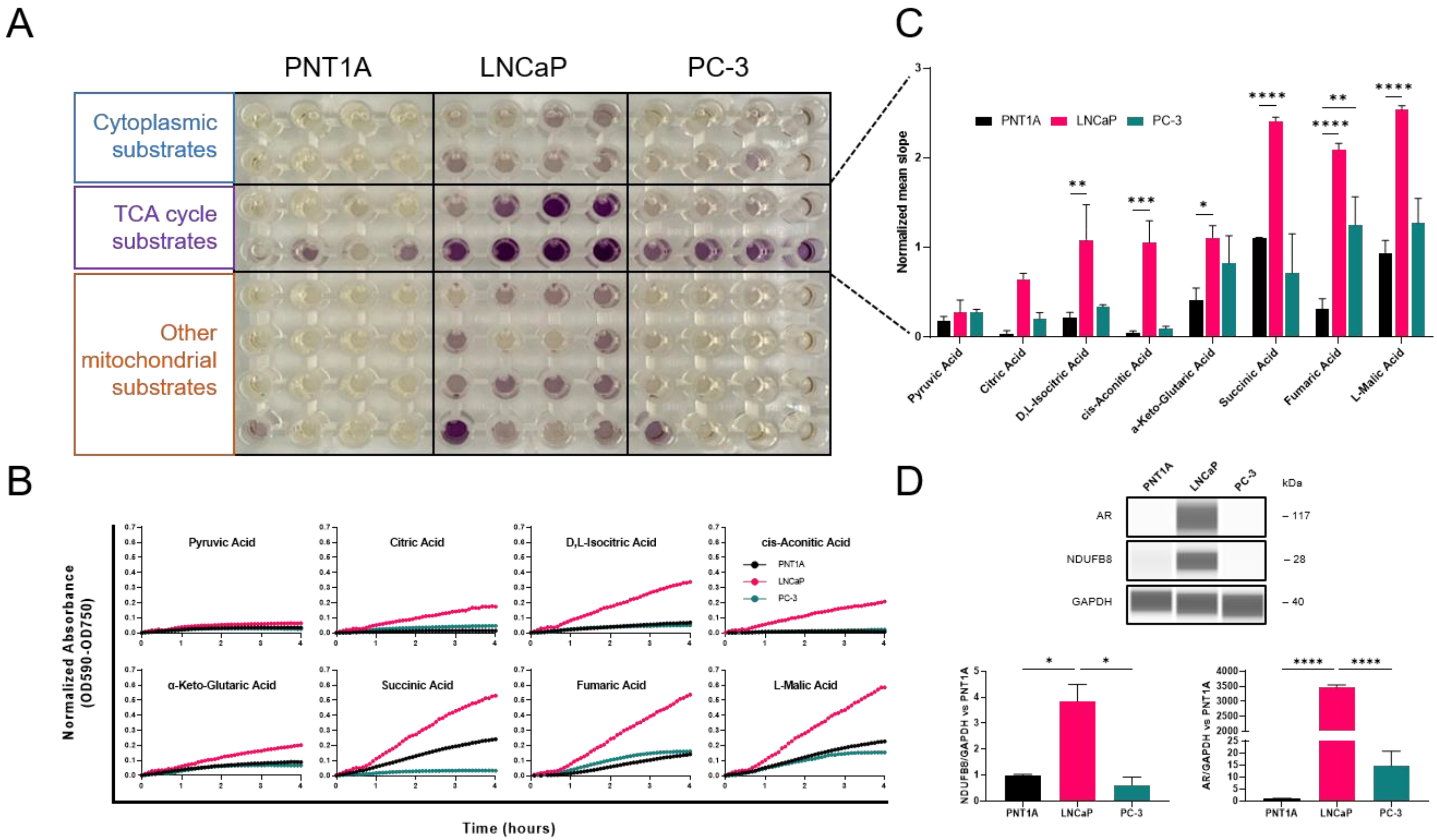
3.1. Subtype-Specific Mitochondrial Activity in Prostate Cancer Cell Lines
3.2. Synergistic Antiproliferative Effect of Androgen Receptor and Mitochondrial Complex I Inhibition
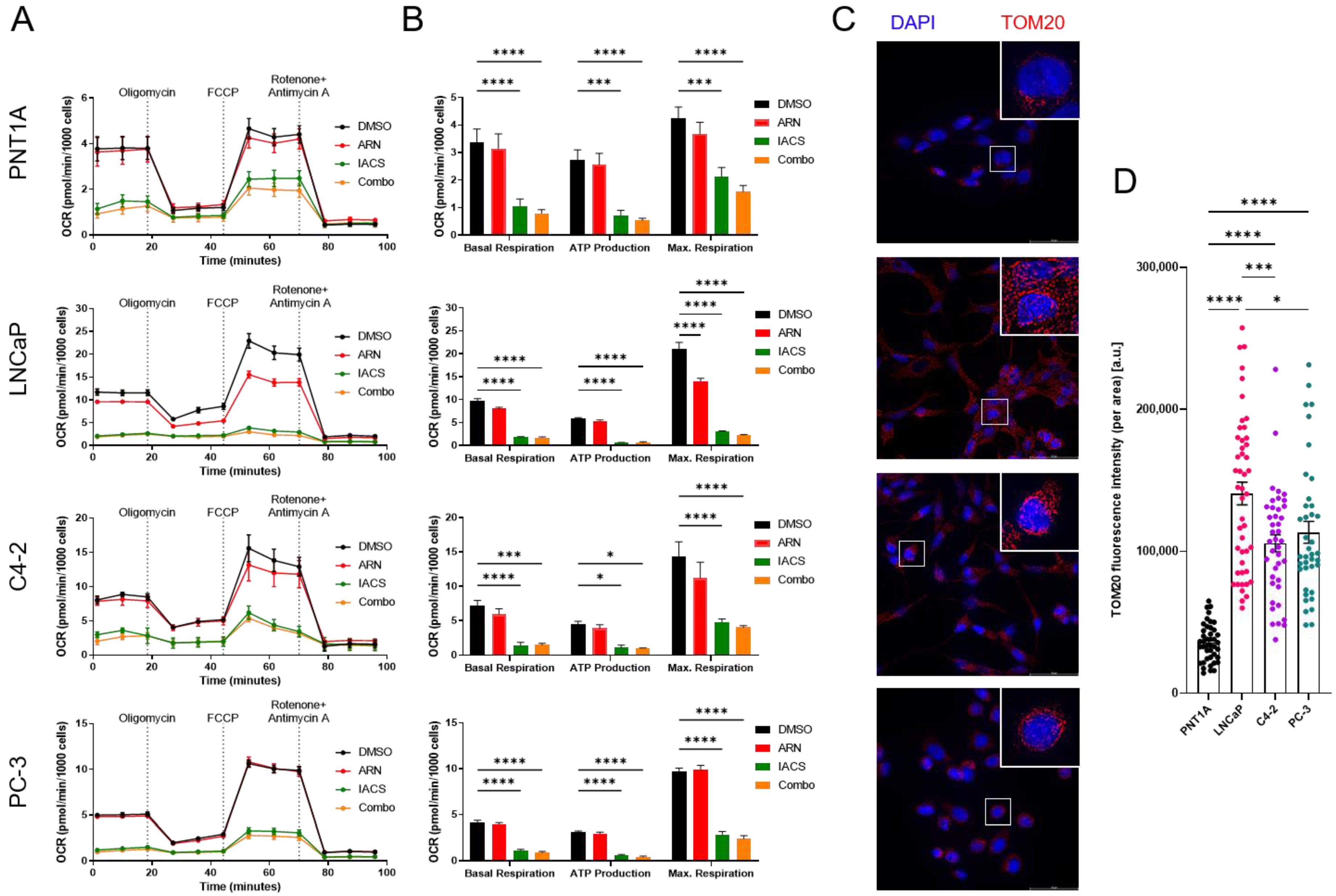
3.3. Cellular Bioenergetics Modulation via Pharmaceutical Inhibition of AR and OXPHOS
3.4. Galactose Supplementation Reveals Metabolic Vulnerabilities of PCa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. Embo J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Antonarakis, E.S.; Luo, J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm. Cancer 2014, 5, 265–273. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Costello, L.C.; Liu, Y.; Franklin, R.B.; Kennedy, M.C. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J. Biol. Chem. 1997, 272, 28875–28881. [Google Scholar] [CrossRef]
- Franklin, R.B.; Costello, L.C. Zinc as an anti-tumor agent in prostate cancer and in other cancers. Arch. Biochem. Biophys. 2007, 463, 211–217. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Richards, J.; de Bono, J.S. New strategies in metastatic prostate cancer: Targeting the androgen receptor signaling pathway. Clin. Cancer Res. 2011, 17, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Shankar, E.; Chan, E.R.; Gupta, S. Metabolic Reprogramming and Predominance of Solute Carrier Genes during Acquired Enzalutamide Resistance in Prostate Cancer. Cells 2020, 9, 2535. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Garcia-Smith, R.; Trujillo, K.A.; Bisoffi, M. Tumor necrosis factor alpha increases aerobic glycolysis and reduces oxidative metabolism in prostate epithelial cells. Prostate 2013, 73, 1538–1546. [Google Scholar] [CrossRef]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Roche, M.E.; Lin, Z.; Whitaker-Menezes, D.; Zhan, T.; Szuhai, K.; Bovee, J.; Abraham, J.A.; Jiang, W.; Martinez-Outschoorn, U.; Basu-Mallick, A. Translocase of the outer mitochondrial membrane complex subunit 20 (TOMM20) facilitates cancer aggressiveness and therapeutic resistance in chondrosarcoma. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165962. [Google Scholar] [CrossRef]
- Carew, J.S.; Huang, P. Mitochondrial defects in cancer. Mol. Cancer 2002, 1, 9. [Google Scholar] [CrossRef]
- Bajpai, P.; Koc, E.; Sonpavde, G.; Singh, R.; Singh, K.K. Mitochondrial localization, import, and mitochondrial function of the androgen receptor. J. Biol. Chem. 2019, 294, 6621–6634. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Wang, D.; Jiang, J.; Huang, W.H.; Li, D.J.; Luo, J.; Gu, W.; Mo, W.J.; Wang, C.J.; Li, Y.; et al. Integrative analysis of AR-mediated transcriptional regulatory network reveals IRF1 as an inhibitor of prostate cancer progression. Prostate 2020, 80, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Newell-Fugate, A.E. Role of androgens and androgen receptor in control of mitochondrial function. Am. J. Physiol. Cell Physiol. 2022, 323, C835–C846. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzola, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Tsouko, E.; Khan, A.S.; White, M.A.; Han, J.J.; Shi, Y.; Merchant, F.A.; Sharpe, M.A.; Xin, L.; Frigo, D.E. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 2014, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Daver, N.; Mahendra, M.; Zhang, J.X.; Kamiya-Matsuoka, C.; Meric-Bernstam, F.; Kantarjian, H.M.; Ravandi, F.; Collins, M.E.; Di Francesco, M.E.; et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: Phase I trials. Nat. Med. 2023, 29, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Dowling, C.M.; Hollinshead, K.E.R.; Di Grande, A.; Pritchard, J.; Zhang, H.; Dillon, E.T.; Haley, K.; Papadopoulos, E.; Mehta, A.K.; Bleach, R.; et al. Multiple screening approaches reveal HDAC6 as a novel regulator of glycolytic metabolism in triple-negative breast cancer. Sci. Adv. 2021, 7, eabc4897. [Google Scholar] [CrossRef]
- Cooper, M.A.; Menta, B.W.; Perez-Sanchez, C.; Jack, M.M.; Khan, Z.W.; Ryals, J.M.; Winter, M.; Wright, D.E. A ketogenic diet reduces metabolic syndrome-induced allodynia and promotes peripheral nerve growth in mice. Exp. Neurol. 2018, 306, 149–157. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Palmer, P.G.; Smith, M.A.; Gevorgyan, V.; Wilson, G.N.; Crish, S.D.; Inman, D.M. Structural and Functional Rescue of Chronic Metabolically Stressed Optic Nerves through Respiration. J. Neurosci. 2018, 38, 5122–5139. [Google Scholar] [CrossRef]
- de Kok, M.J.C.; Schaapherder, A.F.; Wust, R.C.I.; Zuiderwijk, M.; Bakker, J.A.; Lindeman, J.H.N.; Le Devedec, S.E. Circumventing the Crabtree effect in cell culture: A systematic review. Mitochondrion 2021, 59, 83–95. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Hadi, N.A.; Gicquel, T.; El Kaoutari, A.; Dieme, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumgartner, V.; Schaer, D.; Eberli, D.; Salemi, S. Targeting Metabolic Vulnerabilities to Overcome Prostate Cancer Resistance: Dual Therapy with Apalutamide and Complex I Inhibition. Cancers 2023, 15, 5612. https://doi.org/10.3390/cancers15235612
Baumgartner V, Schaer D, Eberli D, Salemi S. Targeting Metabolic Vulnerabilities to Overcome Prostate Cancer Resistance: Dual Therapy with Apalutamide and Complex I Inhibition. Cancers. 2023; 15(23):5612. https://doi.org/10.3390/cancers15235612
Chicago/Turabian StyleBaumgartner, Valentin, Dominik Schaer, Daniel Eberli, and Souzan Salemi. 2023. "Targeting Metabolic Vulnerabilities to Overcome Prostate Cancer Resistance: Dual Therapy with Apalutamide and Complex I Inhibition" Cancers 15, no. 23: 5612. https://doi.org/10.3390/cancers15235612
APA StyleBaumgartner, V., Schaer, D., Eberli, D., & Salemi, S. (2023). Targeting Metabolic Vulnerabilities to Overcome Prostate Cancer Resistance: Dual Therapy with Apalutamide and Complex I Inhibition. Cancers, 15(23), 5612. https://doi.org/10.3390/cancers15235612