
POLD1 DEDD Motif Mutation Confers Hypermutation in Endometrial Cancer and Durable Response to Pembrolizumab



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Institutional Study Approval
2.2. Systematic Analysis of Genotypic-Phenotypic Association of Germline POLD1 Mutations Involving the DEDD Motif
2.3. In Silico Analysis Predicting Functional Effect of DEDD Domain Alteration
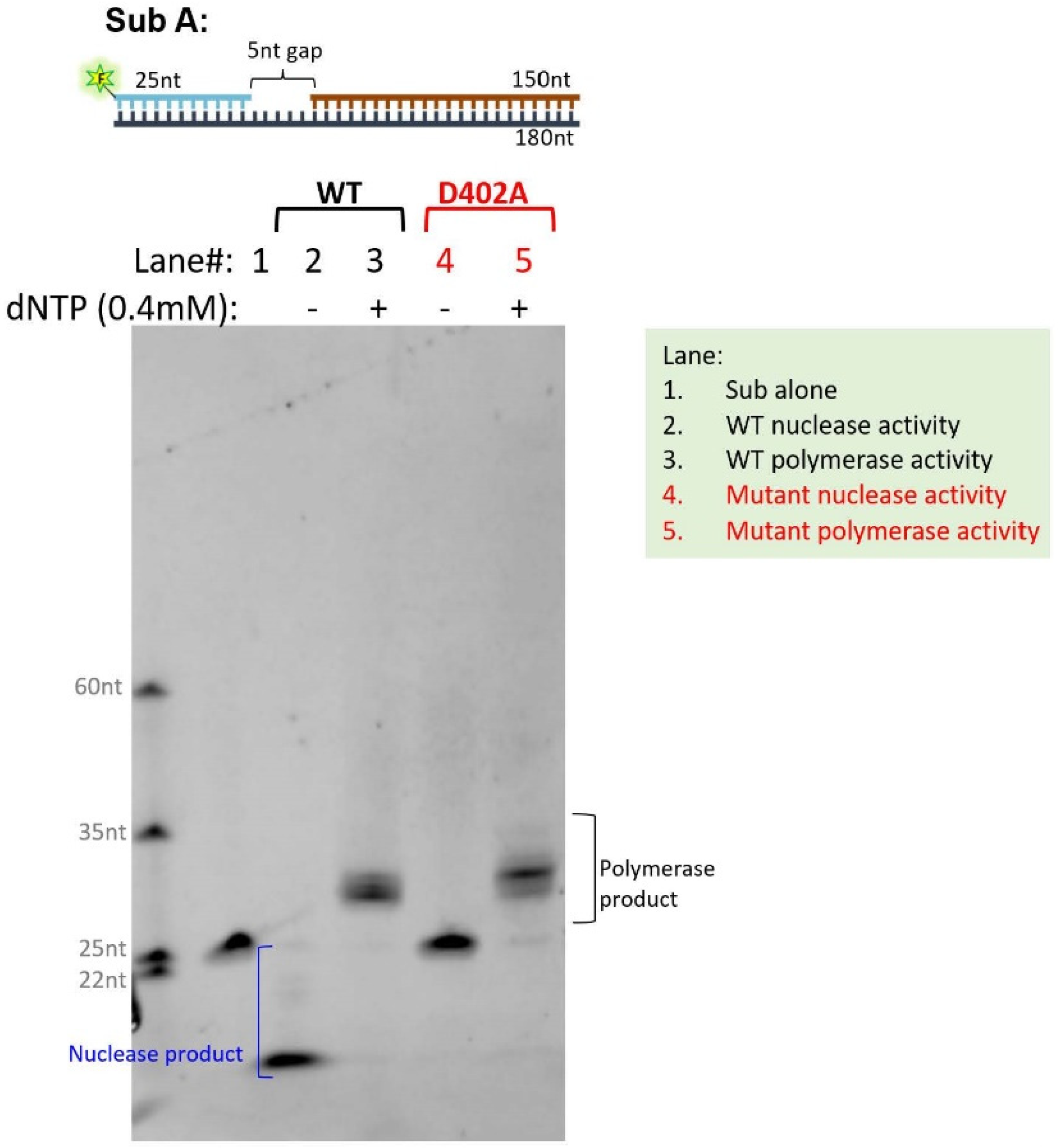
2.4. Human POLδ Expression, Purification, and In Vitro Polymerase and Exonuclease Assay
2.5. Generating Endometrial Cell Line Overexpressing POLD1 p.D402N
2.6. Whole-Exome Sequencing (WES)
2.7. Whole-Transcriptome Sequencing
2.8. Somatic Mutation Calling
2.9. T Cell-Inflamed GEP Score Calculation
3. Results
3.1. Case Control Study
3.2. The Genotypic–Phenotypic Association of Germline POLD1 Mutations Involving the DEDD Motif from Existing Literature Revealed These Affected Individuals Present with Cancers in Colon, Endometrium, and Breast
3.3. In Silico Analysis Predicted Deleterious Functional Effect of Charge-Discordant AA Substitutions in the DEDD Domain
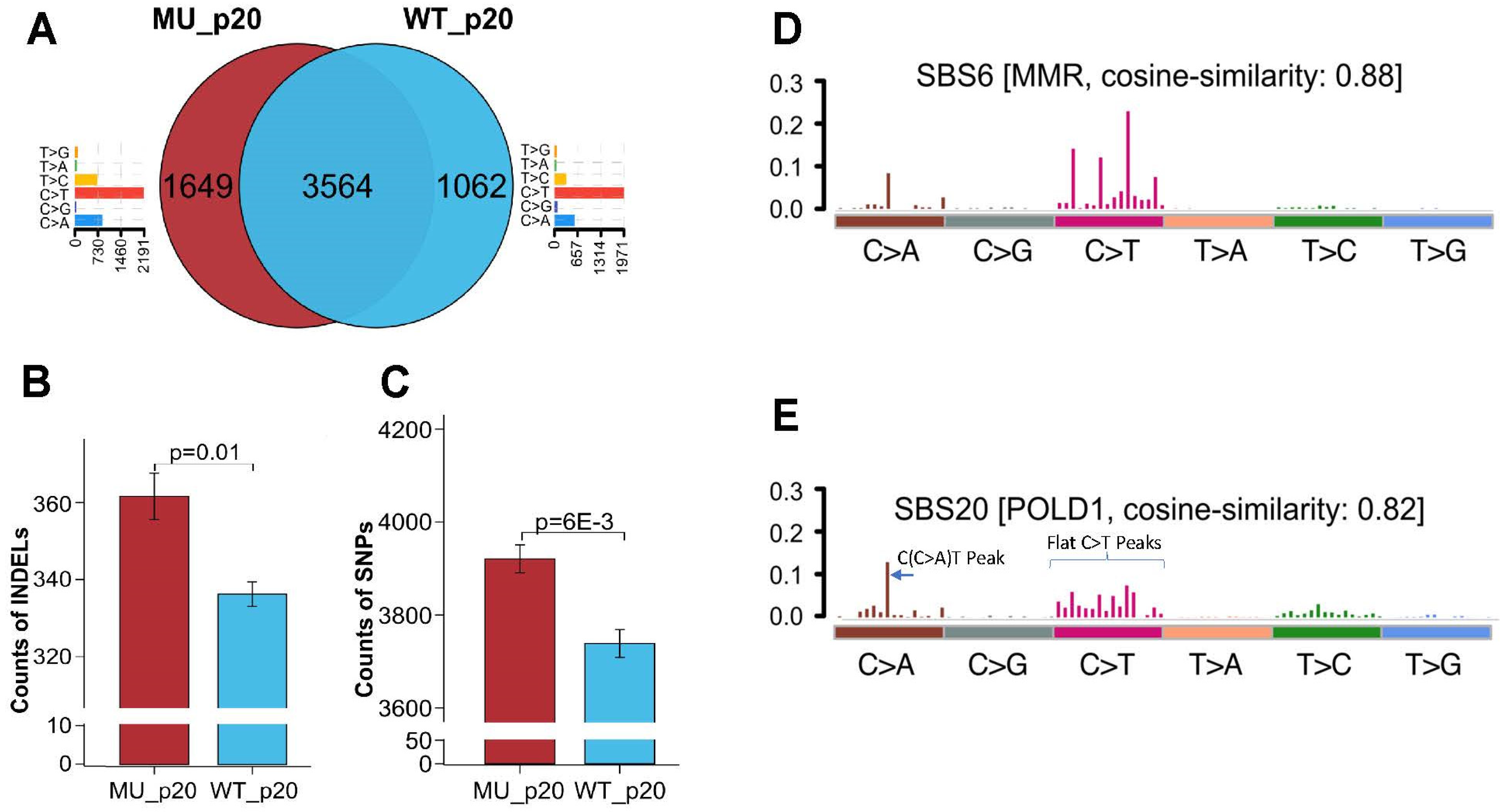
3.4. Whole-Exosome Sequencing of Endometrial Tumors with POLD1 p.D402N Mutation Revealed POLD1 DNA Proofreading Deficiency-Specific Mutational Signature and a Pan-Tumor Genomic Biomarker for Immunotherapy
3.5. Elucidating the Impact of p.D402N Mutation on Cancer Genome Using Stably Transfected Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Lu, Z.; Singh, A.; Zhou, Y.; Zheng, E.; Zhou, M.; Wang, J.; Wu, X.; Hu, Z.; Gu, H.; et al. Error-prone, stress-induced 3′ flap-based Okazaki fragment maturation supports cell survival. Science 2021, 374, 1252–1258. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, Q.; Wang, Y.-N.; Jin, Y.; He, M.-M.; Liu, Z.-X.; Xu, R.-H. Evaluation of POLE and POLD1 Mutations as Biomarkers for Immunotherapy Outcomes Across Multiple Cancer Types. JAMA Oncol. 2019, 5, 1504–1506. [Google Scholar] [CrossRef]
- Nicolas, E.; Golemis, E.A.; Arora, S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016, 590, 128–141. [Google Scholar] [CrossRef]
- Lancey, C.; Tehseen, M.; Raducanu, V.-S.; Rashid, F.; Merino, N.; Ragan, T.J.; Savva, C.G.; Zaher, M.S.; Shirbini, A.; Blanco, F.J.; et al. Structure of the processive human Pol delta holoenzyme. Nat. Commun. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Zuo, Y.; Deutscher, M.P. Exoribonuclease superfamilies: Structural analysis and phylogenetic distribution. Nucleic Acids Res. 2001, 29, 1017–1026. [Google Scholar] [CrossRef]
- Goldsby, R.E.; Hays, L.E.; Chen, X.; Olmsted, E.A.; Slayton, W.B.; Spangrude, G.J.; Preston, B.D. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc. Natl. Acad. Sci. USA 2002, 99, 15560–15565. [Google Scholar] [CrossRef]
- Kokoska, R.J.; Stefanovic, L.; DeMai, J.; Petes, T.D. Increased rates of genomic deletions generated by mutations in the yeast gene encoding DNA polymerase delta or by decreases in the cellular levels of DNA polymerase delta. Mol. Cell. Biol. 2000, 20, 7490–7504. [Google Scholar] [CrossRef]
- Murphy, K.; Darmawan, H.; Schultz, A.; da Silva, E.F.; Reha-Kratz, L.J. A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome 2006, 49, 403–410. [Google Scholar] [CrossRef]
- Robinson, P.S.; Coorens, T.H.H.; Palles, C.; Mitchell, E.; Abascal, F.; Olafsson, S.; Lee, B.C.H.; Lawson, A.R.J.; Lee-Six, H.; Moore, L.; et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat. Genet. 2021, 53, 1434–1442. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, F.; Presnell, S.R.; Tian, K.; Gao, Y.; Tomkinson, A.E.; Gu, L.; Li, G.-M. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell 2005, 122, 693–705. [Google Scholar] [CrossRef]
- Zhou, Y.; Meng, X.; Zhang, S.; Lee, E.Y.C.; Lee, M.Y.W.T. Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS ONE 2012, 7, e39156. [Google Scholar] [CrossRef]
- Dhoonmoon, A.; Nicolae, C.M.; Moldovan, G.L. The KU-PARP14 axis differentially regulates DNA resection at stalled replication forks by MRE11 and EXO1. Nat. Commun. 2022, 13, 5063. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Szelinger, S.; LoBello, J.; King, A.; Aldrich, J.; Garinger, N.; Halbert, M.; Richholt, R.F.; Mastrian, S.D.; Babb, C.; et al. Analytic validation and clinical utilization of the comprehensive genomic profiling test, GEM ExTra((R)). Oncotarget 2021, 12, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–33. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef]
- Palles, C.; Martin, L.; Domingo, E.; Chegwidden, L.; McGuire, J.; Cuthill, V.; Heitzer, E.; Kerr, R.; Kerr, D.; Kearsey, S.; et al. The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management. Fam. Cancer 2022, 21, 197–209. [Google Scholar] [CrossRef]
- Chang, S.C.; Lan, Y.; Lin, P.; Yang, S.; Lin, C.; Liang, W.; Chen, W.; Jiang, J.; Lin, J. Patterns of germline and somatic mutations in 16 genes associated with mismatch repair function or containing tandem repeat sequences. Cancer Med. 2020, 9, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Kim, J.; Maruvka, Y.E.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.M.; Leshchiner, I.; Kamburov, A.; Mouw, K.W.; et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018, 9, 1746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hou, D.; Annis, J.; Sargolzaeiaval, F.; Appelbaum, J.; Takahashi, E.; Martin, G.M.; Herr, A.; Oshima, J. Inactivating Mutations in Exonuclease and Polymerase Domains in DNA Polymerase Delta Alter Sensitivities to Inhibitors of dNTP Synthesis. DNA Cell Biol. 2020, 39, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Makhnoon, S.; Shirts, B.H.; Bowen, D.J. Patients’ perspectives of variants of uncertain significance and strategies for uncertainty management. J. Genet. Couns. 2019, 28, 313–325. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ma, X.; Riaz, N.; Samstein, R.M.; Lee, M.; Makarov, V.; Valero, C.; Chowell, D.; Kuo, F.; Hoen, D.; Fitzgerald, C.W.R.; et al. Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity. Nat. Genet. 2022, 54, 996–1012. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| POLD1 Variant (Male/Female) | Cancer Type | Country/Consortium | Source | Cancer Age | Colonic Polyps |
|---|---|---|---|---|---|
| p.D316G (female) | Colorectal cancer, Endometrial cancer | Spain | Bellido [28] | 44, 54 | Yes |
| p.D316G; p.V295M (female) | Endometrial cancer, Breast Cancer | Spain | Bellido [28] | 57 | N/A |
| p.D316H (female) | Breast Cancer | Spain | Bellido [28] | 64 | Yes |
| p.D316H (male) | Colorectal cancer, angiomyolipoma, mesothelioma | Spain | Bellido [28] | 58 | Yes |
| p.D316N (female) | Breast cancer | United Kingdom/CORGI | Palles [29] | 52 | No |
| p.D316N (female) | Endometrial cancer | United Kingdom/CORGI | Palles [29], Robinson [9] | 54 | Yes |
| p.E318K (not reported) | Colorectal cancer | Taiwan | Chang [30] | N/A | N/A |
| p.D402N (not reported) | Colorectal cancer | Taiwan | Chang [30] | N/A | N/A |
| p.D402N (female; current case) | Endometrial cancer | USA | Current study | 32 | N/A |
| Name/Swissport P28340 | PolyPhen-2 | Mutation Taster | Panther | Mutation Assessor | SIFT | MutPred2 Score * |
|---|---|---|---|---|---|---|
| NM_002691.4(POLD1):c.946G > C (p.Asp316His) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.875 |
| NM_002691.4(POLD1):c.946G > A (p.Asp316Asn) | Probably damaging | Disease causing | Probably damaging | Medium functional impact | Not tolerated | 0.744 |
| NM_002691.4(POLD1):c.947A > G (p.Asp316Gly) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.882 |
| NM_002691.4(POLD1):c.952G > C (p.Glu318Gln) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.782 |
| NM_002691.4(POLD1):c.952G > A (p.Glu318Lys) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.885 |
| NM_002691.4(POLD1):c.953A > G (p.Glu318Gly) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.898 |
| NM_002691.4(POLD1):c.953A > C (p.Glu318Ala) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.878 |
| NM_002691.4(POLD1):c.1204G > T (p.Asp402Tyr) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.932 |
| NM_002691.4(POLD1):c.1204G > A (p.Asp402Asn) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.884 |
| NM_002691.4(POLD1):c.1205A > T (p.Asp402Val) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.924 |
| NM_002691.4(POLD1):c.1205A > G (p.Asp402Gly) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.926 |
| NM_002691.4(POLD1):c.1543G > A (p.Asp515Asn) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.779 |
| NM_002691.4(POLD1):c.1544A > G (p.Asp515Gly) | Probably damaging | Disease causing | Probably damaging | High functional impact | Not tolerated | 0.877 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, C.H.; Wang, E.W.; Ma, L.; Zhou, Y.; Zheng, L.; Hampel, H.; Shehayeb, S.; Lee, S.; Cohen, J.; Kohut, A.; et al. POLD1 DEDD Motif Mutation Confers Hypermutation in Endometrial Cancer and Durable Response to Pembrolizumab. Cancers 2023, 15, 5674. https://doi.org/10.3390/cancers15235674
Wei CH, Wang EW, Ma L, Zhou Y, Zheng L, Hampel H, Shehayeb S, Lee S, Cohen J, Kohut A, et al. POLD1 DEDD Motif Mutation Confers Hypermutation in Endometrial Cancer and Durable Response to Pembrolizumab. Cancers. 2023; 15(23):5674. https://doi.org/10.3390/cancers15235674
Chicago/Turabian StyleWei, Christina Hsiao, Edward Wenge Wang, Lingzi Ma, Yajing Zhou, Li Zheng, Heather Hampel, Susan Shehayeb, Stephen Lee, Joshua Cohen, Adrian Kohut, and et al. 2023. "POLD1 DEDD Motif Mutation Confers Hypermutation in Endometrial Cancer and Durable Response to Pembrolizumab" Cancers 15, no. 23: 5674. https://doi.org/10.3390/cancers15235674
APA StyleWei, C. H., Wang, E. W., Ma, L., Zhou, Y., Zheng, L., Hampel, H., Shehayeb, S., Lee, S., Cohen, J., Kohut, A., Fan, F., Rosen, S., Wu, X., Shen, B., & Zhao, Y. (2023). POLD1 DEDD Motif Mutation Confers Hypermutation in Endometrial Cancer and Durable Response to Pembrolizumab. Cancers, 15(23), 5674. https://doi.org/10.3390/cancers15235674