

Radiation-Induced Innate Neutrophil Response in Tumor Is Mediated by the CXCLs/CXCR2 Axis

 , ,
, , 
 and
and 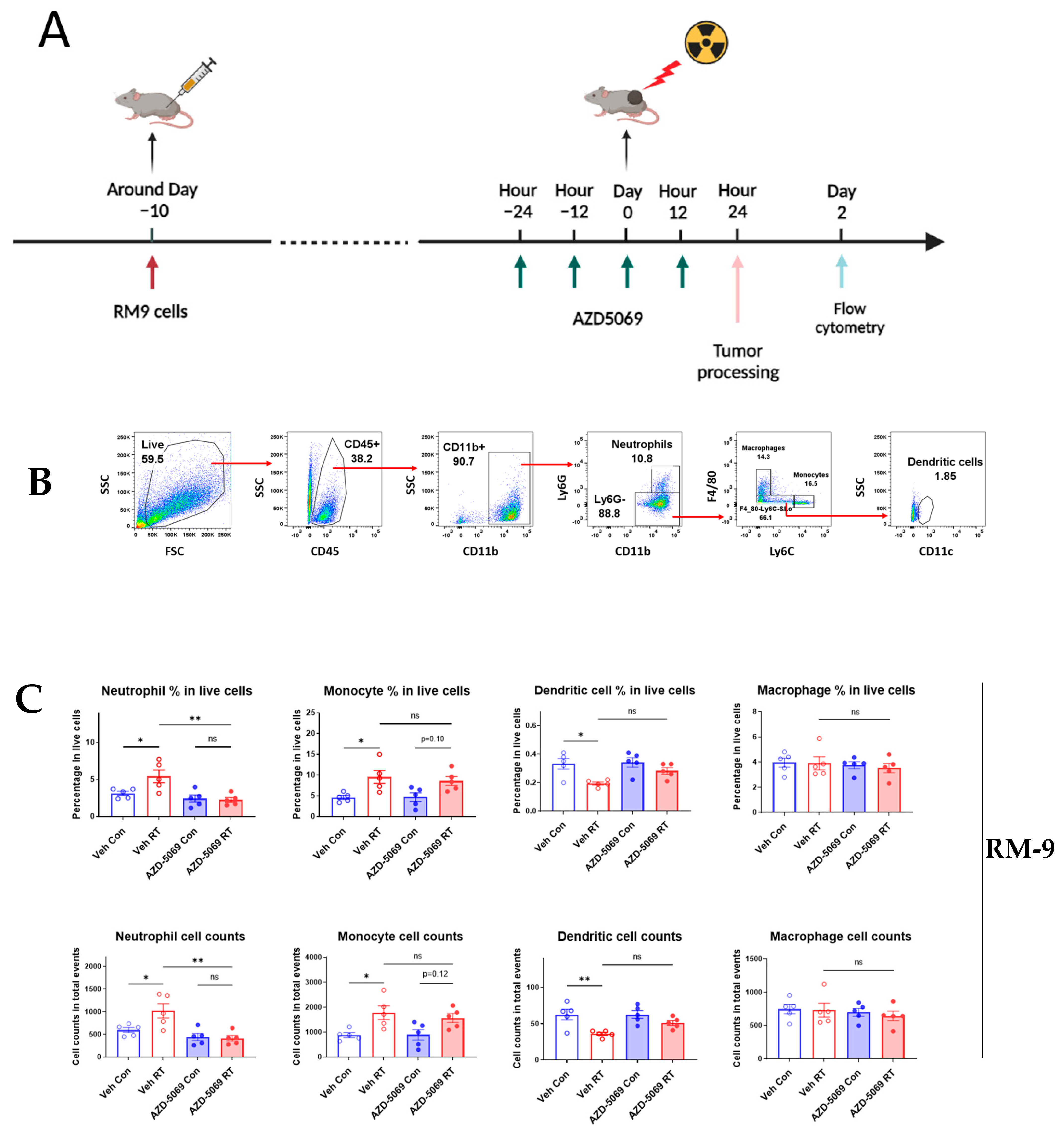{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Lines
2.3. Antibodies
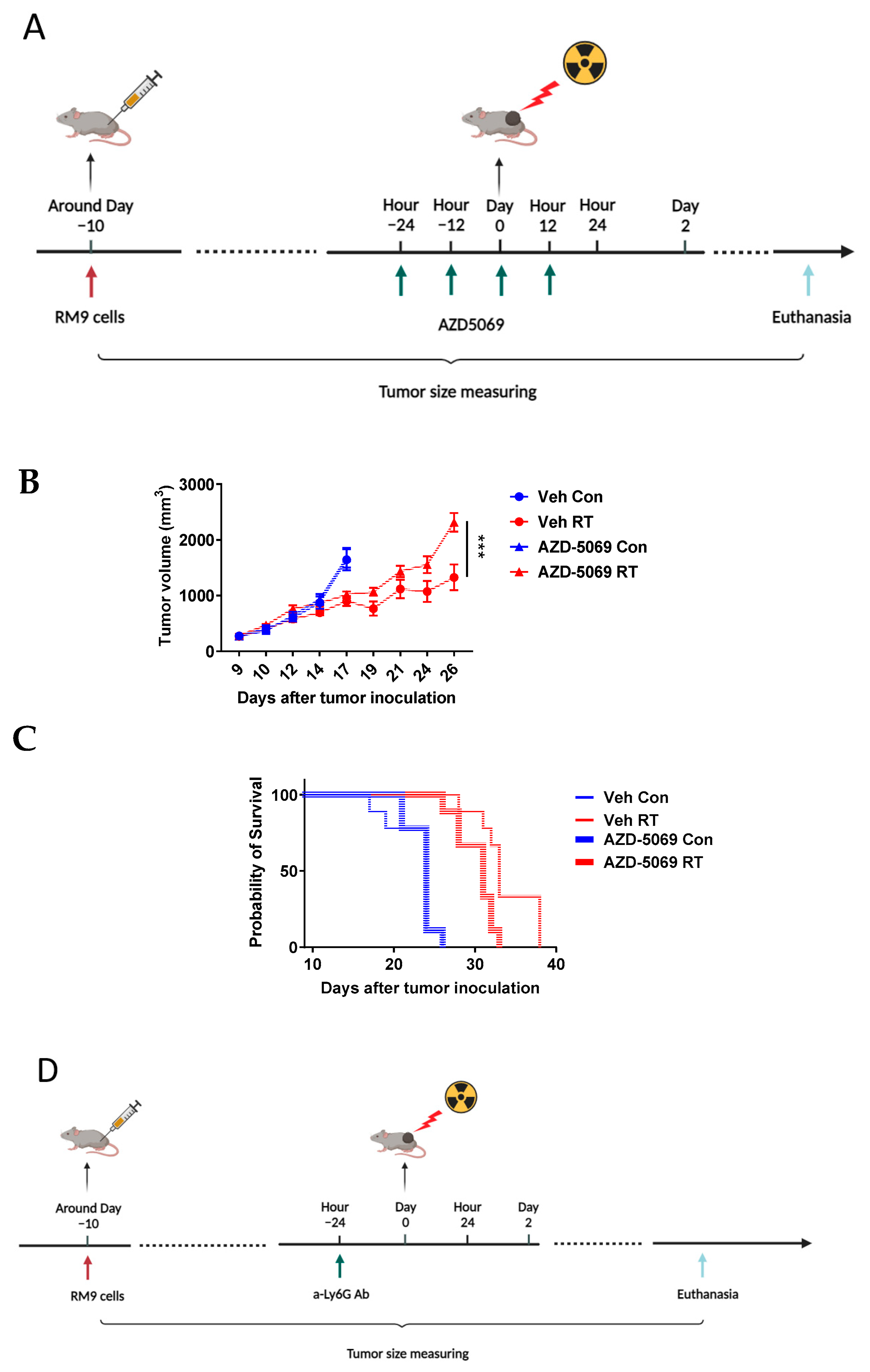
2.4. Tumor Inoculation and Irradiation
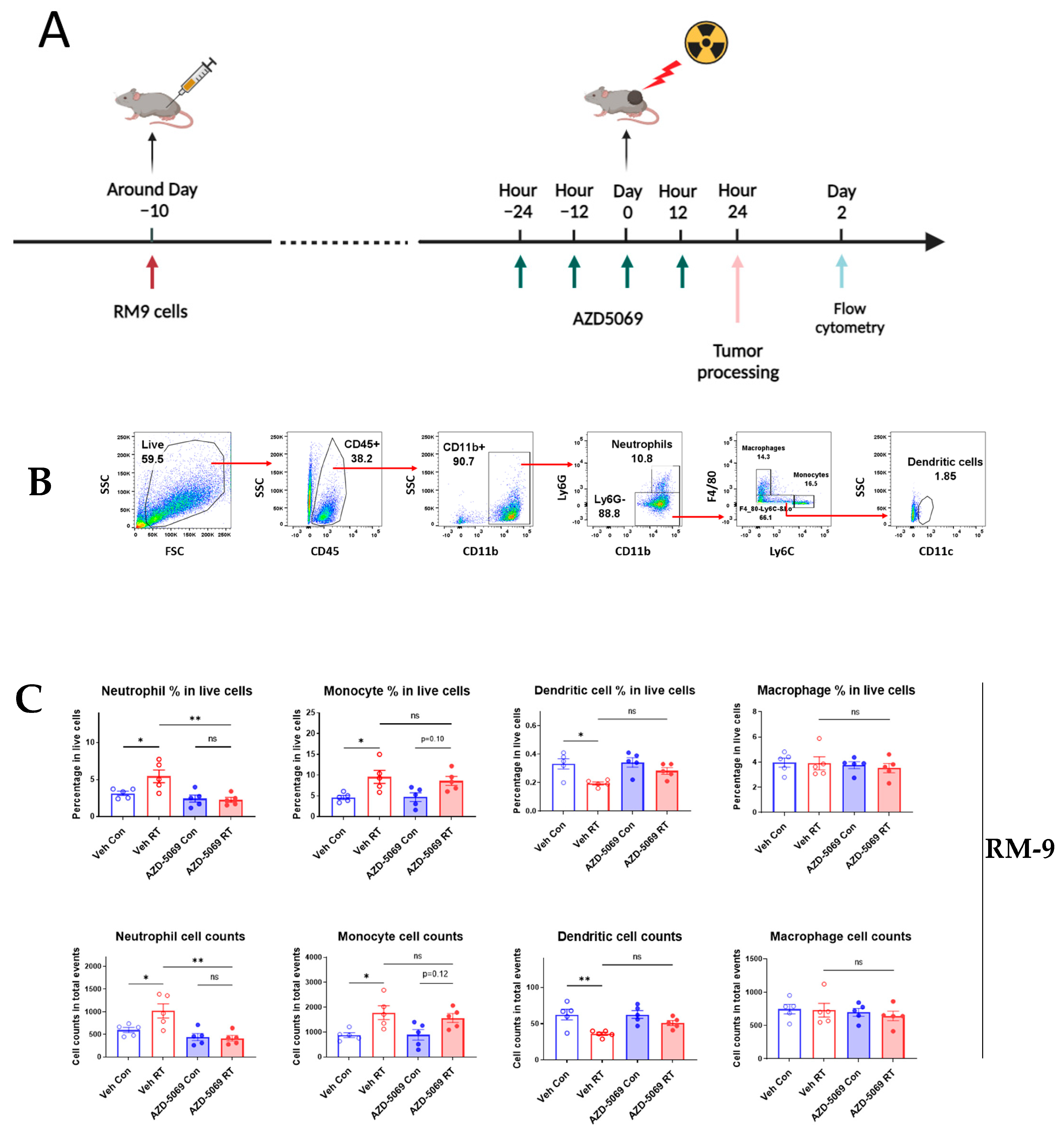
2.5. CXCR2 Antagonism
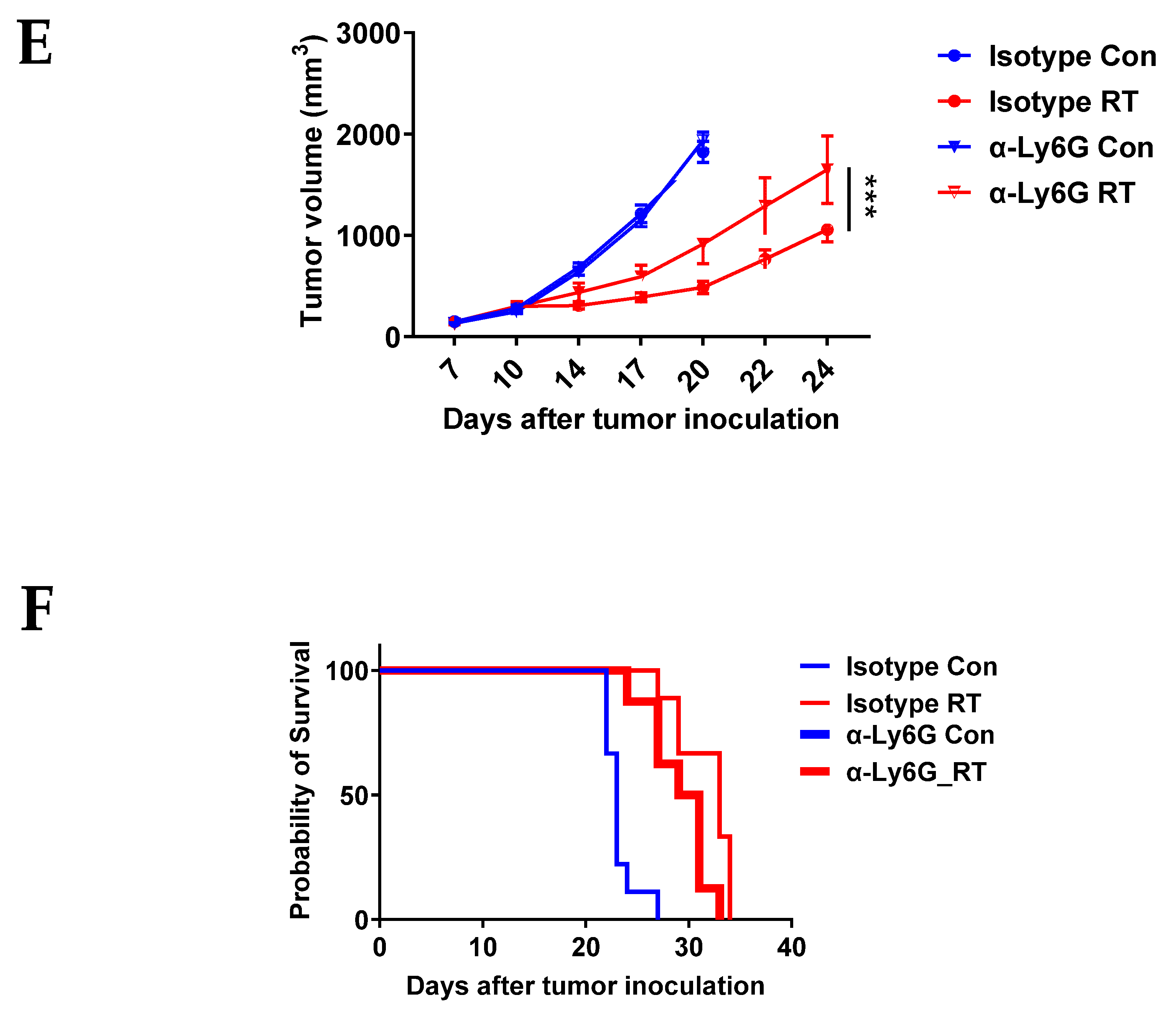
2.6. Neutrophil Depletion
2.7. mRNA-Seq and Data Analysis
2.8. Chemokine Multiplex Assay
2.9. Flow Cytometry Analysis
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
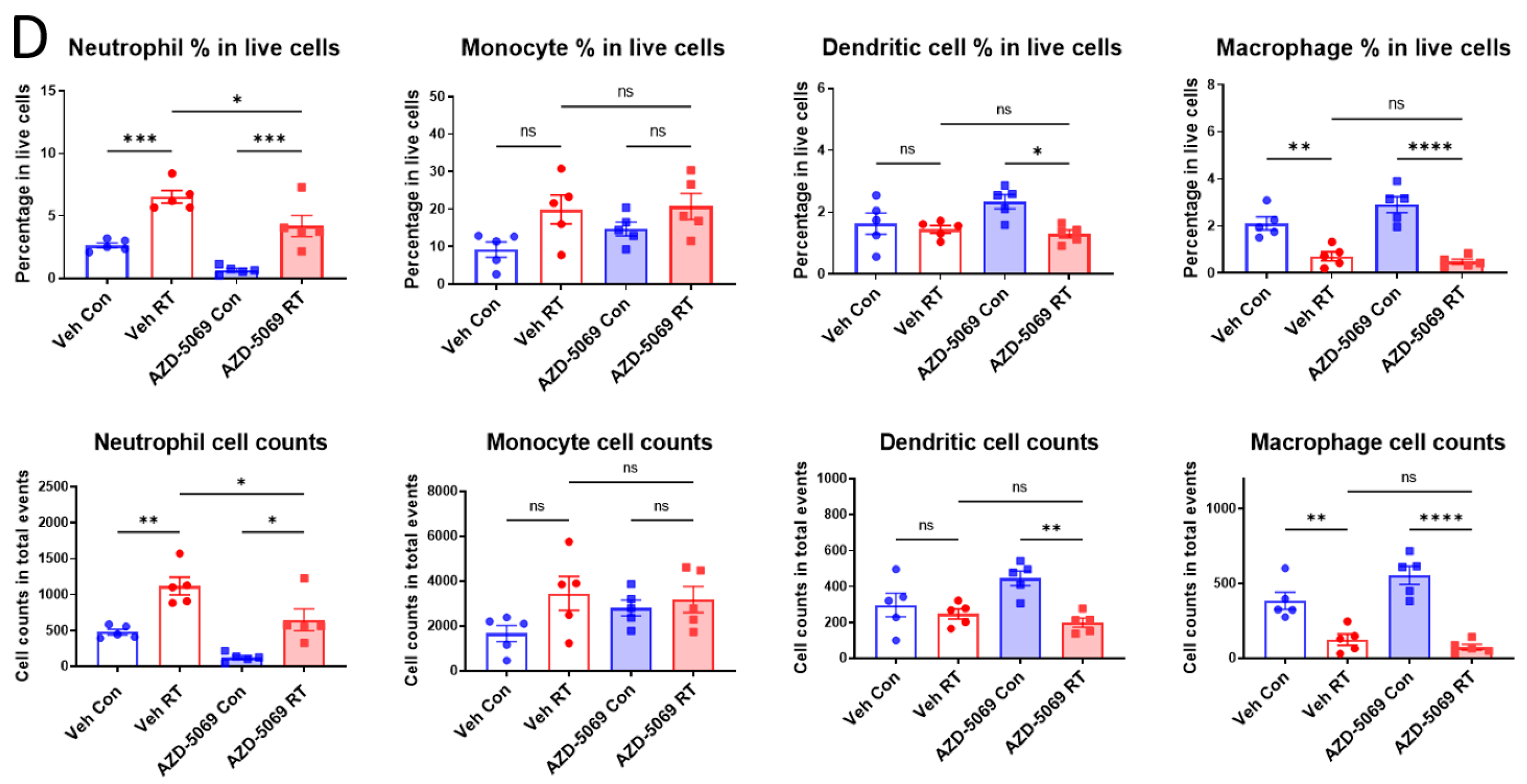
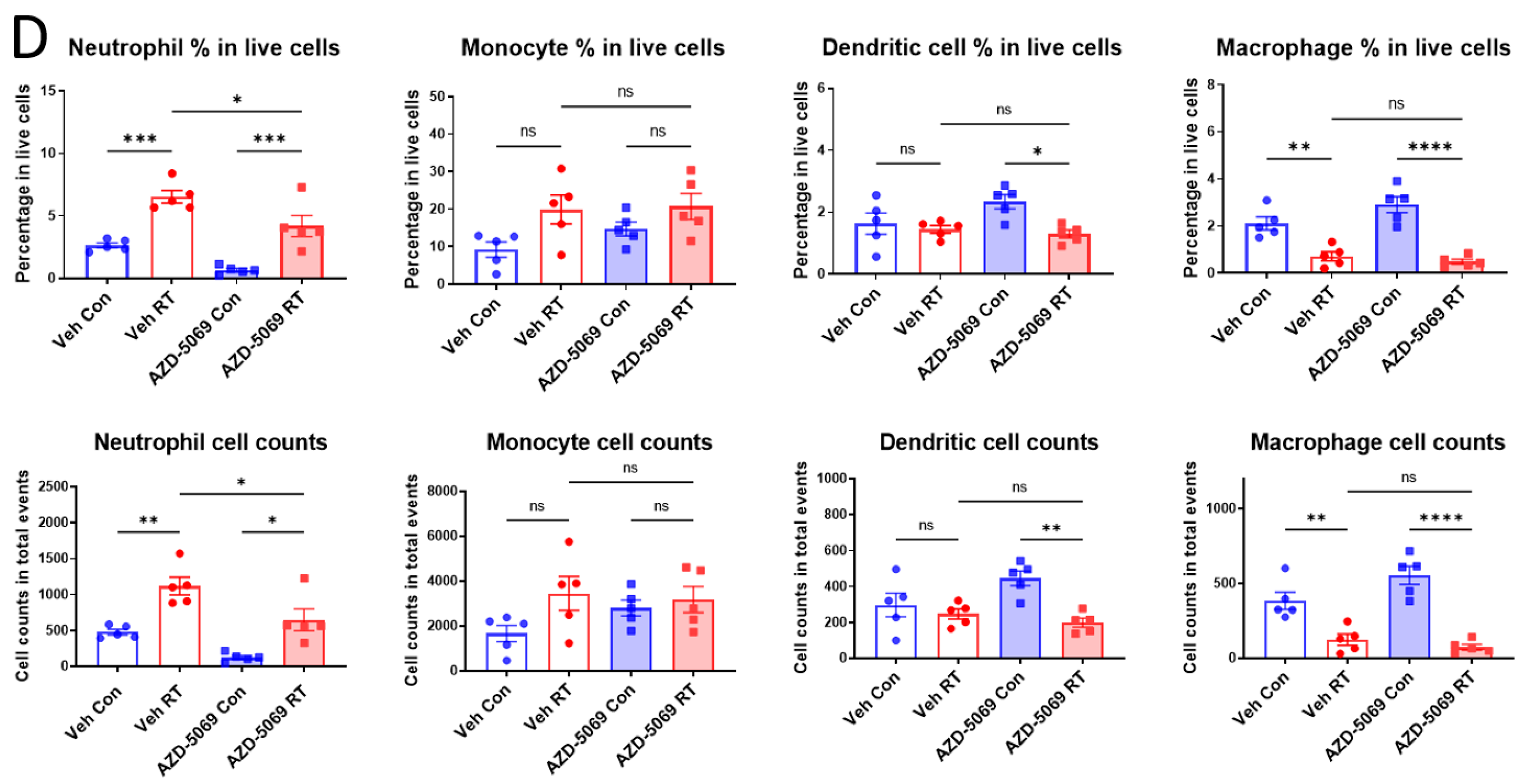
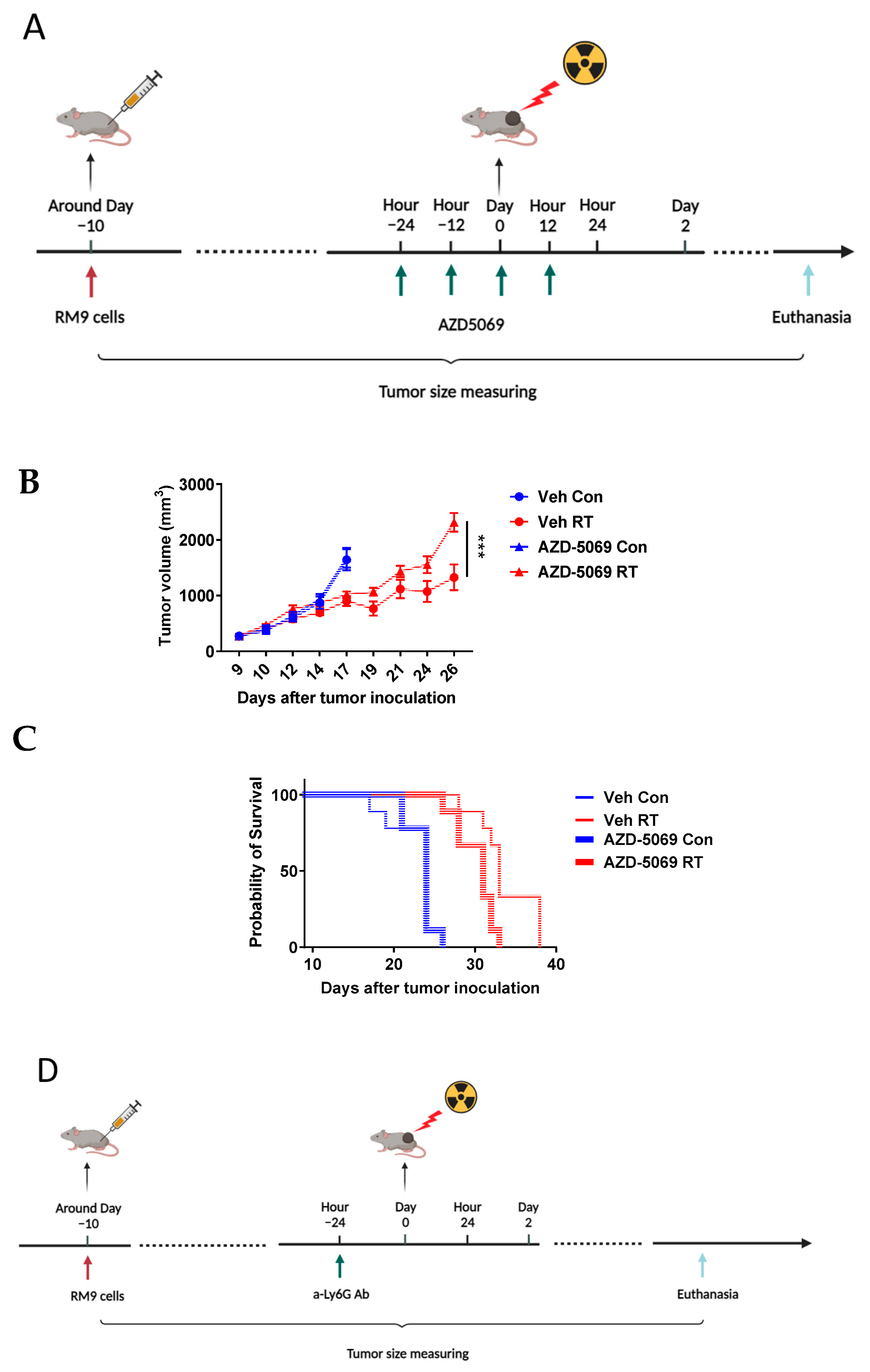
3.1. CXCR2 Blockade Impedes Radiation-Induced Neutrophilic Infiltration into the Tumor
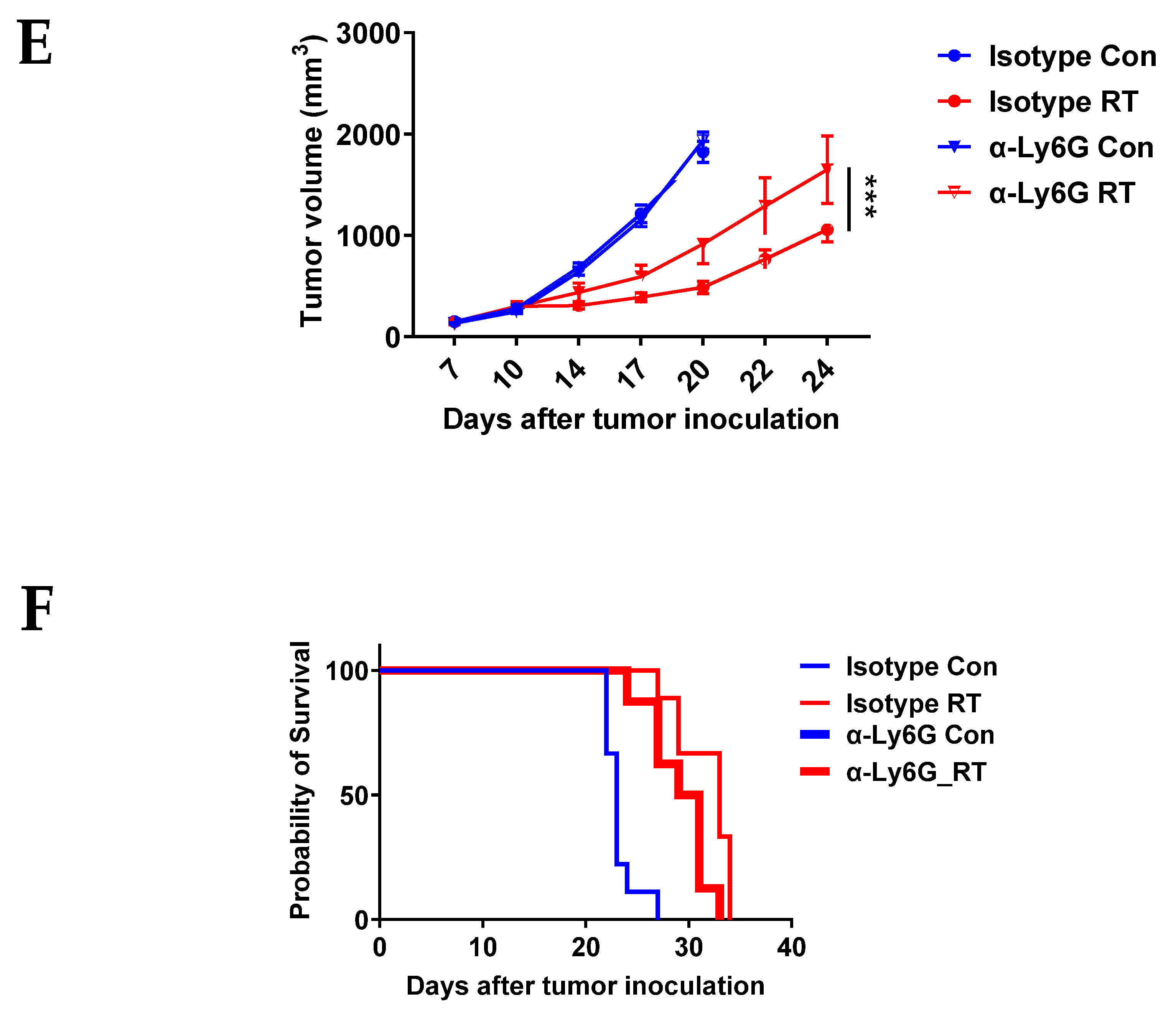
3.2. CXCR2 Blockade Has Similar Effects on RT-Induced Tumor Inhibition and Host Survival as Neutrophil Depletion
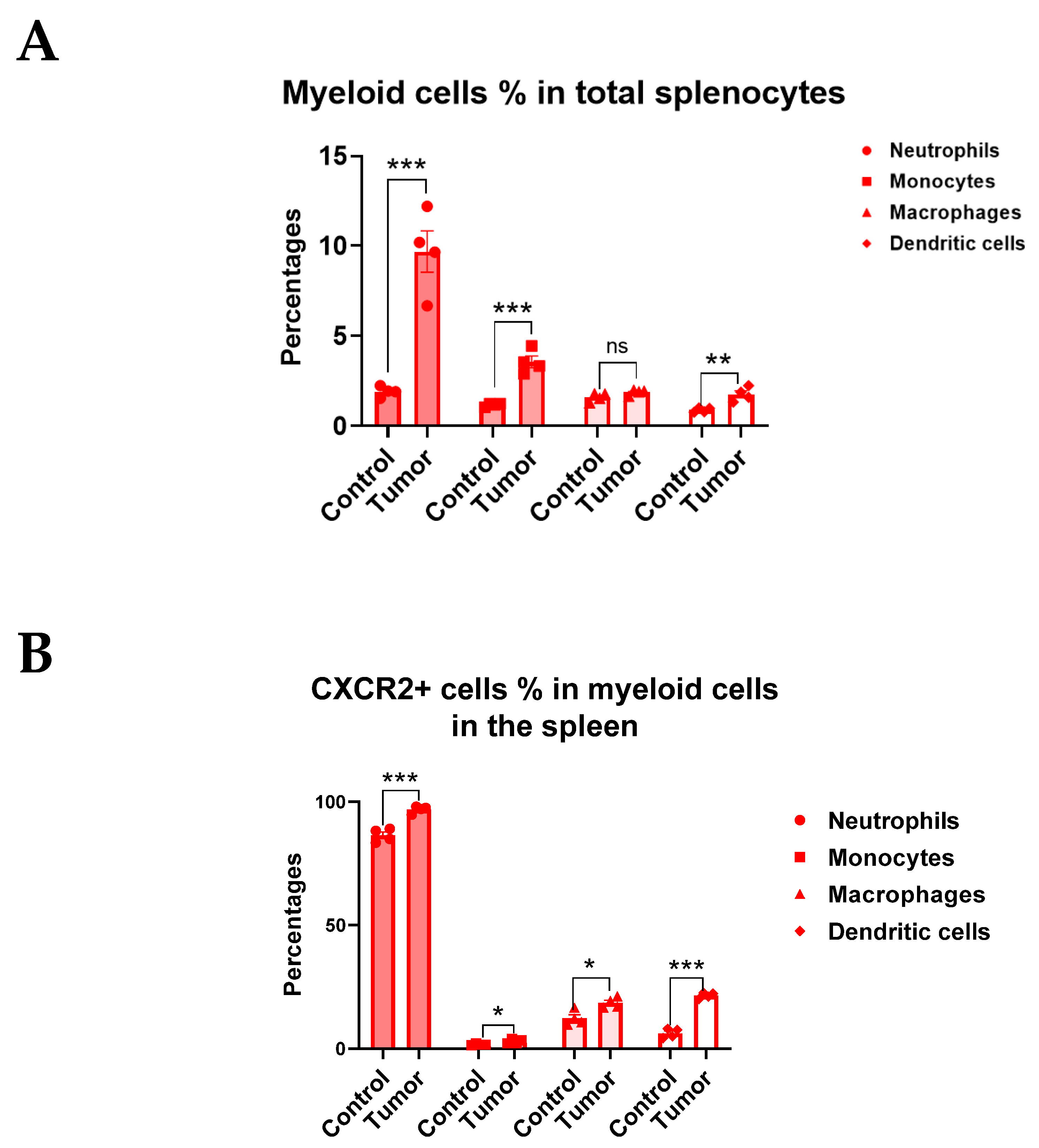
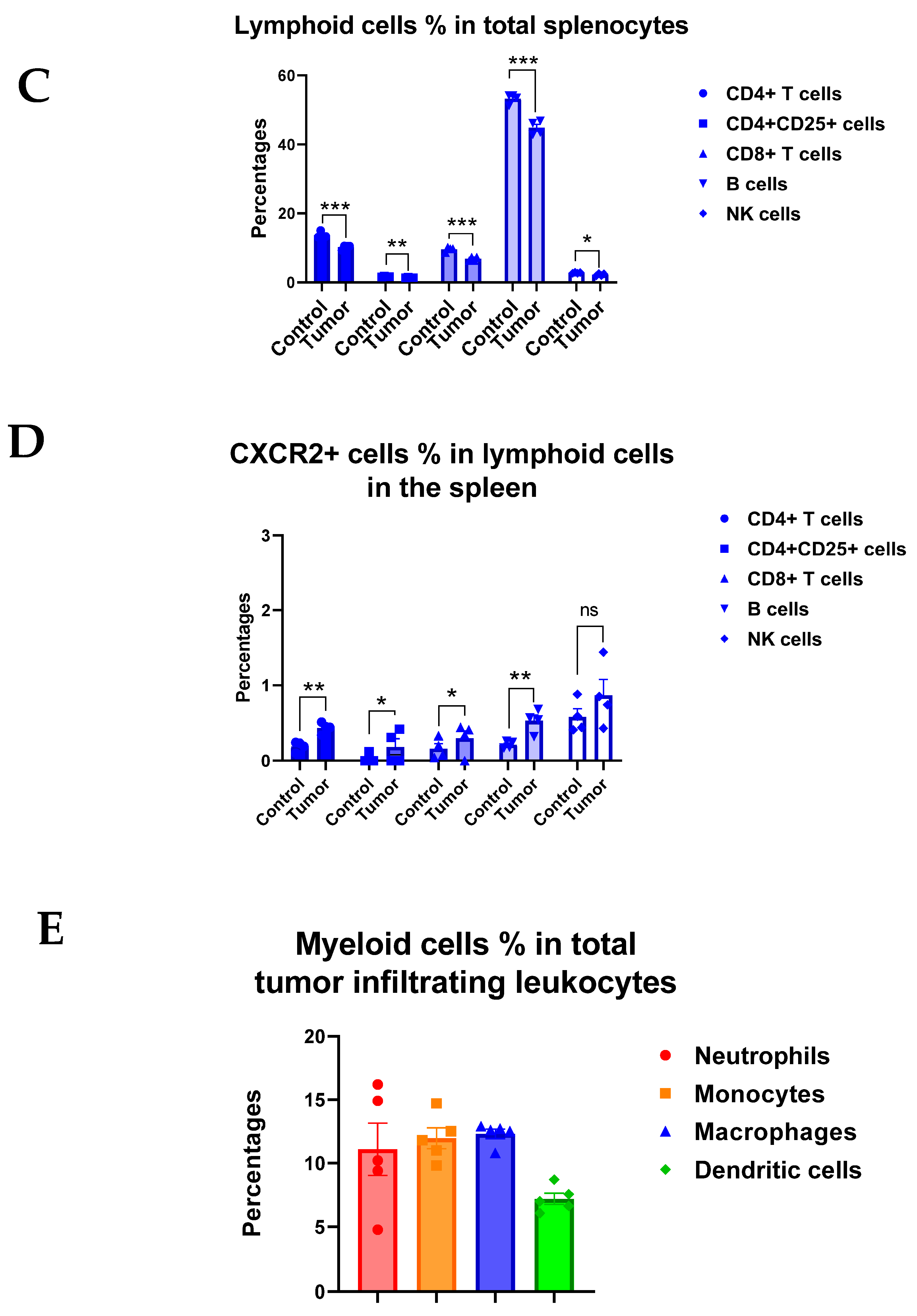
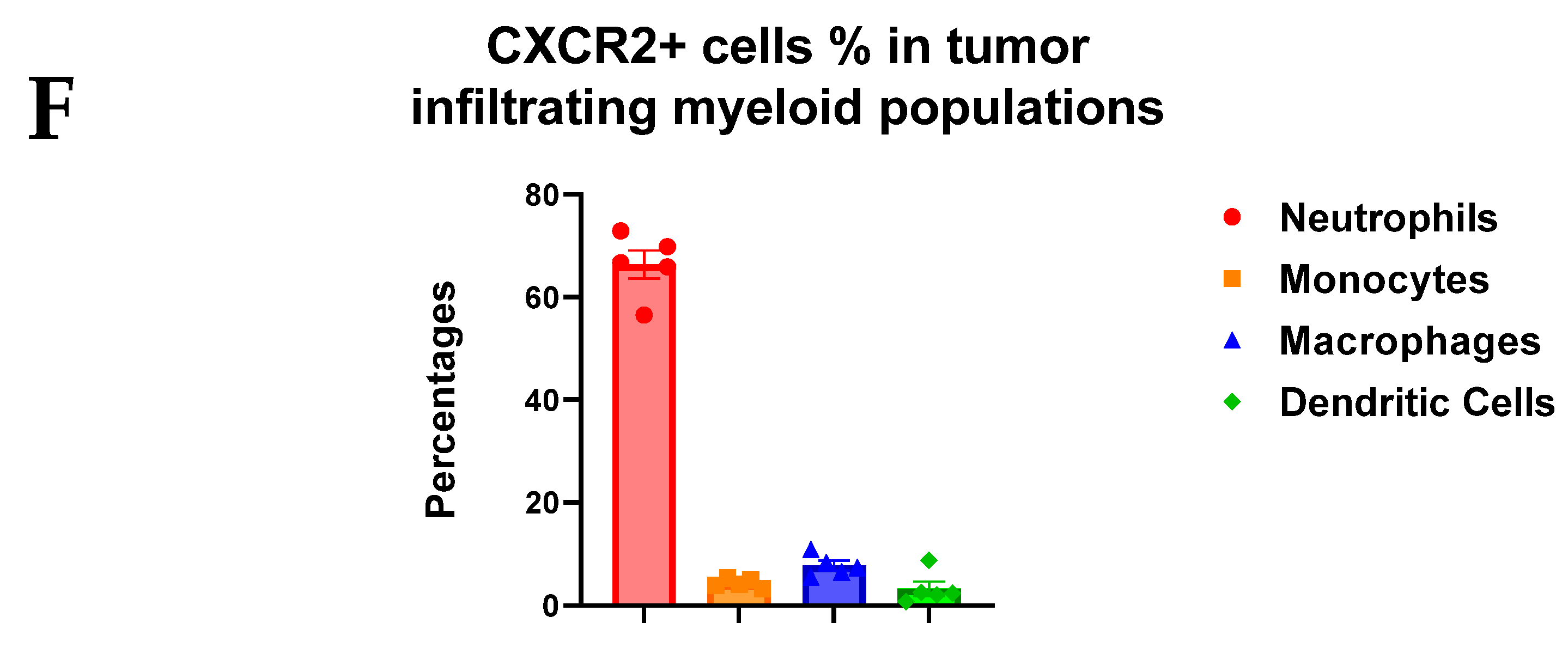
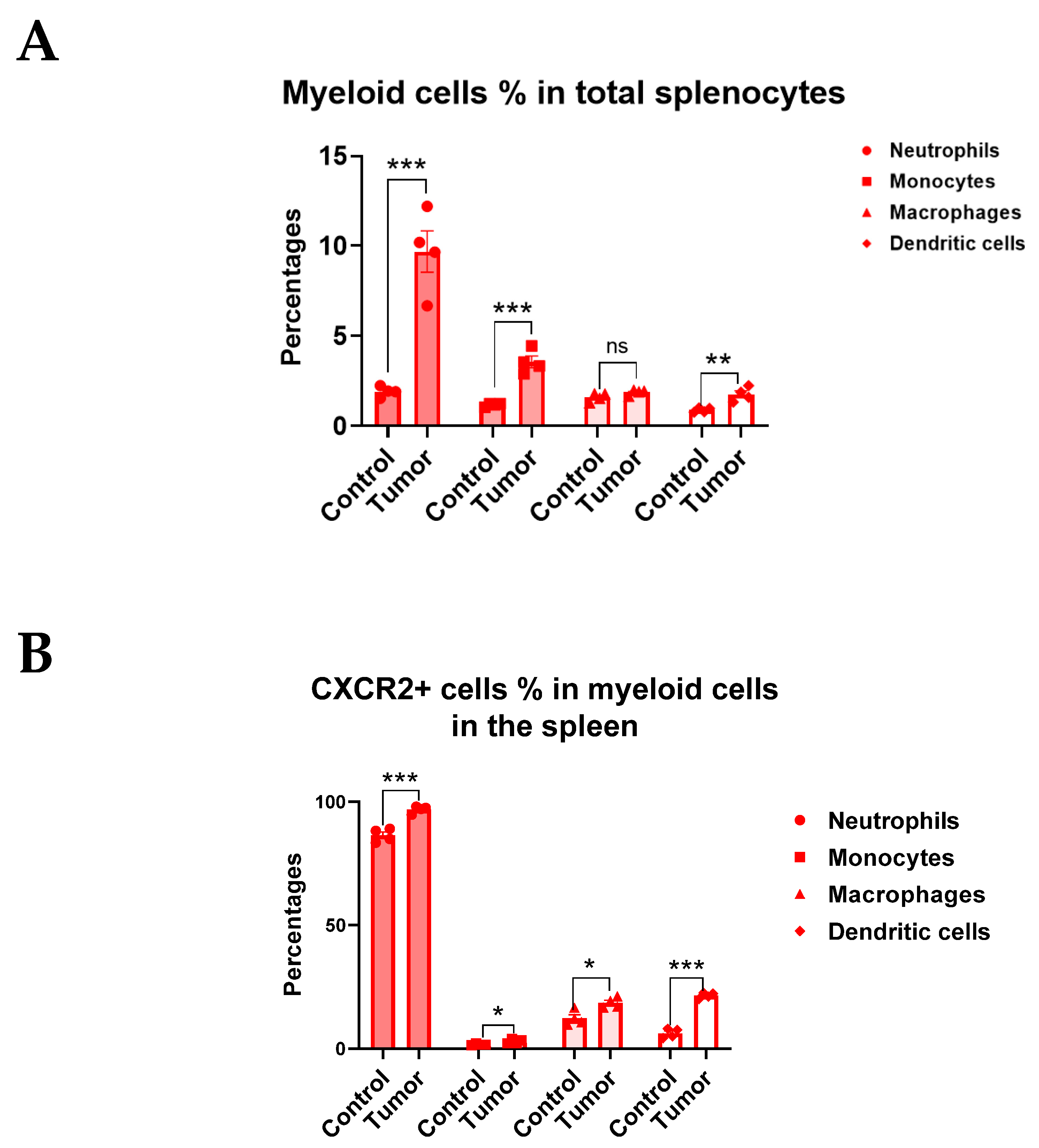
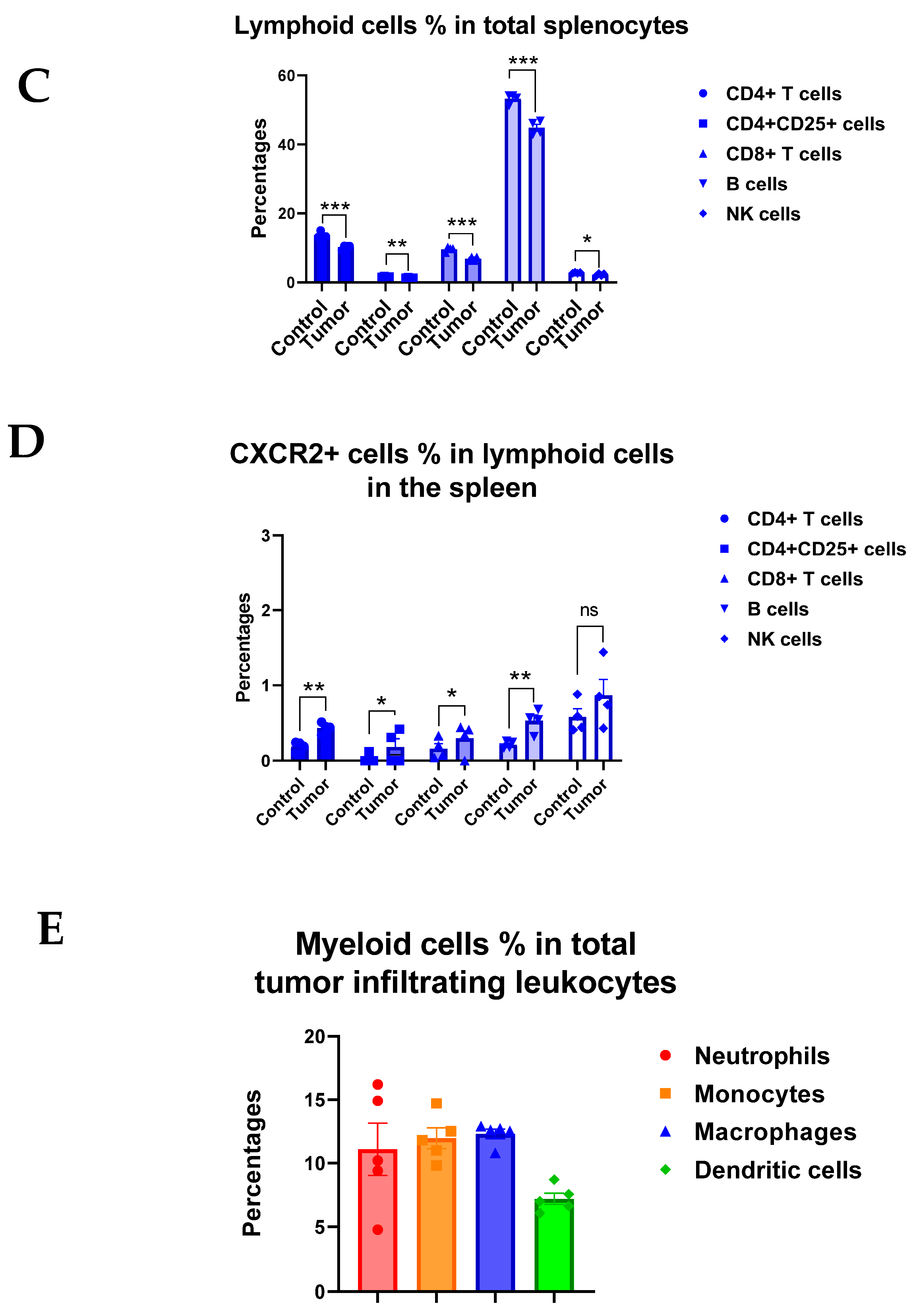
3.3. CXCR2 Is Highly and Preferentially Expressed by Neutrophils
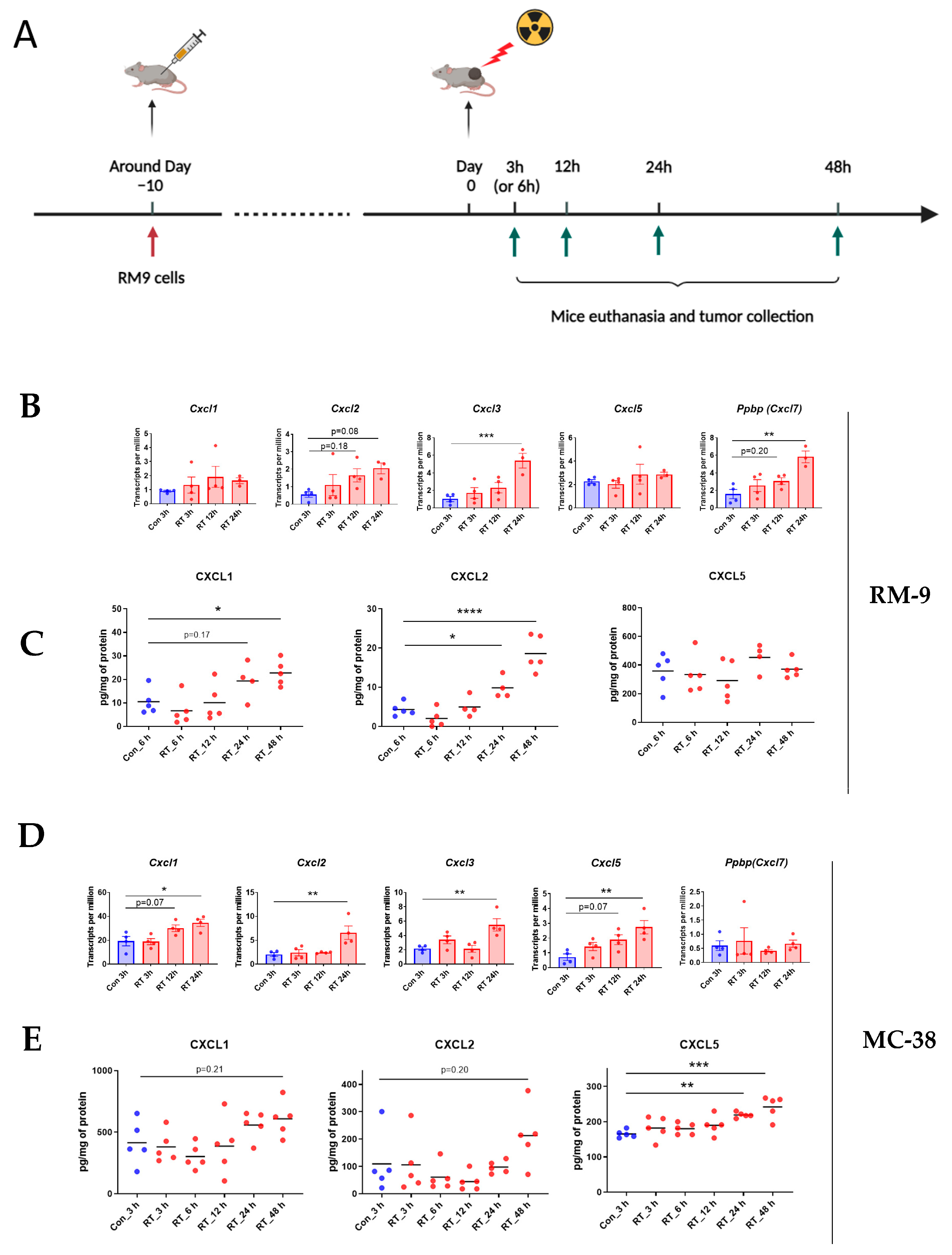
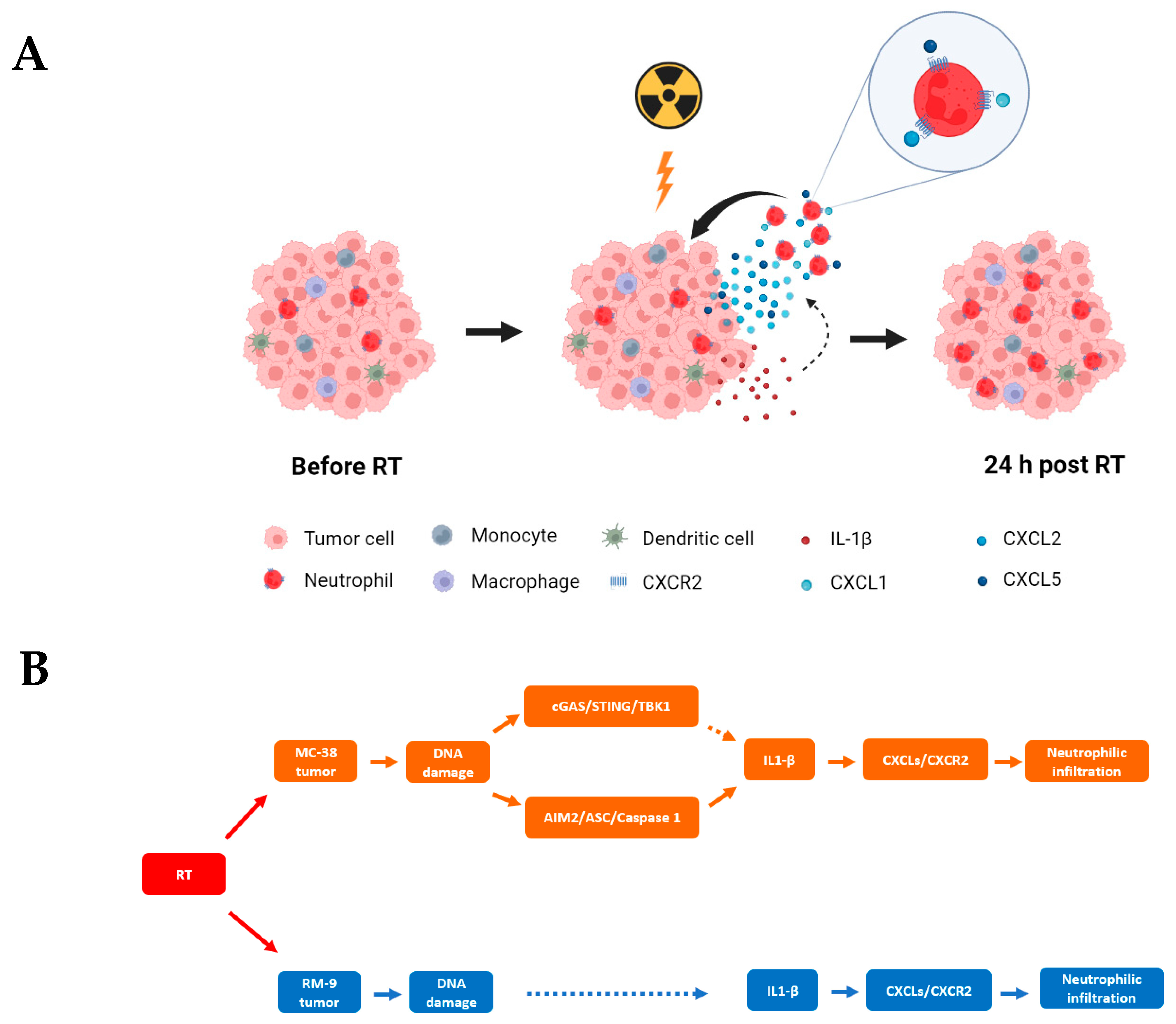
3.4. Radiation Increases CXCR2 Ligands in Tumor Microenvironment
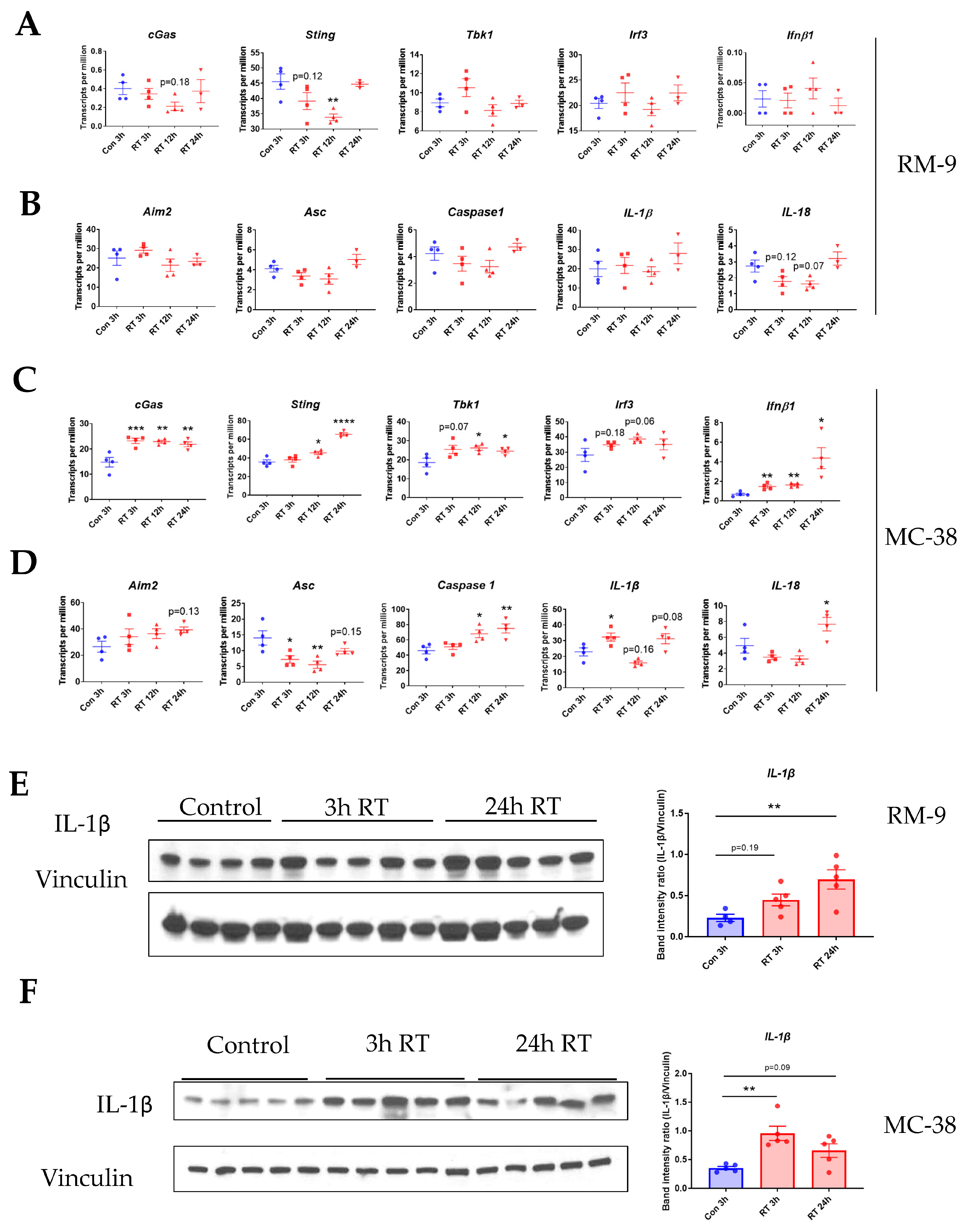
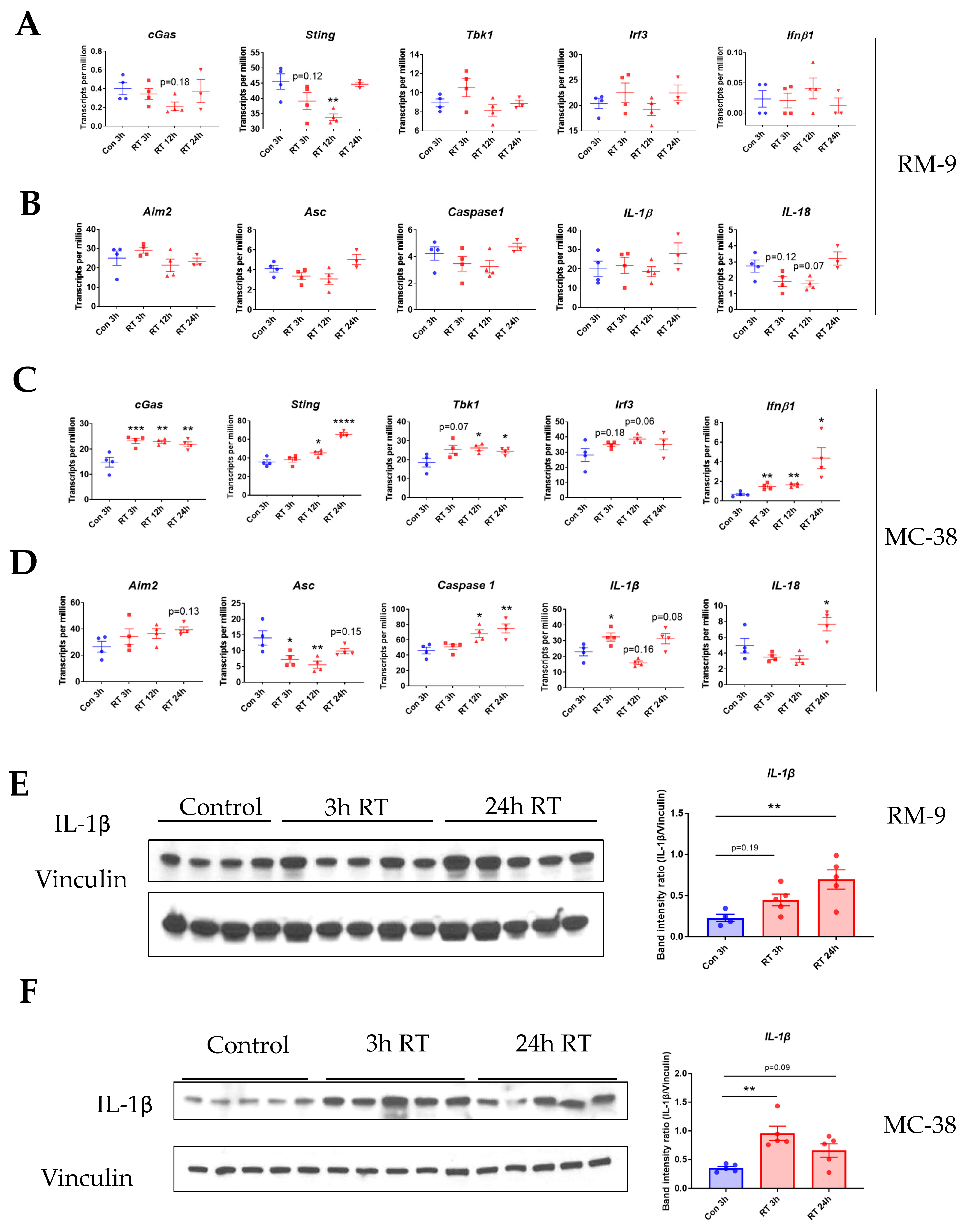
3.5. Radiation Increases the Gene Expression of DNA Sensing Pathways in the Tumor Microenvironment in cGAS-Positive MC-38 Model, but Not cGAS-Negative RM-9 Model
3.6. Radiation Increases IL-1β Expression in Tumor Microenvironment
3.7. Gene Ontology Analysis Shows Gene Enrichment in Related Biological Processes
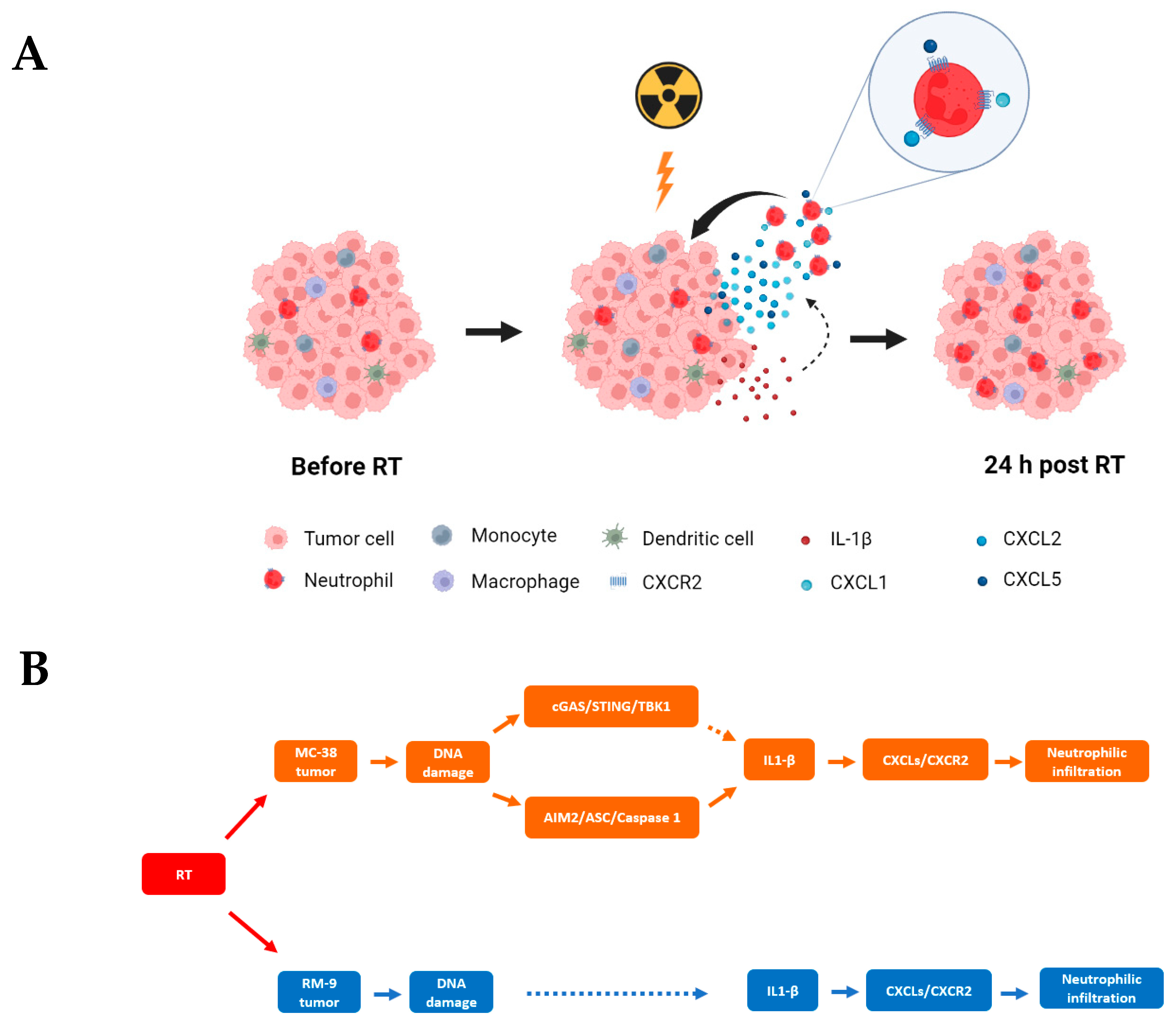
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Borad Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnett, G.C.; West, C.M.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Delaney, G.P.; Barton, M.B. Evidence-based estimates of the demand for radiotherapy. Clin. Oncol. 2015, 27, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10, 1758834017742575. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, T.; Pop, L.M.; Laine, A.; Iyengar, P.; Vitetta, E.S.; Hannan, R. Key role for neutrophils in radiation-induced antitumor immune responses: Potentiation with G-CSF. Proc. Natl. Acad. Sci. USA 2016, 113, 11300–11305. [Google Scholar] [CrossRef]
- Wisdom, A.J.; Hong, C.S.; Lin, A.J.; Xiang, Y.; Cooper, D.E.; Zhang, J.; Xu, E.S.; Kuo, H.C.; Mowery, Y.M.; Carpenter, D.J.; et al. Neutrophils promote tumor resistance to radiation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 18584–18589. [Google Scholar] [CrossRef]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef]
- Zhang, F.; Manna, S.; Pop, L.M.; Chen, Z.J.; Fu, Y.X.; Hannan, R. Type I Interferon Response in Radiation-Induced Anti-Tumor Immunity. Semin. Radiat. Oncol. 2020, 30, 129–138. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Godfrey, V.; Liu, Z.; Han, Y.; Liu, L.; Peng, H.; Weichselbaum, R.R.; Zaki, H.; Fu, Y.X. The AIM2 and NLRP3 inflammasomes trigger IL-1-mediated antitumor effects during radiation. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef]
- Hyun, Y.M.; Hong, C.W. Deep insight into neutrophil trafficking in various organs. J. Leukoc. Biol. 2017, 102, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef]
- Korbecki, J.; Barczak, K.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CXCL1: Gene, Promoter, Regulation of Expression, mRNA Stability, Regulation of Activity in the Intercellular Space. Int. J. Mol. Sci. 2022, 23, 792. [Google Scholar] [CrossRef]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef]
- Biondo, C.; Mancuso, G.; Midiri, A.; Signorino, G.; Domina, M.; Lanza Cariccio, V.; Mohammadi, N.; Venza, M.; Venza, I.; Teti, G.; et al. The interleukin-1beta/CXCL1/2/neutrophil axis mediates host protection against group B streptococcal infection. Infect. Immun. 2014, 82, 4508–4517. [Google Scholar] [CrossRef]
- Liu, W.; Ding, I.; Chen, K.; Olschowka, J.; Xu, J.; Hu, D.; Morrow, G.R.; Okunieff, P. Interleukin 1beta (IL1B) signaling is a critical component of radiation-induced skin fibrosis. Radiat. Res. 2006, 165, 181–191. [Google Scholar] [CrossRef]
- Janko, M.; Ontiveros, F.; Fitzgerald, T.J.; Deng, A.; DeCicco, M.; Rock, K.L. IL-1 generated subsequent to radiation-induced tissue injury contributes to the pathogenesis of radiodermatitis. Radiat. Res. 2012, 178, 166–172. [Google Scholar] [CrossRef]
- Liu, Y.G.; Chen, J.K.; Zhang, Z.T.; Ma, X.J.; Chen, Y.C.; Du, X.M.; Liu, H.; Zong, Y.; Lu, G.C. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 2017, 8, e2579. [Google Scholar] [CrossRef]
- Kang, A.R.; Cho, J.H.; Lee, N.G.; Kwon, J.H.; Song, J.Y.; Hwang, S.G.; Jung, I.S.; Kim, J.S.; Um, H.D.; Oh, S.C.; et al. Radiation-induced IL-1beta expression and secretion promote cancer cell migration/invasion via activation of the NF-kappaB-RIP1 pathway. Biochem. Biophys. Res. Commun. 2021, 534, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Sody, S.; Uddin, M.; Gruneboom, A.; Gorgens, A.; Giebel, B.; Gunzer, M.; Brandau, S. Distinct Spatio-Temporal Dynamics of Tumor-Associated Neutrophils in Small Tumor Lesions. Front. Immunol. 2019, 10, 1419. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Little, A.; Zhang, H. Chronic alcohol consumption inhibits peripheral NK cell development and maturation by decreasing the availability of IL-15. J. Leucoc. Biol. 2017, 101, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013, 41, D1040–D1046. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Elvington, M.; Scheiber, M.; Yang, X.; Lyons, K.; Jacqmin, D.; Wadsworth, C.; Marshall, D.; Vanek, K.; Tomlinson, S. Complement-dependent modulation of antitumor immunity following radiation therapy. Cell Rep. 2014, 8, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Filatenkov, A.; Baker, J.; Mueller, A.M.; Kenkel, J.; Ahn, G.O.; Dutt, S.; Zhang, N.; Kohrt, H.; Jensen, K.; Dejbakhsh-Jones, S.; et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin. Cancer Res. 2015, 21, 3727–3739. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Mantovani, A.; Jaillon, S. Chemokines as Regulators of Neutrophils: Focus on Tumors, Therapeutic Targeting, and Immunotherapy. Cancers 2022, 14, 680. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.I.; Kim, D.; Ahn, S.G.; Bae, S.J.; Cha, C.; Park, S.; Park, S.; Kim, S.I.; Lee, H.S.; Park, J.Y.; et al. Radiotherapy-Induced High Neutrophil-to-Lymphocyte Ratio is a Negative Prognostic Factor in Patients with Breast Cancer. Cancers 2020, 12, 1896. [Google Scholar] [CrossRef]
- Wolfe, A.R.; Siedow, M.; Nalin, A.; DiCostanzo, D.; Miller, E.D.; Diaz, D.A.; Arnett, A.; Cloyd, J.M.; Dillhoff, M.; Ejaz, A.; et al. Increasing neutrophil-to-lymphocyte ratio following radiation is a poor prognostic factor and directly correlates with splenic radiation dose in pancreatic cancer. Radiother. Oncol. 2021, 158, 207–214. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, 7. [Google Scholar] [CrossRef]
- Korbecki, J.; Kupnicka, P.; Chlubek, M.; Goracy, J.; Gutowska, I.; Baranowska-Bosiacka, I. CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int. J. Mol. Sci. 2022, 23, 2168. [Google Scholar] [CrossRef]
- Fujimori, A.; Okayasu, R.; Ishihara, H.; Yoshida, S.; Eguchi-Kasai, K.; Nojima, K.; Ebisawa, S.; Takahashi, S. Extremely low dose ionizing radiation up-regulates CXC chemokines in normal human fibroblasts. Cancer Res. 2005, 65, 10159–10163. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, D.M.; Han, R.; Yu, Y.; Deng, S.H.; Liu, T.; Zhang, T.; Xu, Y. Low-Dose Radiation Promotes Invasion and Migration of A549 Cells by Activating the CXCL1/NF-kappaB Signaling Pathway. Onco Targets Ther. 2020, 13, 3619–3629. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Huang, S.; Vallabhaneni, G.; Armstrong, E.; Varambally, S.; Tomlins, S.A.; Chinnaiyan, A.M.; Harari, P.M. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva). Cancer Res. 2005, 65, 3328–3335. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.A.; Moriconi, F.; Sheikh, N.; Naz, N.; Khan, S.; Dudas, J.; Mansuroglu, T.; Hess, C.F.; Rave-Frank, M.; Christiansen, H.; et al. Single-dose gamma-irradiation induces up-regulation of chemokine gene expression and recruitment of granulocytes into the portal area but not into other regions of rat hepatic tissue. Am. J. Pathol. 2010, 176, 1801–1815. [Google Scholar] [CrossRef]
- Hu, J.; Ji, Q.; Chen, F.; Gong, X.; Chen, C.; Zhang, K.; Hua, Y.; Wang, J.; Li, R.; Wang, X.; et al. CXCR2 Is Essential for Radiation-Induced Intestinal Injury by Initiating Neutrophil Infiltration. J. Immunol. Res. 2022, 2022, 7966089. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef]
- Ahn, J.; Son, S.; Oliveira, S.C.; Barber, G.N. STING-Dependent Signaling Underlies IL-10 Controlled Inflammatory Colitis. Cell Rep. 2017, 21, 3873–3884. [Google Scholar] [CrossRef]
- Guerin, M.V.; Finisguerra, V.; Van den Eynde, B.J.; Bercovici, N.; Trautmann, A. Preclinical murine tumor models: A structural and functional perspective. eLife 2020, 9, e50740. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Leask, A. Tumor-associated fibrosis impairs the response to immunotherapy. Matrix Biol. 2023, 119, 125–140. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Mulvaney, O.; Salcedo, E.; Manna, S.; Zhu, J.Z.; Wang, T.; Ahn, C.; Pop, L.M.; Hannan, R. Radiation-Induced Innate Neutrophil Response in Tumor Is Mediated by the CXCLs/CXCR2 Axis. Cancers 2023, 15, 5686. https://doi.org/10.3390/cancers15235686
Zhang F, Mulvaney O, Salcedo E, Manna S, Zhu JZ, Wang T, Ahn C, Pop LM, Hannan R. Radiation-Induced Innate Neutrophil Response in Tumor Is Mediated by the CXCLs/CXCR2 Axis. Cancers. 2023; 15(23):5686. https://doi.org/10.3390/cancers15235686
Chicago/Turabian StyleZhang, Faya, Oscar Mulvaney, Erica Salcedo, Subrata Manna, James Z. Zhu, Tao Wang, Chul Ahn, Laurentiu M. Pop, and Raquibul Hannan. 2023. "Radiation-Induced Innate Neutrophil Response in Tumor Is Mediated by the CXCLs/CXCR2 Axis" Cancers 15, no. 23: 5686. https://doi.org/10.3390/cancers15235686
APA StyleZhang, F., Mulvaney, O., Salcedo, E., Manna, S., Zhu, J. Z., Wang, T., Ahn, C., Pop, L. M., & Hannan, R. (2023). Radiation-Induced Innate Neutrophil Response in Tumor Is Mediated by the CXCLs/CXCR2 Axis. Cancers, 15(23), 5686. https://doi.org/10.3390/cancers15235686