

The Involvement of Peroxiporins and Antioxidant Transcription Factors in Breast Cancer Therapy Resistance





Abstract
:Simple Summary
Abstract
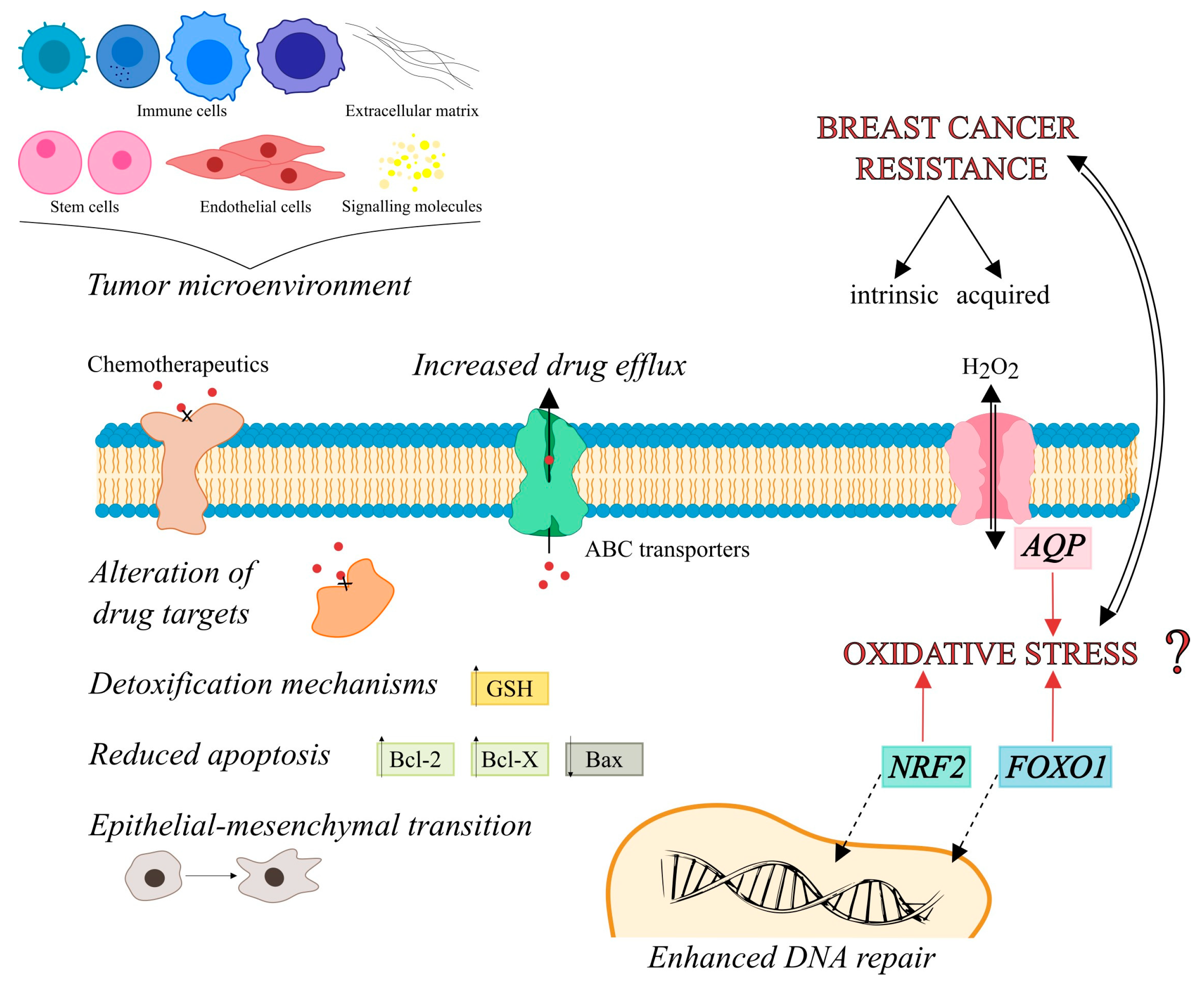
1. Introduction
2. Peroxiporins
2.1. AQP1
2.2. AQP3
2.3. AQP5
2.4. AQP9
2.5. AQP11 & AQP8
3. Transcription Factors
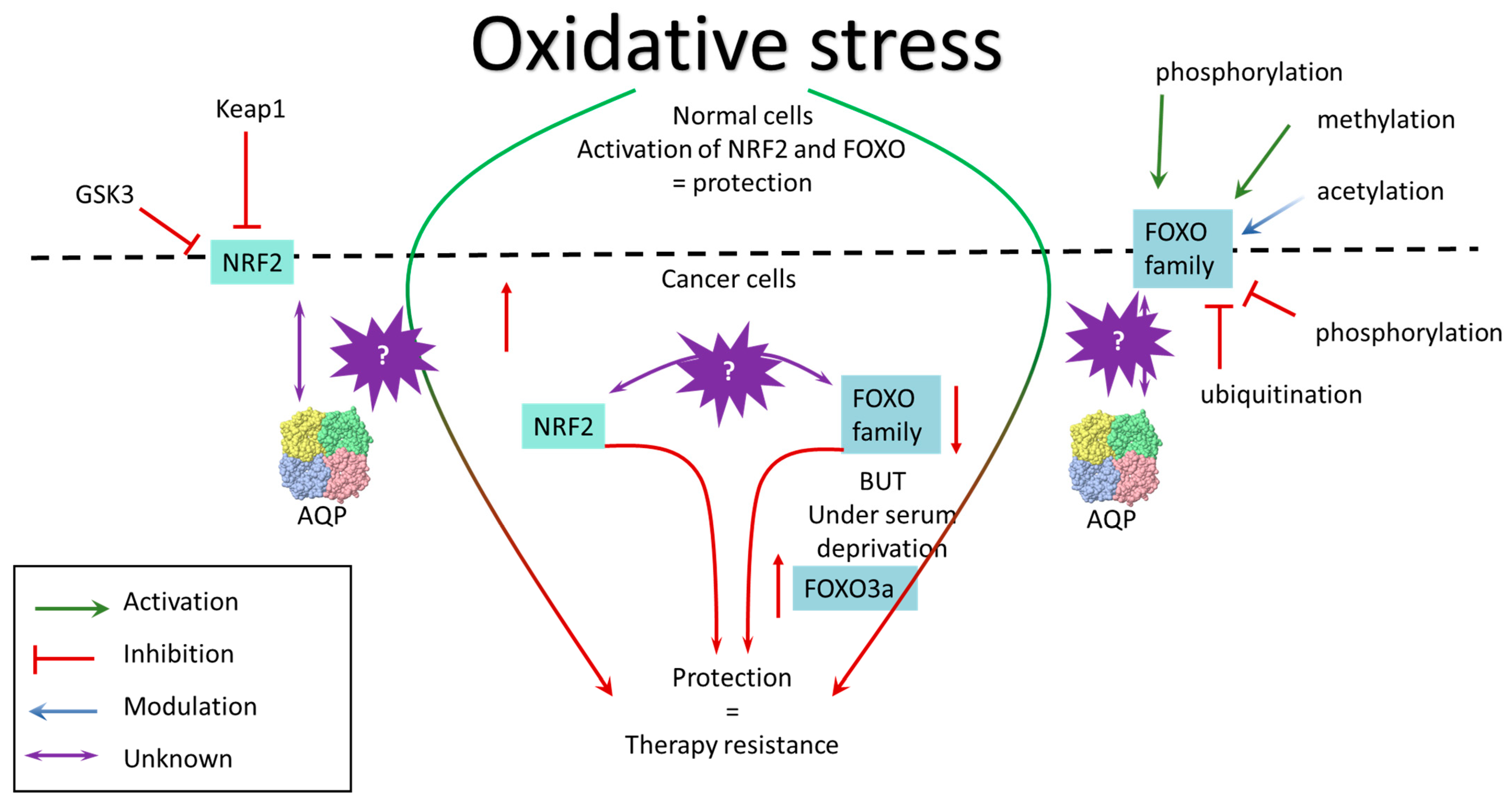
3.1. NRF2
3.2. FOXO
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European Cancer Burden in 2020: Incidence and Mortality Estimates for 40 Countries and 25 Major Cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef] [PubMed]
- Boecker, W. (Ed.) Preneoplasia of the Breast: A New Conceptual Approach to Proliferative Breast Disease; Saunders Elsevier: Amsterdam, The Netherlands, 2006; ISBN 978-0-7020-2892-2. [Google Scholar]
- Guo, L.; Kong, D.; Liu, J.; Zhan, L.; Luo, L.; Zheng, W.; Zheng, Q.; Chen, C.; Sun, S. Breast Cancer Heterogeneity and Its Implication in Personalized Precision Therapy. Exp. Hematol. Oncol. 2023, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Cortes, J.; Kaufman, P.A.; Yelle, L.; Awada, A.; Binder, T.A.; Olivo, M.; Song, J.; O’Shaughnessy, J.A.; Jove, M.; et al. “New” Metastases Are Associated with a Poorer Prognosis than Growth of Pre-Existing Metastases in Patients with Metastatic Breast Cancer Treated with Chemotherapy. Breast Cancer Res. BCR 2015, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- Zagami, P.; Carey, L.A. Triple Negative Breast Cancer: Pitfalls and Progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Grischke, E.-M.; Brucker, S.Y. Endocrine-Resistant Breast Cancer: Mechanisms and Treatment. Breast Care 2020, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and Acquired Resistance to HER2-Targeted Therapies in HER2 Gene-Amplified Breast Cancer: Mechanisms and Clinical Implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef]
- Luque-Bolivar, A.; Pérez-Mora, E.; Villegas, V.E.; Rondón-Lagos, M. Resistance and Overcoming Resistance in Breast Cancer. Breast Cancer Targets Ther. 2020, 12, 211–229. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Alexa-Stratulat, T.; Pešić, M.; Gašparović, A.Č.; Trougakos, I.P.; Riganti, C. What Sustains the Multidrug Resistance Phenotype beyond ABC Efflux Transporters? Looking beyond the Tip of the Iceberg. Drug Resist. Updat. 2019, 46, 100643. [Google Scholar] [CrossRef]
- Amawi, H.; Sim, H.M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef]
- Bueschbell, B.; Caniceiro, A.B.; Suzano, P.M.S.; Machuqueiro, M.; Rosário-Ferreira, N.; Moreira, I.S. Network Biology and Artificial Intelligence Drive the Understanding of the Multidrug Resistance Phenotype in Cancer. Drug Resist. Updat. 2022, 60, 100811. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Palmeira, C.M.; Wallace, K.B. Doxorubicin-Induced Persistent Oxidative Stress to Cardiac Myocytes. Toxicol. Lett. 2001, 121, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Lauriola, A.; Davalli, P.; Marverti, G.; Santi, S.; Caporali, A.; D’Arca, D. Targeting the Interplay of Independent Cellular Pathways and Immunity: A Challenge in Cancer Immunotherapy. Cancers 2023, 15, 3009. [Google Scholar] [CrossRef] [PubMed]
- Orešić, T.; Bubanović, S.; Ramić, S.; Šarčević, B.; Gašparović, A.Č. Nuclear Localization of NRF2 in Stroma of HER2 Positive and Triple-Negative Breast Cancer. Pathol.-Res. Pract. 2023, 248, 154662. [Google Scholar] [CrossRef]
- Madeira, A.; Moura, T.F.; Soveral, G. Detecting Aquaporin Function and Regulation. Front. Chem. 2016, 4, 3. [Google Scholar] [CrossRef]
- Wagner, K.; Unger, L.; Salman, M.M.; Kitchen, P.; Bill, R.M.; Yool, A.J. Signaling Mechanisms and Pharmacological Modulators Governing Diverse Aquaporin Functions in Human Health and Disease. Int. J. Mol. Sci. 2022, 23, 1388. [Google Scholar] [CrossRef]
- Dai, C.; Charlestin, V.; Wang, M.; Walker, Z.T.; Miranda-Vergara, M.C.; Facchine, B.A.; Wu, J.; Kaliney, W.J.; Dovichi, N.J.; Li, J.; et al. Aquaporin-7 Regulates the Response to Cellular Stress in Breast Cancer. Cancer Res. 2020, 80, 4071–4086. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, J.; Li, Y.; Hu, H.; Ye, Q.; Yang, C.; Yang, L.; Zhang, B.; Ma, T. Aquaporin-7 Facilitates Proliferation and Adipogenic Differentiation of Mouse Bone Marrow Mesenchymal Stem Cells by Regulating Hydrogen Peroxide Transport. Stem Cell Rev. Rep. 2023, 19, 2378–2390. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen Peroxide—Production, Fate and Role in Redox Signaling of Tumor Cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]
- Yusupov, M.; Yan, D.; Cordeiro, R.M.; Bogaerts, A. Atomic Scale Simulation of H2O2 Permeation through Aquaporin: Toward the Understanding of Plasma Cancer Treatment. J. Phys. Appl. Phys. 2018, 51, 125401. [Google Scholar] [CrossRef]
- Wragg, D.; Leoni, S.; Casini, A. Aquaporin-Driven Hydrogen Peroxide Transport: A Case of Molecular Mimicry? RSC Chem. Biol. 2020, 1, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 Mediates Hydrogen Peroxide Uptake to Regulate Downstream Intracellular Signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Moniaga, C.S.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 Facilitates Membrane Transport of Hydrogen Peroxide in Mammalian Cells. Biochem. Biophys. Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; Pimpão, C.; Mósca, A.F.; Coxixo, A.S.; Lopes, D.; da Silva, I.V.; Pedersen, P.A.; Antunes, F.; Soveral, G. Human Aquaporin-5 Facilitates Hydrogen Peroxide Permeation Affecting Adaption to Oxidative Stress and Cancer Cell Migration. Cancers 2019, 11, 932. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; Mósca, A.; Martins, A.; Nobre, T.; Prista, C.; Antunes, F.; Gasparovic, A.C.; Soveral, G. Rat Aquaporin-5 Is pH-Gated Induced by Phosphorylation and Is Implicated in Oxidative Stress. Int. J. Mol. Sci. 2016, 17, 2090. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medraño-Fernandez, I. Human Aquaporin-11 Guarantees Efficient Transport of H2O2 across the Endoplasmic Reticulum Membrane. Redox Biol. 2019, 28, 101326. [Google Scholar] [CrossRef]
- Sorrentino, I.; Galli, M.; Medraño-Fernandez, I.; Sitia, R. Transfer of H2O2 from Mitochondria to the Endoplasmic Reticulum via Aquaporin-11. Redox Biol. 2022, 55, 102410. [Google Scholar] [CrossRef]
- Zhu, Z.; Jiao, L.; Li, T.; Wang, H.; Wei, W.; Qian, H. Expression of AQP3 and AQP5 as a Prognostic Marker in Triple-Negative Breast Cancer. Oncol. Lett. 2018, 16, 2661–2667. [Google Scholar] [CrossRef]
- Ahmad, A.E.; Khajah, M.A.; Khushaish, S.; Luqmani, Y.A. Aquaporin Expression in Breast Cancer and Their Involvement in Bleb Formation, Cell Motility and Invasion in Endocrine Resistant Variant Cells. Int. J. Oncol. 2020, 56, 1014–1024. [Google Scholar] [CrossRef]
- Edamana, S.; Pedersen, S.F.; Nejsum, L.N. Aquaporin Water Channels Affect the Response of Conventional Anticancer Therapies of 3D Grown Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2023, 639, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, P.K.; Cittelly, D.M.; Robin, T.P.; Carlson, J.A.; Stuhr, K.A.; Contreras-Zarate, M.J.; Lai, S.; Ormond, D.R.; Rusthoven, C.G.; Gaspar, L.E.; et al. Combination of Trastuzumab Emtansine and Stereotactic Radiosurgery Results in High Rates of Clinically Significant Radionecrosis and Dysregulation of Aquaporin-4. Clin. Cancer Res. 2019, 25, 3946–3953. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Zhang, H.; Shao, Y.; Liu, X.; Yang, L.; Huang, Y.; Fu, L.; Gu, F.; Ma, Y. Expression of Aquaporin1, a Water Channel Protein, in Cytoplasm Is Negatively Correlated with Prognosis of Breast Cancer Patients. Oncotarget 2016, 7, 8143–8154. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, H.; Liu, X.; Zhao, Y.; Chen, Y.; Jin, J.; Guo, C.; Zhang, M.; Gu, F.; Ma, Y. Water Channel Protein AQP1 in Cytoplasm Is a Critical Factor in Breast Cancer Local Invasion. J. Exp. Clin. Cancer Res. 2023, 42, 49. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Chen, W.; Yin, J.; Sun, J.; Xie, Q.; Wu, M.; Zeng, F.; Ren, H. RIPK1 Is a Negative Mediator in Aquaporin 1-Driven Triple-Negative Breast Carcinoma Progression and Metastasis. npj Breast Cancer 2021, 7, 53. [Google Scholar] [CrossRef]
- Ji, Y.; Liao, X.; Jiang, Y.; Wei, W.; Yang, H. Aquaporin 1 Knockdown Inhibits Triple-Negative Breast Cancer Cell Proliferation and Invasion in Vitro and in Vivo. Oncol. Lett. 2021, 21, 437. [Google Scholar] [CrossRef]
- Zou, L.-B.; Shi, S.; Zhang, R.-J.; Wang, T.-T.; Tan, Y.-J.; Zhang, D.; Fei, X.-Y.; Ding, G.-L.; Gao, Q.; Chen, C.; et al. Aquaporin-1 Plays a Crucial Role in Estrogen-Induced Tubulogenesis of Vascular Endothelial Cells. J. Clin. Endocrinol. Metab. 2013, 98, E672–E682. [Google Scholar] [CrossRef]
- Luo, L.; Yang, R.; Zhao, S.; Chen, Y.; Hong, S.; Wang, K.; Wang, T.; Cheng, J.; Zhang, T.; Chen, D. Decreased miR-320 Expression Is Associated with Breast Cancer Progression, Cell Migration, and Invasiveness via Targeting Aquaporin 1. Acta Biochim. Biophys. Sin. 2018, 50, 473–480. [Google Scholar] [CrossRef]
- Chong, W.; Zhang, H.; Guo, Z.; Yang, L.; Shao, Y.; Liu, X.; Zhao, Y.; Wang, Z.; Zhang, M.; Guo, C.; et al. Aquaporin 1 Promotes Sensitivity of Anthracycline Chemotherapy in Breast Cancer by Inhibiting β-Catenin Degradation to Enhance TopoIIα Activity. Cell Death Differ. 2021, 28, 382–400. [Google Scholar] [CrossRef]
- Oberska, P.; Jedrzejczak-Silicka, M.; Michałek, K.; Grabowska, M. Initial Assessment of Suitability of MCF-7 and HepG2 Cancer Cell Lines for AQP3 Research in Cancer Biology. Acta Histochem. 2021, 123, 151716. [Google Scholar] [CrossRef]
- Huang, Y.T.; Zhou, J.; Shi, S.; Xu, H.Y.; Qu, F.; Zhang, D.; Chen, Y.D.; Yang, J.; Huang, H.F.; Sheng, J.Z. Identification of Estrogen Response Element in Aquaporin-3 Gene That Mediates Estrogen-Induced Cell Migration and Invasion in Estrogen Receptor-Positive Breast Cancer. Sci. Rep. 2015, 5, srep12484. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-C.; Zhang, W.-R.; Cao, W.-F.; Liu, B.-W.; Zhang, F.; Zhao, H.-M.; Meng, R.; Zhang, L.; Niu, R.-F.; Hao, X.-S.; et al. Aquaporin3 Is Required for FGF-2-Induced Migration of Human Breast Cancers. PLoS ONE 2013, 8, e56735. [Google Scholar] [CrossRef] [PubMed]
- Mlinarić, M.; Lučić, I.; Milković, L.; da Silva, I.V.; Tartaro Bujak, I.; Musani, V.; Soveral, G.; Čipak Gašparović, A. AQP3-Dependent PI3K/Akt Modulation in Breast Cancer Cells. Int. J. Mol. Sci. 2023, 24, 8133. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Kitchen, P.; Conner, M.T.; Hill, E.J.; Nagel, D.; Bill, R.M.; Dunmore, S.J.; Armesilla, A.L.; Gross, S.; Carmichael, A.R.; et al. Downregulation of Aquaporin 3 Inhibits Cellular Proliferation, Migration and Invasion in the MDA-MB-231 Breast Cancer Cell Line. Oncol. Lett. 2018, 16, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Trigueros-Motos, L.; Pérez-Torras, S.; Casado, F.J.; Molina-Arcas, M.; Pastor-Anglada, M. Aquaporin 3 (AQP3) Participates in the Cytotoxic Response to Nucleoside-Derived Drugs. BMC Cancer 2012, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Yellapu, N.K.; Ly, T.; Sardiu, M.E.; Pei, D.; Welch, D.R.; Thompson, J.A.; Koestler, D.C. Synergistic Anti-Proliferative Activity of JQ1 and GSK2801 in Triple-Negative Breast Cancer. BMC Cancer 2022, 22, 627. [Google Scholar] [CrossRef]
- Satooka, H.; Hara-Chikuma, M. Aquaporin-3 Controls Breast Cancer Cell Migration by Regulating Hydrogen Peroxide Transport and Its Downstream Cell Signaling. Mol. Cell. Biol. 2016, 36, 1206–1218. [Google Scholar] [CrossRef]
- Zhong, L.; Xia, Y.; He, T.; Wenjie, S.; Jinxia, A.; Lijun, Y.; Hui, G. Polymeric Photothermal Nanoplatform with the Inhibition of Aquaporin 3 for Anti-Metastasis Therapy of Breast Cancer. Acta Biomater. 2022, 153, 505–517. [Google Scholar] [CrossRef]
- Dai, X.; Cai, D.; Wang, P.; Nan, N.; Yu, L.; Zhang, Z.; Zhou, R.; Hua, D.; Zhang, J.; Ostrikov, K.; et al. Cold Atmospheric Plasmas Target Breast Cancer Stemness via Modulating AQP3-19Y Mediated AQP3-5K and FOXO1 K48-Ubiquitination. Int. J. Biol. Sci. 2022, 18, 3544–3561. [Google Scholar] [CrossRef]
- Login, F.H.; Palmfeldt, J.; Cheah, J.S.; Yamada, S.; Nejsum, L.N. Aquaporin-5 Regulation of Cell-Cell Adhesion Proteins: An Elusive “Tail” Story. Am. J. Physiol. Cell Physiol. 2021, 320, C282–C292. [Google Scholar] [CrossRef]
- Edamana, S.; Login, F.H.; Riishede, A.; Dam, V.S.; Tramm, T.; Nejsum, L.N. The Cell Polarity Protein Scribble Is Downregulated by the Water Channel Aquaporin-5 in Breast Cancer Cells. Am. J. Physiol. Cell Physiol. 2023, 324, C307–C319. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of Scribble Promotes Mammary Tumorigenesis and Reveals a Role for Cell Polarity in Carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.J.; Moon, C. Aquaporin 5 (AQP5) Expression in Breast Cancer and Its Clinicopathological Characteristics. PLoS ONE 2023, 18, e0270752. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pei, B.; Wang, H.; Tang, C.; Zhu, W.; Jin, F. Effect of AQP-5 Silencing by siRNA Interference on Chemosensitivity of Breast Cancer Cells. OncoTargets Ther. 2018, 11, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Basal, O.A.; Zahran, R.F.; Saad, E.A. Rifampicin Efficacy against Doxorubicin-Induced Cardiotoxicity in Mice. Egypt. Heart J. 2023, 75, 73. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Jung, H.J.; Choi, H.J.; Jang, H.J.; Park, H.J.; Nejsum, L.N.; Kwon, T.H. Exosomes Co-Expressing AQP5-Targeting miRNAs and IL-4 Receptor-Binding Peptide Inhibit the Migration of Human Breast Cancer Cells. FASEB J. 2020, 34, 3379–3398. [Google Scholar] [CrossRef]
- Liao, S.; Chen, H.; Liu, M.; Gan, L.; Li, C.; Zhang, W.; Lv, L.; Mei, Z. Aquaporin 9 Inhibits Growth and Metastasis of Hepatocellular Carcinoma Cells via Wnt/β-Catenin Pathway. Aging 2020, 12, 1527–1544. [Google Scholar] [CrossRef]
- Yao, D.; Liu, S.; Lian, F.; Xu, X.; Yang, J.; Chen, R.; Cao, Y. AQP9 (Aquaporin 9) Determines Arsenic Uptake and Tolerance in Human Hepatocellular Carcinoma Cells In Vitro. Cureus 2022, 14, e26753. [Google Scholar] [CrossRef]
- Miao, Z.-F.; Chang, E.E.; Tsai, F.-Y.; Yeh, S.-C.; Wu, C.-F.; Wu, K.-Y.; Wang, C.-J.; Tsou, T.-C. Increased Aquaglyceroporin 9 Expression Disrupts Arsenic Resistance in Human Lung Cancer cells. Toxicol. In Vitr. 2009, 23, 209–216. [Google Scholar] [CrossRef]
- Leung, J.; Pang, A.; Yuen, W.-H.; Kwong, Y.-L.; Tse, E.W.C. Relationship of Expression of Aquaglyceroporin 9 with Arsenic Uptake and Sensitivity in Leukemia Cells. Blood 2007, 109, 740–746. [Google Scholar] [CrossRef]
- Iriyama, N.; Yuan, B.; Yoshino, Y.; Hatta, Y.; Horikoshi, A.; Aizawa, S.; Takeuchi, J.; Toyoda, H. Aquaporin 9, a Promising Predictor for the Cytocidal Effects of Arsenic Trioxide in Acute Promyelocytic Leukemia Cell Lines and Primary Blasts. Oncol. Rep. 2013, 29, 2362–2368. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Feng, X.; Liu, Y.; Deng, Y.; Chen, H.; Chen, D.; Fang, L.; Cai, Y.; Liu, H.; Wang, L.; et al. AQP9-Induced Cell Cycle Arrest Is Associated with RAS Activation and Improves Chemotherapy Treatment Efficacy in Colorectal Cancer. Cell Death Dis. 2017, 8, e2894. [Google Scholar] [CrossRef] [PubMed]
- Dou, R.; Deng, Y.; Huang, L.; Fu, S.; Tan, S.; Wang, L.; Lian, L.; Fang, L.; Fan, X.; Jin, G.; et al. Multi-Microarray Identifies Lower AQP9 Expression in Adjuvant Chemotherapy Nonresponders with Stage III colorectal Cancer. Cancer Lett. 2013, 336, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Kozono, S.; Lin, Y.-M.; Seo, H.-S.; Pinch, B.; Lian, X.; Qiu, C.; Herbert, M.K.; Chen, C.-H.; Tan, L.; Gao, Z.J.; et al. Arsenic Targets Pin1 and Cooperates with Retinoic Acid to Inhibit Cancer-Driving Pathways and Tumor-Initiating Cells. Nat. Commun. 2018, 9, 3069. [Google Scholar] [CrossRef] [PubMed]
- King, L.S.; Kozono, D.; Agre, P. From Structure to Disease: The Evolving Tale of Aquaporin Biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 687–698. [Google Scholar] [CrossRef]
- Rodrigues, C.; Milkovic, L.; Bujak, I.T.; Tomljanovic, M.; Soveral, G.; Gasparovic, A.C. Lipid Profile and Aquaporin Expression under Oxidative Stress in Breast Cancer Cells of Different Malignancies. Oxid. Med. Cell. Longev. 2019, 2019, 2061830. [Google Scholar] [CrossRef]
- Zhu, X.; Jin, X.; Li, Z.; Chen, X.; Zhao, J. miR-152-3p Facilitates Cell Adhesion and Hepatic Metastases in Colorectal Cancer via Targeting AQP11. Pathol. Res. Pract. 2023, 244, 154389. [Google Scholar] [CrossRef]
- Chetry, M.; Li, S.; Liu, H.; Hu, X.; Zhu, X. Prognostic Values of Aquaporins mRNA Expression in Human Ovarian Cancer. Biosci. Rep. 2018, 38, BSR20180108. [Google Scholar] [CrossRef]
- Medraño-Fernandez, I.; Bestetti, S.; Bertolotti, M.; Bienert, G.P.; Bottino, C.; Laforenza, U.; Rubartelli, A.; Sitia, R. Stress Regulates Aquaporin-8 Permeability to Impact Cell Growth and Survival. Antioxid. Redox Signal. 2016, 24, 1031–1044. [Google Scholar] [CrossRef]
- Huang, J.; Yue, S.; Ke, B.; Zhu, J.; Shen, X.; Zhai, Y.; Yamamoto, M.; Busuttil, R.W.; Kupiec-Weglinski, J.W. NRF2 regulates TLR4 innate responses in mouse liver ischemia/reperfusion injury via akt/foxo1 signaling network. Transplantation 2014, 98, 721–728. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-Dependent Proteasomal Degradation of Transcription Factor Nrf2 Contributes to the Negative Regulation of Antioxidant Response Element-Driven Gene Expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The Antioxidant Responsive Element. Activation by Oxidative Stress and Identification of the DNA Consensus Sequence Required for Functional Activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar Basic Leucine Zipper Transcription Factor Nrf2 (NF-E2 P45-Related Factor 2) Controls both Constitutive and Inducible Expression of Intestinal Detoxification and Glutathione Biosynthetic Enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-Regulated Genes Induced by the Chemopreventive Agent Sulforaphane by Oligonucleotide Microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer Chemoprevention Mechanisms Mediated Through the Keap1–Nrf2 Pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef]
- Cuadrado, A. Structural and Functional Characterization of Nrf2 Degradation by Glycogen Synthase Kinase 3/β-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 Gene in Human Lung Cancer Cell Lines and Lung Cancer Tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.-K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to Carcinogenesis Is Increased and Chemoprotective Efficacy of Enzyme Inducers Is Lost in Nrf2 Transcription Factor-Deficient Mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, G.; Sekhar, K.R.; Jowhar, D.; Samson, P.C.; Wikswo, J.P.; Beauchamp, R.D.; Datta, P.K.; Freeman, M.L. Increased Cell Migration and Plasticity in Nrf2-Deficient Cancer Cell Lines. Oncogene 2010, 29, 3703–3714. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-Deficiency Creates a Responsive Microenvironment for Metastasis to the Lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Takahashi, J.; Yamamoto, M. Molecular Basis of the KEAP1-NRF2 Signaling Pathway. Mol. Cells 2023, 46, 133–141. [Google Scholar] [CrossRef]
- Ooi, A.; Dykema, K.; Ansari, A.; Petillo, D.; Snider, J.; Kahnoski, R.; Anema, J.; Craig, D.; Carpten, J.; Teh, B.-T.; et al. CUL3 and NRF2 Mutations Confer an NRF2 Activation Phenotype in a Sporadic Form of Papillary Renal Cell Carcinoma. Cancer Res. 2013, 73, 2044–2051. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.-I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural Basis for Defects of Keap1 Activity Provoked by Its Point Mutations in Lung Cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 Interaction in Non-Small-Cell Lung Cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Bevinakoppamath, S.; Ramachandra, S.C.; Yadav, A.K.; Basavaraj, V.; Vishwanath, P.; Prashant, A. Understanding the Emerging Link Between Circadian Rhythm, Nrf2 Pathway, and Breast Cancer to Overcome Drug Resistance. Front. Pharmacol. 2022, 12, 719631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-Associated Protein 1 Function in Prostate Cancer Cells Causes Chemoresistance and Radioresistance and Promotes Tumor Growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Shim, G.; Manandhar, S.; Shin, D.; Kim, T.-H.; Kwak, M.-K. Acquisition of Doxorubicin Resistance in Ovarian Carcinoma Cells Accompanies Activation of the NRF2 Pathway. Free Radic. Biol. Med. 2009, 47, 1619–1631. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 Function Activates Nrf2 and Provides Advantages for Lung Cancer Cell Growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef]
- Singh, A.; Boldin-Adamsky, S.; Thimmulappa, R.K.; Rath, S.K.; Ashush, H.; Coulter, J.; Blackford, A.; Goodman, S.N.; Bunz, F.; Watson, W.H.; et al. RNAi-Mediated Silencing of Nuclear Factor Erythroid-2–Related Factor 2 Gene Expression in Non–Small Cell Lung Cancer Inhibits Tumor Growth and Increases Efficacy of Chemotherapy. Cancer Res. 2008, 68, 7975–7984. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic Alteration of Keap1 Confers Constitutive Nrf2 Activation and Resistance to Chemotherapy in Gallbladder Cancer. Gastroenterology 2008, 135, 1358–1368. [Google Scholar] [CrossRef]
- Homma, S.; Ishii, Y.; Morishima, Y.; Yamadori, T.; Matsuno, Y.; Haraguchi, N.; Kikuchi, N.; Satoh, H.; Sakamoto, T.; Hizawa, N.; et al. Nrf2 Enhances Cell Proliferation and Resistance to Anticancer Drugs in Human Lung Cancer. Clin. Cancer Res. 2009, 15, 3423–3432. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 Enhances Resistance of Cancer Cells to Chemotherapeutic Drugs, the Dark Side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Hu, L.; Miao, W.; Loignon, M.; Kandouz, M.; Batist, G. Putative Chemopreventive Molecules Can Increase Nrf2-Regulated Cell Defense in Some Human Cancer Cell Lines, Resulting in Resistance to Common Cytotoxic Therapies. Cancer Chemother. Pharmacol. 2009, 66, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.-L. Role of the Keap1–Nrf2 Pathway in Cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2014; pp. 281–320. [Google Scholar] [CrossRef]
- Guo, Y.; Shen, L. Overexpression of NRF2 Is Correlated with Prognoses of Patients with Malignancies: A Meta-Analysis. Thorac. Cancer 2017, 8, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Soares, M.; Ramalhinho, A.C.; Moutinho, J.F.; Breitenfeld, L.; Pereira, L. The Prognostic Value of NRF2 in Breast Cancer Patients: A Systematic REVIEW with meta-Analysis. Breast Cancer Res. Treat. 2019, 179, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Loignon, M.; Miao, W.; Hu, L.; Bier, A.; Bismar, T.A.; Scrivens, P.J.; Mann, K.; Basik, M.; Bouchard, A.; Fiset, P.O.; et al. Cul3 Overexpression Depletes Nrf2 in Breast Cancer and Is Associated with Sensitivity to Carcinogens, to OXIDATIVE stress, and to Chemotherapy. Mol. Cancer Ther. 2009, 8, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; De Blasio, A.; Drago-Ferrante, R.; Di Fiore, R.; Buttitta, G.; Morreale, M.; Scerri, C.; Vento, R.; Tesoriere, G. Parthenolide Prevents Resistance of MDA-MB231 Cells to Doxorubicin and Mitoxantrone: The Role of Nrf2. Cell Death Discov. 2017, 3, 17078. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, D.; Jin, X.; Gao, P.; Wang, Q.; Zhang, J.; Zhang, N. PA-MSHA Inhibits the Growth of Doxorubicin-Resistant MCF-7/ADR Human Breast Cancer Cells by Downregulating Nrf2/p62. Cancer Med. 2016, 5, 3520–3531. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, F.; Sun, Z.; Zhou, W.; Li, Z.; You, Q.; Guo, Q.; Hu, R. Drug Resistance Associates with Activation of Nrf2 in MCF-7/ DOX Cells, and Wogonin Reverses it by Down-Regulating Nrf2-Mediated Cellular Defense Response. Mol. Carcinog. 2012, 52, 824–834. [Google Scholar] [CrossRef]
- Wu, T.; Harder, B.G.; Wong, P.K.; Lang, J.E.; Zhang, D.D. Oxidative Stress, Mammospheres and Nrf2-New Implication for Breast Cancer Therapy? Mol. Carcinog. 2014, 54, 1494–1502. [Google Scholar] [CrossRef]
- Syu, J.-P.; Chi, J.-T.; Kung, H.-N. Nrf2 Is the Key to Chemotherapy Resistance in MCF7 Breast Cancer Cells under Hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef]
- Kim, S.K.; Yang, J.W.; Kim, M.R.; Roh, S.H.; Kim, H.G.; Lee, K.Y.; Jeong, H.G.; Kang, K.W. Increased Expression of Nrf2/ARE-Dependent Anti-Oxidant Proteins in Tamoxifen-Resistant Breast Cancer Cells. Free Radic. Biol. Med. 2008, 45, 537–546. [Google Scholar] [CrossRef]
- Bekele, R.T.; Venkatraman, G.; Liu, R.-Z.; Tang, X.; Mi, S.; Benesch, M.G.K.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.W.; et al. Oxidative Stress Contributes to the Tamoxifen-Induced Killing of Breast Cancer Cells: Implications for Tamoxifen Therapy and Resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.A. Combination of siRNA-Directed Gene Silencing with Epigallocatechin-3-Gallate (EGCG) Reverses Drug Resistance in Human Breast Cancer Cells. J. Chem. Biol. 2015, 9, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tian, Z.; Guo, R.; Ren, F. Nrf2 Inhibitor, Brusatol in Combination with Trastuzumab Exerts Synergistic Antitumor Activity in HER2-Positive Cancers by Inhibiting Nrf2/HO-1 and HER2-AKT/ERK1/2 Pathways. Oxid. Med. Cell. Longev. 2020, 2020, 9867595. [Google Scholar] [CrossRef]
- Kamble, D.; Mahajan, M.; Dhat, R.; Sitasawad, S. Keap1-Nrf2 Pathway Regulates ALDH and Contributes to Radioresistance in Breast Cancer Stem Cells. Cells 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. Pharmacological Applications of Nrf2 Inhibitors as Potential Antineoplastic Drugs. Int. J. Mol. Sci. 2019, 20, 2025. [Google Scholar] [CrossRef] [PubMed]
- Link, W.; Fernandez-Marcos, P.J. FOXO Transcription Factors at the Interface of Metabolism and Cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO Code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef]
- Obsil, T.; Obsilova, V. Structure/Function Relationships Underlying Regulation of FOXO Transcription Factors. Oncogene 2008, 27, 2263–2275. [Google Scholar] [CrossRef]
- Spreitzer, E.; Alderson, T.R.; Bourgeois, B.; Eggenreich, L.; Habacher, H.; Bramerdorfer, G.; Pritišanac, I.; Sánchez-Murcia, P.A.; Madl, T. FOXO Transcription Factors Differ in Their Dynamics and Intra/Intermolecular Interactions. Curr. Res. Struct. Biol. 2022, 4, 118–133. [Google Scholar] [CrossRef]
- Brown, A.K.; Webb, A.E. Chapter Seven—Regulation of FOXO Factors in Mammalian Cells. In Current Topics in Developmental Biology; Forkhead FOXO Transcription Factors in Development and Disease; Ghaffari, S., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 127, pp. 165–192. [Google Scholar]
- Kim, J.; Ahn, D.; Park, C.-J. FOXO4 Transactivation Domain Interaction with Forkhead DNA Binding Domain and Effect on Selective DNA Recognition for Transcription Initiation. J. Mol. Biol. 2021, 433, 166808. [Google Scholar] [CrossRef]
- Bourgeois, B.; Gui, T.; Hoogeboom, D.; Hocking, H.G.; Richter, G.; Spreitzer, E.; Viertler, M.; Richter, K.; Madl, T.; Burgering, B.M.T. Multiple Regulatory Intrinsically Disordered Motifs Control FOXO4 Transcription Factor Binding and Function. Cell Rep. 2021, 36, 109446. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox Regulation of FoxO Transcription Factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.-K.; McGuire, V.A.; Koo, C.-Y.; Muir, K.W.; de Olano, N.; Maifoshie, E.; Kelly, D.J.; McGovern, U.B.; Monteiro, L.J.; Gomes, A.R.; et al. Phosphorylation of FOXO3a on Ser-7 by P38 Promotes Its Nuclear Localization in Response to Doxorubicin. J. Biol. Chem. 2012, 287, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Dengler, H.S.; Baracho, G.V.; Omori, S.A.; Bruckner, S.; Arden, K.; Castrillon, D.H.; DePinho, R.A.; Rickert, R.C. Distinct Roles for Foxo1 at Multiple Stages of B Cell Differentiation. Nat. Immunol. 2008, 9, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cheng, X.; Tian, Y.; Yuan, Z.; Fan, X.; Yang, D.; Yang, M. Nutritional Programming of the Lifespan of Male Drosophila by Activating FOXO on Larval Low-Nutrient Diet. Nutrients 2023, 15, 1840. [Google Scholar] [CrossRef]
- Li, Z.; Song, J.; Jiang, G.; Shang, Y.; Jiang, Y.; Zhang, J.; Xiao, L.; Chen, M.; Tang, D.; Tong, X.; et al. Juvenile Hormone Suppresses the FoxO-Takeout Axis to Shorten Longevity in Male Silkworm. Pestic. Biochem. Physiol. 2023, 192, 105388. [Google Scholar] [CrossRef]
- Morris, B.J.; Willcox, D.C.; Donlon, T.A.; Willcox, B.J. FOXO3: A Major Gene for Human Longevity—A Mini-Review. Gerontology 2015, 61, 515–525. [Google Scholar] [CrossRef]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1α-O-GlcNAc Transferase Complex Regulates FoxO Transcription Factor Activity in Response to Glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef]
- Nakae, J.; Biggs, W.H.; Kitamura, T.; Cavenee, W.K.; Wright, C.V.E.; Arden, K.C.; Accili, D. Regulation of Insulin Action and Pancreatic Beta-Cell Function by Mutated Alleles of the Gene Encoding Forkhead Transcription Factor Foxo1. Nat. Genet. 2002, 32, 245–253. [Google Scholar] [CrossRef]
- Dansen, T.B.; Burgering, B.M.T. Unravelling the Tumor-Suppressive Functions of FOXO Proteins. Trends Cell Biol. 2008, 18, 421–429. [Google Scholar] [CrossRef]
- Dong, T.; Zhang, Y.; Chen, Y.; Liu, P.; An, T.; Zhang, J.; Yang, H.; Zhu, W.; Yang, X. FOXO1 Inhibits the Invasion and Metastasis of Hepatocellular Carcinoma by Reversing ZEB2-Induced Epithelial-Mesenchymal Transition. Oncotarget 2016, 8, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhao, H.; Wei, S.; Du, Q.; Dong, K.; Yan, Y.; Geller, D.A. Hepatocellular Carcinoma-Derived FOXO1 Inhibits Tumor Progression by Suppressing IL-6 Secretion from Macrophages. Neoplasia 2023, 40, 100900. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.Y.; Jeong, S.Y.; Lee, J.W.; Kim, J.; Kim, J.H.; Chu, H.S.; Jeong, W.J.; Lee, B.J.; Ahn, B.; Kim, J.; et al. FOXO1 Is a Key Mediator of Glucocorticoid-Induced Expression of Tristetraprolin in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 13673. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Rodríguez, J.A.; Puente-Rivera, J.; Córdova-Esparza, D.M.; Nuñez-Olvera, S.I.; Silva-Cázares, M.B. Bioinformatic miRNA-mRNAs Analysis Revels to miR-934 as a Potential Regulator of the Epithelial–Mesenchymal Transition in Triple-Negative Breast Cancer. Cells 2023, 12, 834. [Google Scholar] [CrossRef]
- Rani, M.; Kumari, R.; Singh, S.P.; Devi, A.; Bansal, P.; Siddiqi, A.; Alsahli, M.A.; Almatroodi, S.A.; Rahmani, A.H.; Rizvi, M.M.A. MicroRNAs as Master Regulators of FOXO Transcription Factors in Cancer Management. Life Sci. 2023, 321, 121535. [Google Scholar] [CrossRef]
- Santos, J.C.; Lima, N.d.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-Mediated Breast Cancer Chemoresistance via miR-155 Transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, Z.; Qiu, N.; Ling, L.; Jia, X.; Song, Y.; Li, H.; Li, J.; Lyu, H.; Liu, H.; et al. Disruption of FOXO3a-miRNA Feedback Inhibition of IGF2/IGF-1R/IRS1 Signaling Confers Herceptin Resistance in HER2-Positive Breast Cancer. Nat. Commun. 2021, 12, 2699. [Google Scholar] [CrossRef]
- Li, J.; Peng, S.; Zou, X.; Geng, X.; Wang, T.; Zhu, W.; Xia, T. Value of Negatively Correlated miR-205-5p/HMGB3 and miR-96-5p/FOXO1 on the Diagnosis of Breast Cancer and Benign Breast Diseases. Cancer Pathog. Ther. 2023, 1, 159–167. [Google Scholar] [CrossRef]
- Yin, Z.; Wang, W.; Qu, G.; Wang, L.; Wang, X.; Pan, Q. MiRNA-96-5p Impacts the Progression of Breast Cancer through Targeting FOXO3. Thorac. Cancer 2020, 11, 956–963. [Google Scholar] [CrossRef]
- Matkar, S.; Sharma, P.; Gao, S.; Gurung, B.; Katona, B.W.; Liao, J.; Muhammad, A.B.; Kong, X.-C.; Wang, L.; Jin, G.; et al. An Epigenetic Pathway Regulates Sensitivity of Breast Cancer Cells to HER2 Inhibition via FOXO/c-Myc Axis. Cancer Cell 2015, 28, 472–485. [Google Scholar] [CrossRef]
- Storz, P.; Döppler, H.; Copland, J.A.; Simpson, K.J.; Toker, A. FOXO3a Promotes Tumor Cell Invasion through the Induction of Matrix Metalloproteinases. Mol. Cell. Biol. 2009, 29, 4906–4917. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wu, Z.; Wu, Y.; Hankey, W.; Prior, T.W.; Li, L.; Ganju, R.K.; Shen, R.; Zou, X. Cdc25A Regulates Matrix Metalloprotease 1 through Foxo1 and Mediates Metastasis of Breast Cancer Cells. Mol. Cell. Biol. 2011, 31, 3457–3471. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Duan, M. Downregulation of FOXO6 in Breast Cancer Promotes Epithelial–Mesenchymal Transition and Facilitates Migration and Proliferation of Cancer Cells. Cancer Manag. Res. 2018, 10, 5145–5156. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Lee, S.W.; Kang, M.; Choi, J.K.; Park, K.; Byun, J.S.; Kim, D.Y. FoxO1 as a Regulator of Aquaporin 5 Expression in the Salivary Gland. J. Dent. Res. 2021, 100, 002203452110034. [Google Scholar] [CrossRef]
- Lee, S.J.; Chae, Y.S.; Kim, J.G.; Kim, W.W.; Jung, J.H.; Park, H.Y.; Jeong, J.Y.; Park, J.Y.; Jung, H.J.; Kwon, T.H. AQP5 Expression Predicts Survival in Patients with Early Breast Cancer. Ann. Surg. Oncol. 2014, 21, 375–383. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Studies | Peroxiporins | Models | Findings |
|---|---|---|---|
| [18] | AQP7 | Mouse models Cell lines (4T1, EpH4, NMuMG) Samples of breast cancer patients |
|
| [30] | AQP3 and AQP5 | TNBC patients |
|
| [31] | AQP1, AQP3, and AQP5 | Cell lines: endocrine-sensitive (YS1.2) endocrine-resistant (pII and MDA-MB-231) normal breast epithelial cells (MCF10A) |
|
| [32] | AQP1,AQP3, and AQP5 | Cell lines (MCF7 and MDA-MB-231) as sferoids | The effect of AQP1, AQP3, and AQP5 on conventional anticancer chemotherapies (cisplatin, 5-fluorouracil (5-FU), doxorubicin, and their combination alone or with the Ras inhibitor salirasib):
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milković, L.; Mlinarić, M.; Lučić, I.; Čipak Gašparović, A. The Involvement of Peroxiporins and Antioxidant Transcription Factors in Breast Cancer Therapy Resistance. Cancers 2023, 15, 5747. https://doi.org/10.3390/cancers15245747
Milković L, Mlinarić M, Lučić I, Čipak Gašparović A. The Involvement of Peroxiporins and Antioxidant Transcription Factors in Breast Cancer Therapy Resistance. Cancers. 2023; 15(24):5747. https://doi.org/10.3390/cancers15245747
Chicago/Turabian StyleMilković, Lidija, Monika Mlinarić, Ivan Lučić, and Ana Čipak Gašparović. 2023. "The Involvement of Peroxiporins and Antioxidant Transcription Factors in Breast Cancer Therapy Resistance" Cancers 15, no. 24: 5747. https://doi.org/10.3390/cancers15245747
APA StyleMilković, L., Mlinarić, M., Lučić, I., & Čipak Gašparović, A. (2023). The Involvement of Peroxiporins and Antioxidant Transcription Factors in Breast Cancer Therapy Resistance. Cancers, 15(24), 5747. https://doi.org/10.3390/cancers15245747