

Establishment and Thorough Characterization of Xenograft (PDX) Models Derived from Patients with Pancreatic Cancer for Molecular Analyses and Chemosensitivity Testing

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Pancreas Carcinoma Tissues
2.2. Establishment of Human Pancreas Carcinoma Patient-Derived Xenograft (PDX) Models
2.3. Tumor Histology and Immunohistochemistry
2.4. In Vivo Chemosensitivity Testing of Pancreas Carcinoma PDX Models
2.5. In Vivo Ultrasound-Based Tumor Measurement of Orthotopic PDAC
2.6. Total RNA Isolation and cDNA Synthesis
2.7. Quantitative Real-Time PCR (qPCR) for Gene Expression Analysis
2.8. Molecular Characterization of PDX Models by RNA Sequencing
2.8.1. Total RNA Isolation and Sequencing
2.8.2. Data Processing and Mutational Analysis
2.8.3. Data Processing and Gene Expression Analysis
2.8.4. Human Leukocyte Antigen (HLA) Typing
2.9. Statistical Analyses
3. Results
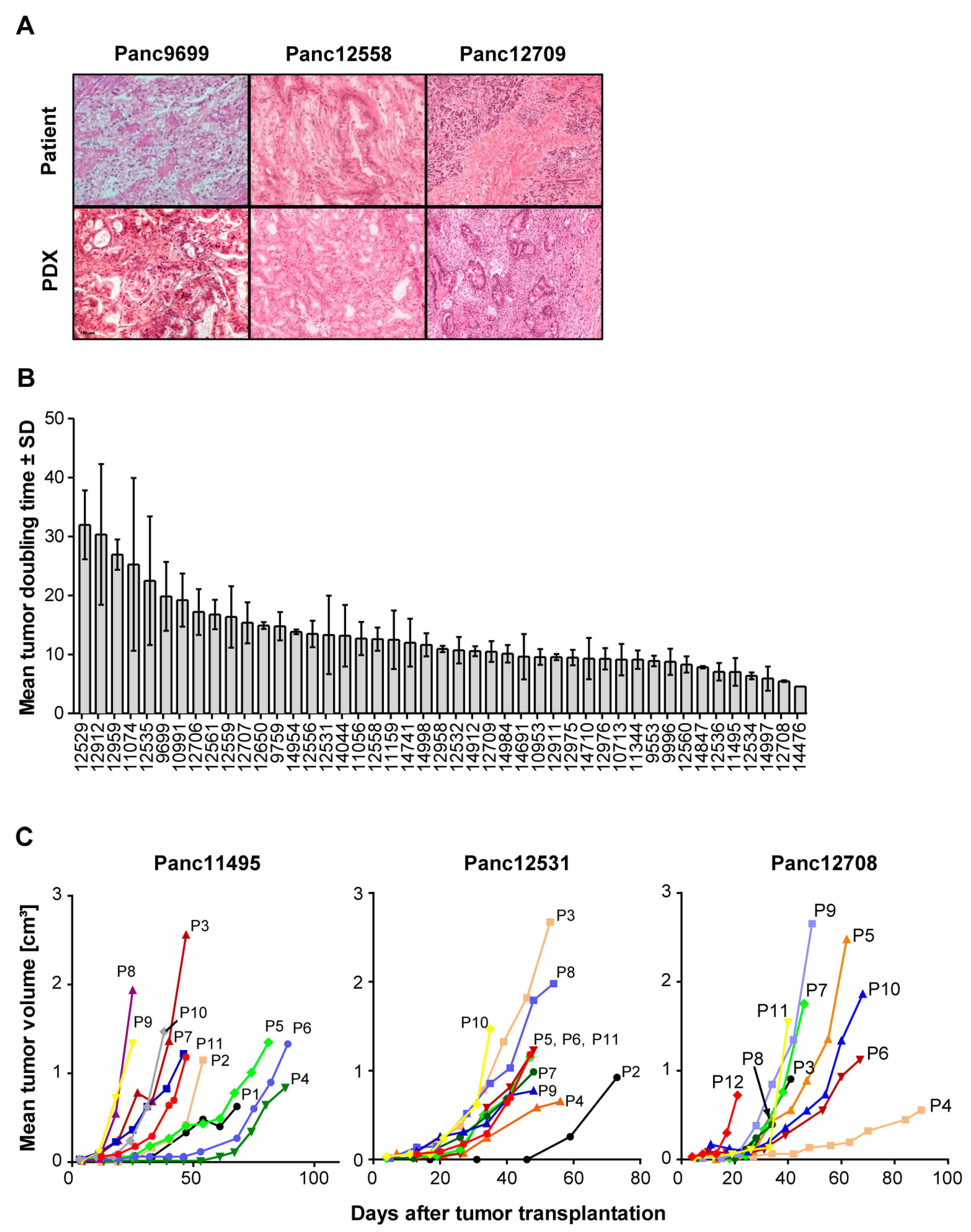
3.1. Patient Tumor Characteristics and Histology of Pancreas Carcinoma PDX Models
3.2. Growth Characteristics of PDX Models
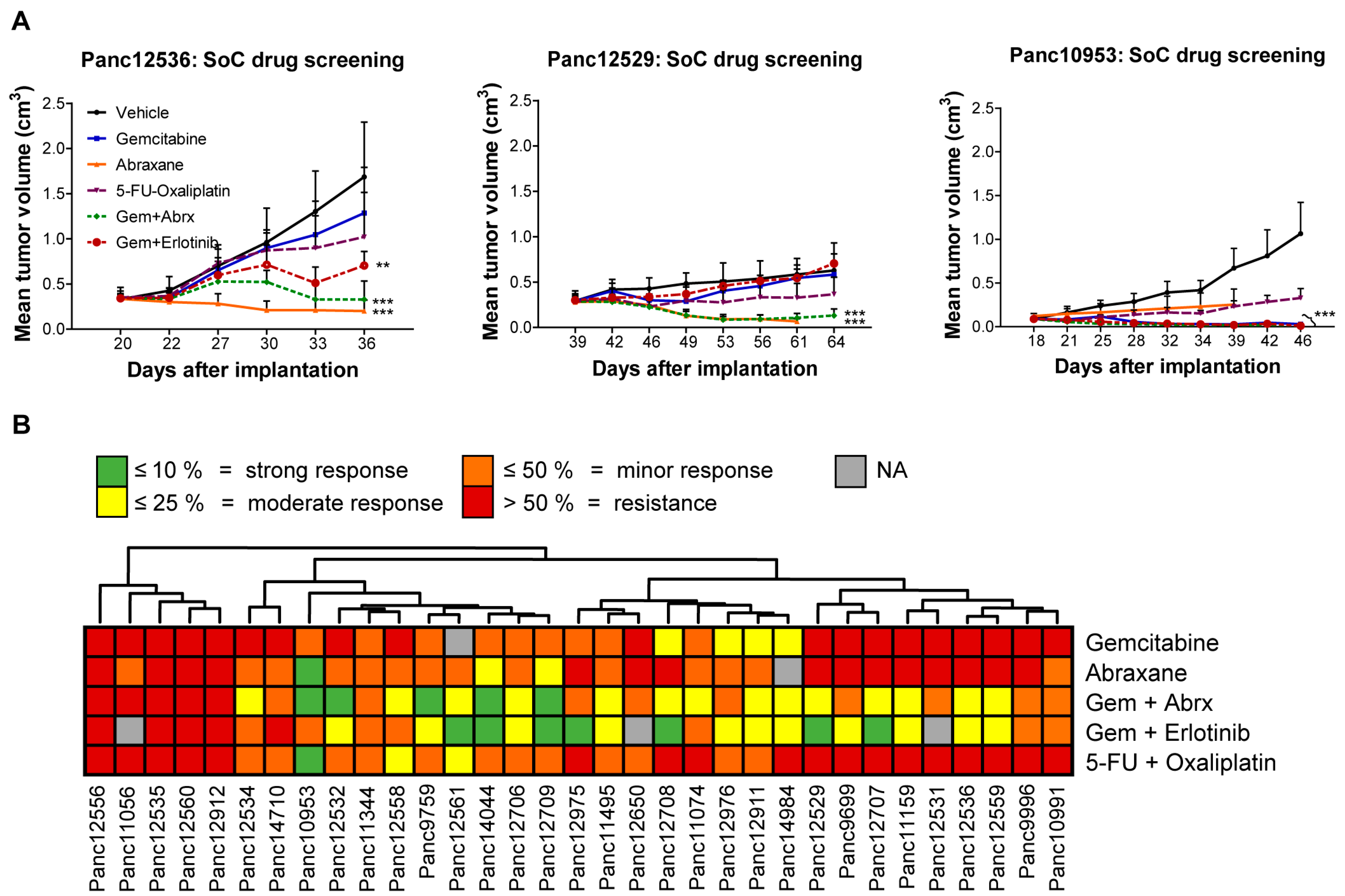
3.3. Chemosensitivity of PDX Models
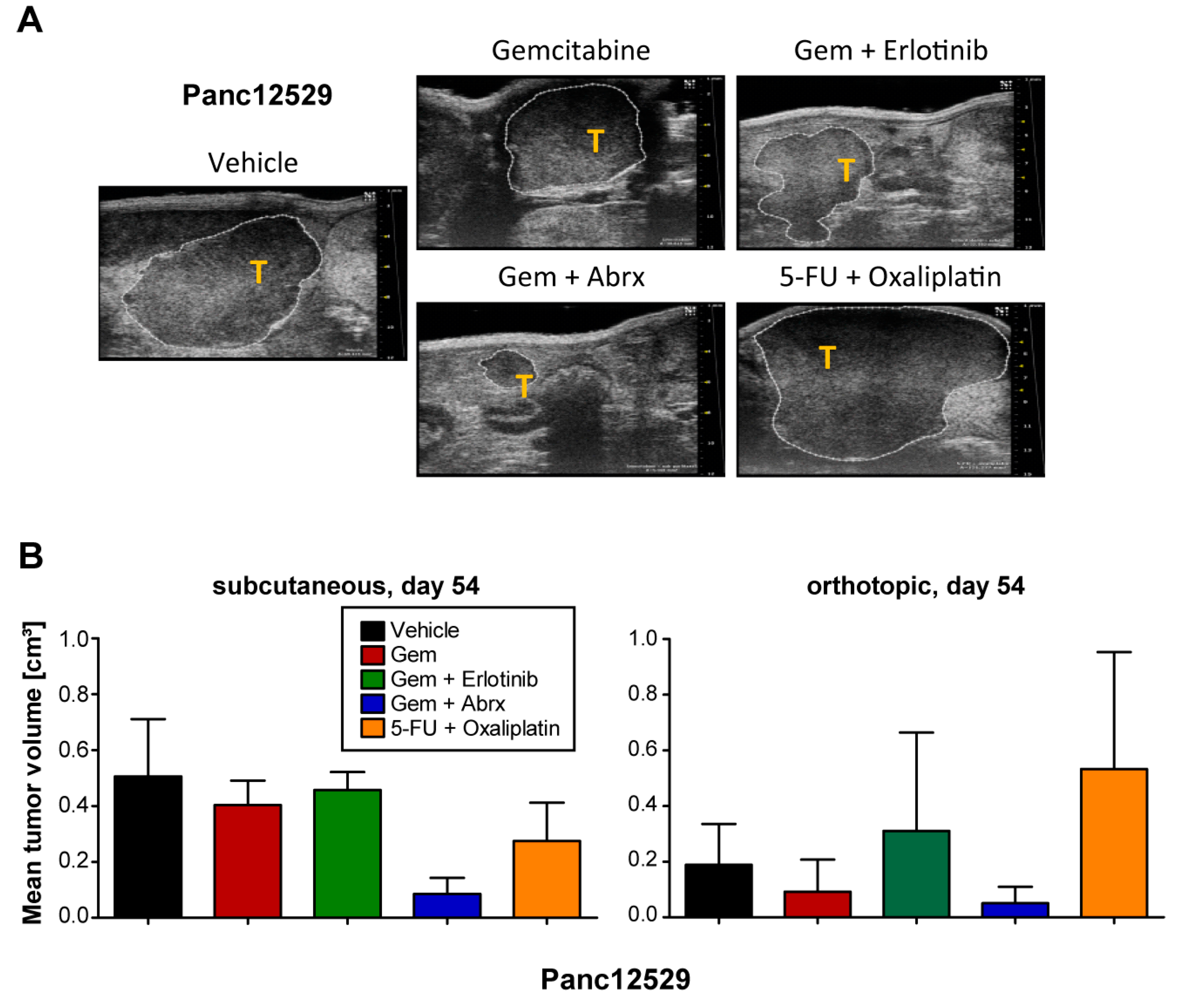
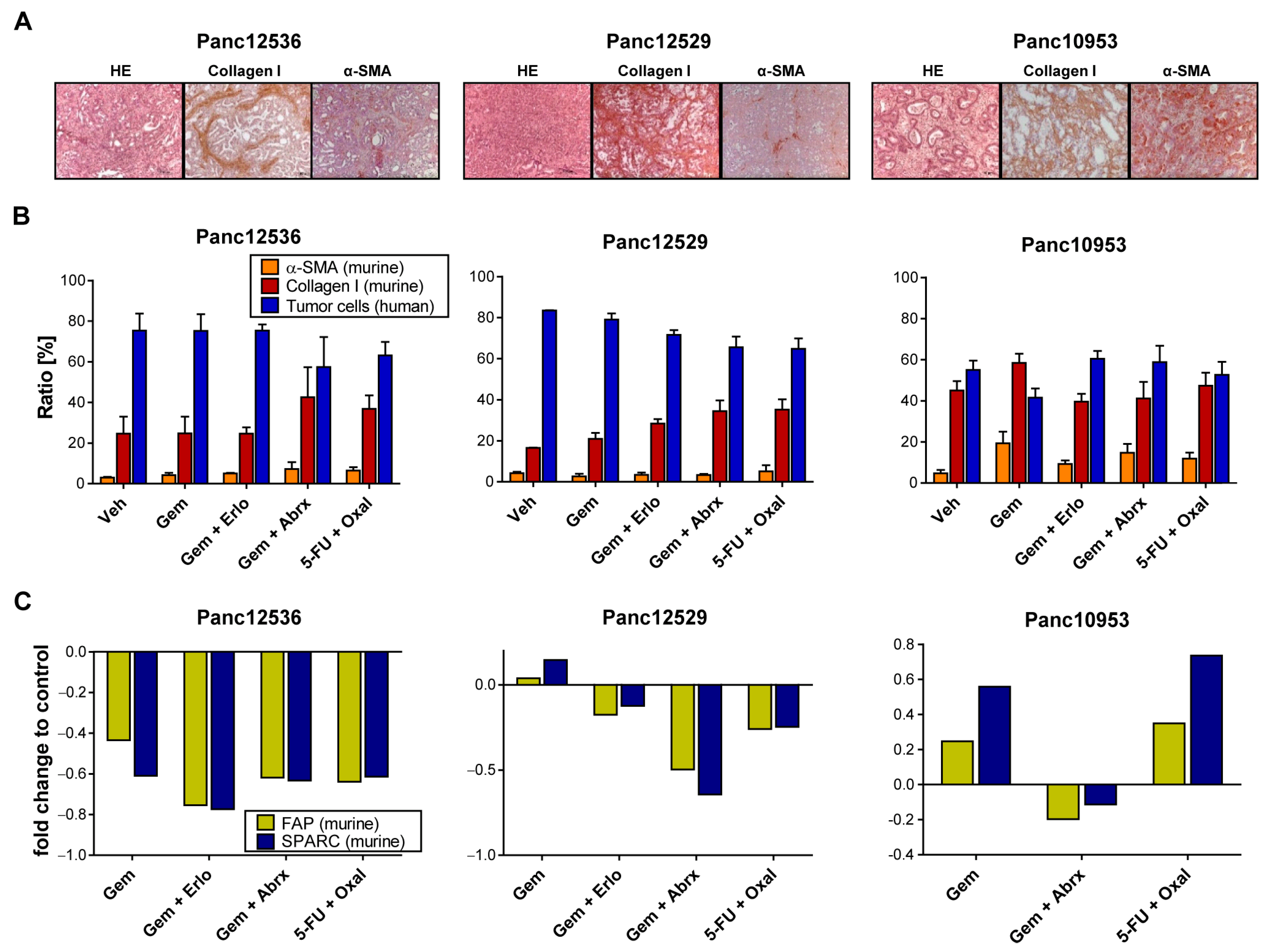
3.4. Impact of Transplantation Route and Stroma on Chemosensitivity
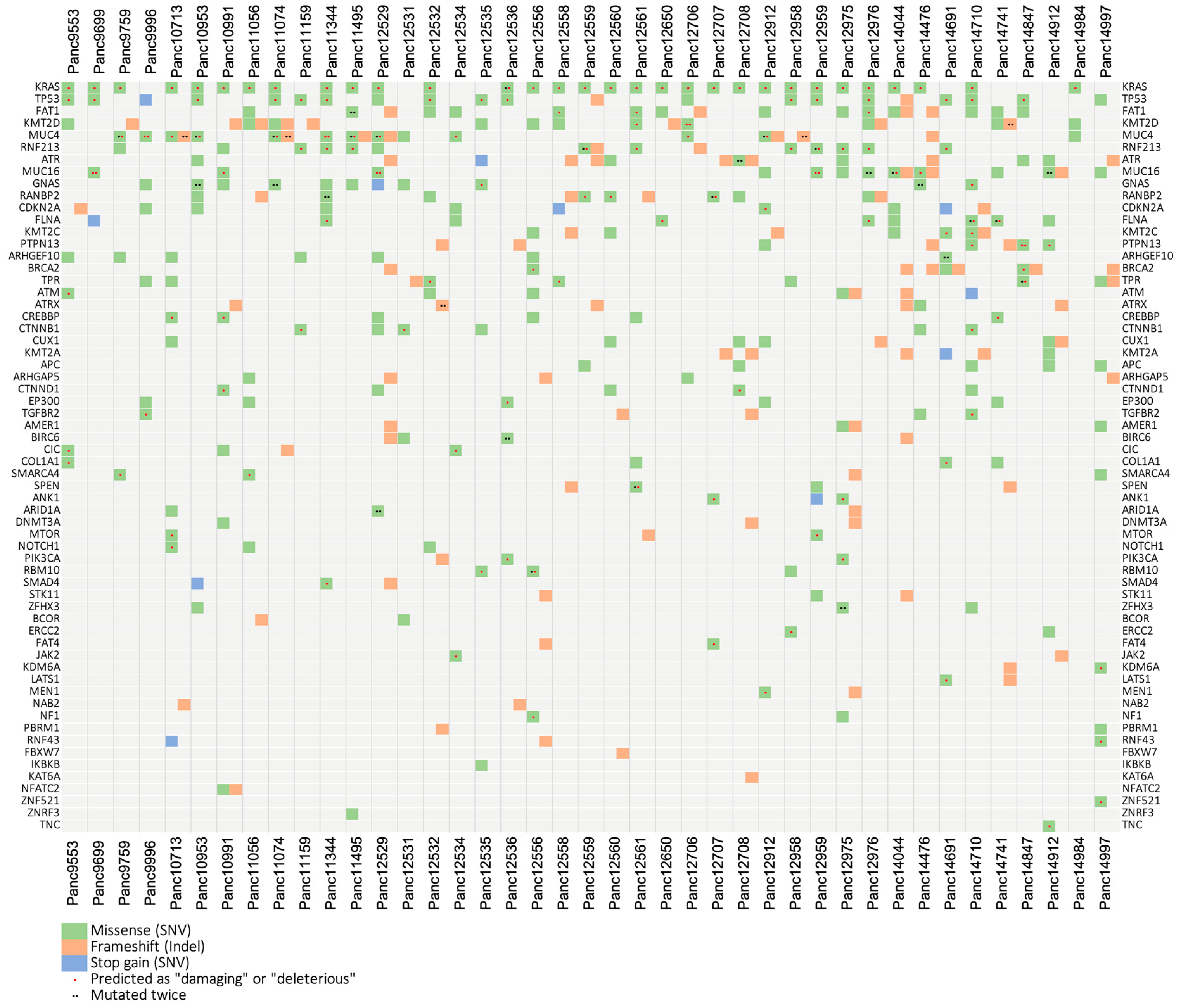
3.5. Mutational Analysis
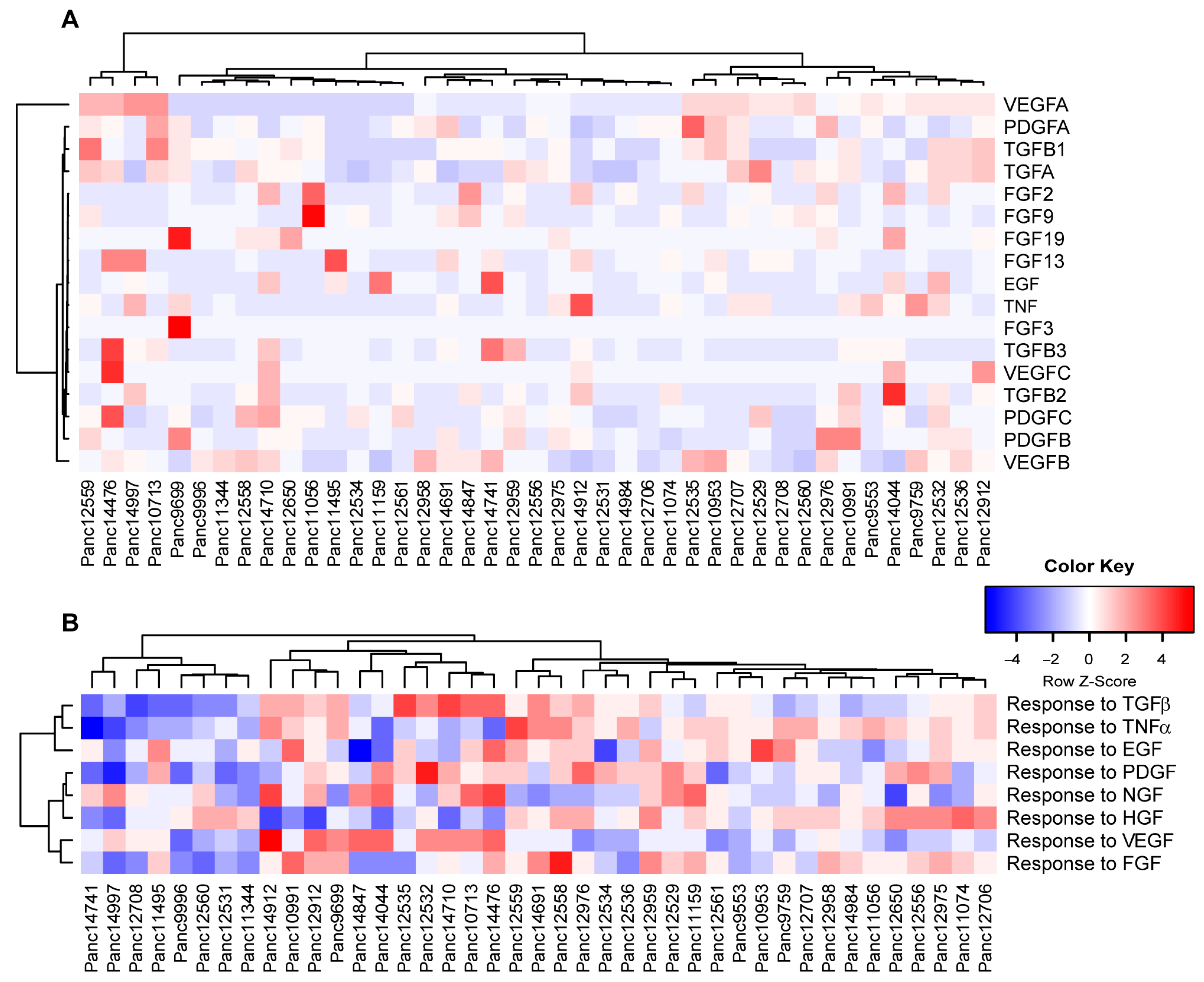
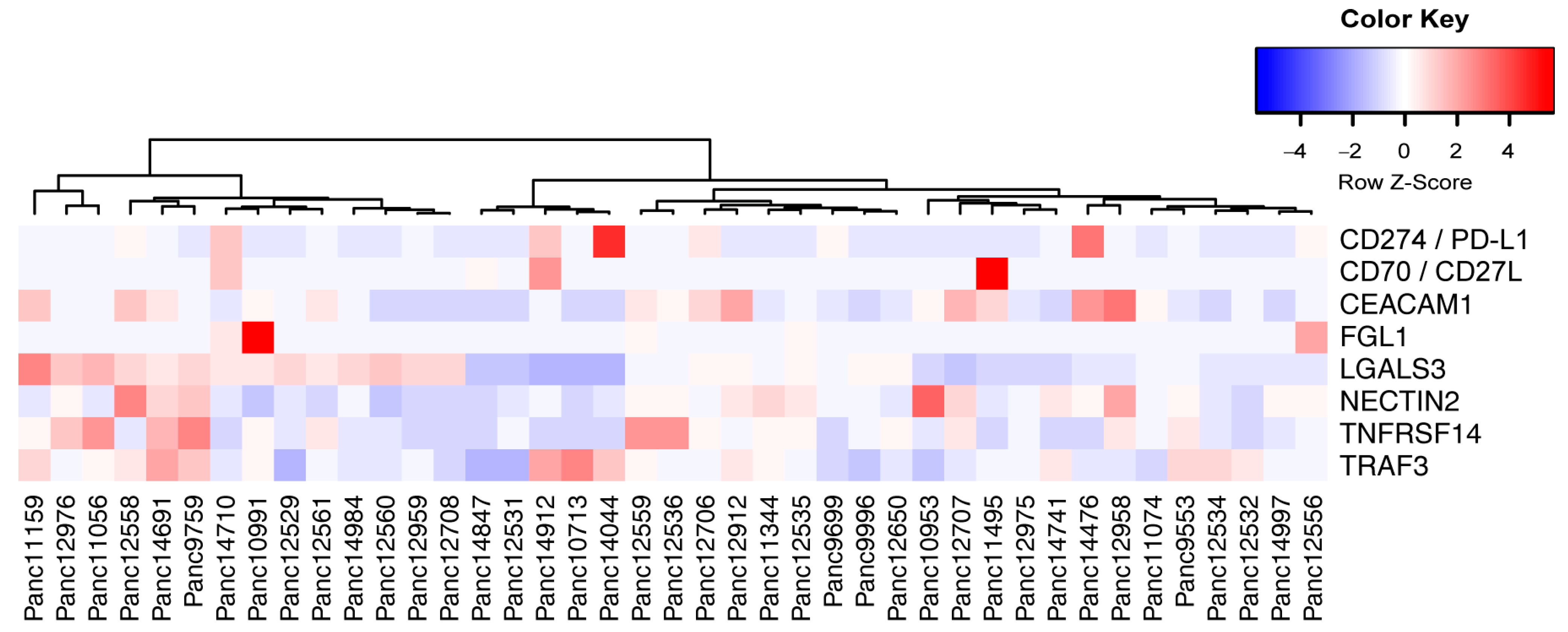
3.6. Expression Analysis and Pathway Activities
3.7. Human Leukocyte Antigen (HLA) Typing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.-M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the Challenges of Pancreatic Cancer: Future Directions for Improving Outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic Cancer in Chronic Pancreatitis; Aetiology, Incidence, and Early Detection. Best. Pract. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Porta, M.; Fabregat, X.; Malats, N.; Guarner, L.; Carrato, A.; de Miguel, A.; Ruiz, L.; Jariod, M.; Costafreda, S.; Coll, S.; et al. Exocrine Pancreatic Cancer: Symptoms at Presentation and Their Relation to Tumour Site and Stage. Clin. Transl. Oncol. 2005, 7, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.B.; Folloder, J. Clinical Management of Pancreatic Cancer. J. Adv. Pract. Oncol. 2014, 5, 356–364. [Google Scholar] [PubMed]
- Rishi, A.; Goggins, M.; Wood, L.D.; Hruban, R.H. Pathological and Molecular Evaluation of Pancreatic Neoplasms. Semin. Oncol. 2015, 42, 28–39. [Google Scholar] [CrossRef]
- Hruban, R.H.; Klimstra, D.S. Adenocarcinoma of the Pancreas. Semin. Diagn. Pathol. 2014, 31, 443–451. [Google Scholar] [CrossRef]
- Hruban, R.H.; Canto, M.I.; Goggins, M.; Schulick, R.; Klein, A.P. Update on Familial Pancreatic Cancer. Adv. Surg. 2010, 44, 293–311. [Google Scholar] [CrossRef]
- Rustgi, A.K. Familial Pancreatic Cancer: Genetic Advances. Genes Dev. 2014, 28, 1–7. [Google Scholar] [CrossRef]
- Ghiorzo, P. Genetic Predisposition to Pancreatic Cancer. World J. Gastroenterol. 2014, 20, 10778–10789. [Google Scholar] [CrossRef] [PubMed]
- Yeo, T.P. Demographics, Epidemiology, and Inheritance of Pancreatic Ductal Adenocarcinoma. Semin. Oncol. 2015, 42, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Das, S.; Brand, R.; Whitcomb, D.C. Inherited Pancreatic Cancer Syndromes. Cancer J. 2012, 18, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Williamson, L.M.; Topham, J.T.; Lee, M.K.C.; Goytain, A.; Ho, J.; Denroche, R.E.; Jang, G.; Pleasance, E.; Shen, Y.; et al. NRG1 Gene Fusions Are Recurrent, Clinically Actionable Gene Rearrangements in KRAS Wild-Type Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 4674–4681. [Google Scholar] [CrossRef] [PubMed]
- Heining, C.; Horak, P.; Uhrig, S.; Codo, P.L.; Klink, B.; Hutter, B.; Fröhlich, M.; Bonekamp, D.; Richter, D.; Steiger, K.; et al. NRG1 Fusions in KRAS Wild-Type Pancreatic Cancer. Cancer Discov. 2018, 8, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, H.; Gu, J.; Zheng, L. Prognostic Role of HER2 Amplification Based on Fluorescence in Situ Hybridization (FISH) in Pancreatic Ductal Adenocarcinoma (PDAC): A Meta-Analysis. World J. Surg. Oncol. 2016, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From State-of-the-Art Treatments to Novel Therapies for Advanced-Stage Pancreatic Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular Pharmacology of Gemcitabine. Ann. Oncol. 2006, 17 (Suppl. S5), v7–v12. [Google Scholar] [CrossRef]
- Renouf, D.; Moore, M. Evolution of Systemic Therapy for Advanced Pancreatic Cancer. Expert. Rev. Anticancer. Ther. 2010, 10, 529–540. [Google Scholar] [CrossRef]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III Randomized Comparison of Gemcitabine versus Gemcitabine plus Capecitabine in Patients with Advanced Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Louvet, C.; Labianca, R.; Hammel, P.; Lledo, G.; Zampino, M.G.; André, T.; Zaniboni, A.; Ducreux, M.; Aitini, E.; Taïeb, J.; et al. Gemcitabine in Combination with Oxaliplatin Compared with Gemcitabine Alone in Locally Advanced or Metastatic Pancreatic Cancer: Results of a GERCOR and GISCAD Phase III Trial. J. Clin. Oncol. 2005, 23, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schönekäs, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized Phase III Trial of Gemcitabine plus Cisplatin Compared with Gemcitabine Alone in Advanced Pancreatic Cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- De Vita, F.; Ventriglia, J.; Febbraro, A.; Laterza, M.M.; Fabozzi, A.; Savastano, B.; Petrillo, A.; Diana, A.; Giordano, G.; Troiani, T.; et al. NAB-Paclitaxel and Gemcitabine in Metastatic Pancreatic Ductal Adenocarcinoma (PDAC): From Clinical Trials to Clinical Practice. BMC Cancer 2016, 16, 709. [Google Scholar] [CrossRef]
- Gupta, M.; Iyer, R.; Fountzilas, C. Poly(ADP-Ribose) Polymerase Inhibitors in Pancreatic Cancer: A New Treatment Paradigms and Future Implications. Cancers 2019, 11, 1980. [Google Scholar] [CrossRef]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude De Almeida, F.; Baptista Simões, C.; Wiesmüller, L.; Seufferlein, T.; Kleger, A. DNA Damage Repair as a Target in Pancreatic Cancer: State-of-the-Art and Future Perspectives. Gut 2021, 70, 606–617. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Stossel, C.; Raitses-Gurevich, M.; Atias, D.; Beller, T.; Glick Gorman, Y.; Halperin, S.; Peer, E.; Denroche, R.E.; Zhang, A.; Notta, F.; et al. Spectrum of Response to Platinum and PARP Inhibitors in Germline BRCA–Associated Pancreatic Cancer in the Clinical and Preclinical Setting. Cancer Discov. 2023, 13, 1826–1843. [Google Scholar] [CrossRef]
- Rivera, M.; Fichtner, I.; Wulf-Goldenberg, A.; Sers, C.; Merk, J.; Patone, G.; Alp, K.M.; Kanashova, T.; Mertins, P.; Hoffmann, J.; et al. Patient-Derived Xenograft (PDX) Models of Colorectal Carcinoma (CRC) as a Platform for Chemosensitivity and Biomarker Analysis in Personalized Medicine. Neoplasia 2021, 23, 21–35. [Google Scholar] [CrossRef]
- Gürgen, D.; Becker, M.; Dahlmann, M.; Flechsig, S.; Schaeffeler, E.; Büttner, F.A.; Schmees, C.; Bohnert, R.; Bedke, J.; Schwab, M.; et al. A Molecularly Characterized Preclinical Platform of Subcutaneous Renal Cell Carcinoma (RCC) Patient-Derived Xenograft Models to Evaluate Novel Treatment Strategies. Front. Oncol. 2022, 12, 889789. [Google Scholar] [CrossRef] [PubMed]
- Alcaniz, J.; Winkler, L.; Dahlmann, M.; Becker, M.; Orthmann, A.; Haybaeck, J.; Krassnig, S.; Skofler, C.; Kratzsch, T.; Kuhn, S.A.; et al. Clinically Relevant Glioblastoma Patient-Derived Xenograft Models to Guide Drug Development and Identify Molecular Signatures. Front. Oncol. 2023, 13, 1129627. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-Throughput Screening Using Patient-Derived Tumor Xenografts to Predict Clinical Trial Drug Response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, R.; Juan, D.; Rausell, A.; Muñoz, M.; Baños, N.; Menéndez, C.; Lopez-Casas, P.P.; Rico, D.; Valencia, A.; Hidalgo, M. Transcriptional Dissection of Pancreatic Tumors Engrafted in Mice. Genome Med. 2014, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Mattie, M.; Christensen, A.; Chang, M.S.; Yeh, W.; Said, S.; Shostak, Y.; Capo, L.; Verlinsky, A.; An, Z.; Joseph, I.; et al. Molecular Characterization of Patient-Derived Human Pancreatic Tumor Xenograft Models for Preclinical and Translational Development of Cancer Therapeutics. Neoplasia 2013, 15, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Musteanu, M.; Lopez-Casas, P.P.; Shields, D.J.; Olson, P.; Rejto, P.A.; Hidalgo, M. Whole Exome Sequencing of Rapid Autopsy Tumors and Xenograft Models Reveals Possible Driver Mutations Underlying Tumor Progression. PLoS ONE 2015, 10, e0142631. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.-A.; Radulovich, N.; Ibrahimov, E.; Martins-Filho, S.N.; Li, Q.; Pintilie, M.; Weiss, J.; Raghavan, V.; Cabanero, M.; Denroche, R.E.; et al. Patient-Derived Tumor Xenograft and Organoid Models Established from Resected Pancreatic, Duodenal and Biliary Cancers. Sci. Rep. 2021, 11, 10619. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Sooraj, D.; Williams, E.D.; Beresford-Smith, B. Xenome—A Tool for Classifying Reads from Xenograft Samples. Bioinformatics 2012, 28, i172–i178. [Google Scholar] [CrossRef]
- DePristo, M.; Banks, E.; Poplin, R.; Garimella, K.; Maguire, J.; Hartl, C.; Philippakis, A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.gov/tcga (accessed on 15 August 2023).
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; MacArthur, D.G. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 July 2023).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, G.; Girotti, M.R.; Smowton, C.; Taylor, S.; Wirth, C.; Dynowski, M.; Frese, K.K.; Brady, G.; Dive, C.; Marais, R.; et al. Next-Generation Sequencing Analysis and Algorithms for PDX and CDX Models. Mol. Cancer Res. 2017, 15, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Seal, R.L.; Braschi, B.; Gray, K.; Jones, T.E.M.; Tweedie, S.; Haim-Vilmovsky, L.; Bruford, E.A. Genenames.Org: The HGNC Resources in 2023. Nucleic Acids Res. 2023, 51, D1003–D1009. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Warnes, G.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A. Gplots: Various R Programming Tools for Plotting Data. R Package Version 2009, 2, 1. [Google Scholar]
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes. R Package Version 2022, 1, 1–3. [Google Scholar]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version. 2020. Available online: https://rpkgs.datanovia.com/factoextra/index.html (accessed on 15 August 2023).
- Krug, K.; Mertins, P.; Zhang, B.; Hornbeck, P.; Raju, R.; Ahmad, R.; Szucs, M.; Mundt, F.; Forestier, D.; Jane-Valbuena, J.; et al. A Curated Resource for Phosphosite-Specific Signature Analysis. Mol. Cell. Proteom. 2019, 18, 576–593. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug Signatures Database for Gene Set Analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Boegel, S.; Löwer, M.; Schäfer, M.; Bukur, T.; de Graaf, J.; Boisguérin, V.; Türeci, O.; Diken, M.; Castle, J.C.; Sahin, U. HLA Typing from RNA-Seq Sequence Reads. Genome Med. 2012, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.M.D.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A.; Digklia, A. The Landscape and Therapeutic Targeting of BRCA1, BRCA2 and Other DNA Damage Response Genes in Pancreatic Cancer. Curr. Issues Mol. Biol. 2023, 45, 2105–2120. [Google Scholar] [CrossRef]
- Stoof, J.; Harrold, E.; Mariottino, S.; Lowery, M.A.; Walsh, N. DNA Damage Repair Deficiency in Pancreatic Ductal Adenocarcinoma: Preclinical Models and Clinical Perspectives. Front. Cell Dev. Biol. 2021, 9, 749490. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel Immune Checkpoint Targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Magouliotis, D.E.; Lafazanis, K.; Koutsougianni, F.; Sakellaridis, N.; Ioannou, M.; Zacharoulis, D.; Dimas, K. Establishment of Patient-Derived Orthotopic Xenografts (PDX) as Models for Pancreatic Ductal Adenocarcinoma. In Vivo 2022, 36, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-Y. Patient-Derived Xenografts as Compatible Models for Precision Oncology. Lab. Anim. Res. 2020, 36, 14. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, Y.; Jia, R.; Fan, X. The Fidelity of Cancer Cells in PDX Models: Characteristics, Mechanism and Clinical Significance. Int. J. Cancer 2020, 146, 2078–2088. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-Derived Xenograft Models in Cancer Therapy: Technologies and Applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-Derived Xenograft (PDX) Models, Applications and Challenges in Cancer Research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Wennerström, A.B.; Lothe, I.M.B.; Sandhu, V.; Kure, E.H.; Myklebost, O.; Munthe, E. Generation and Characterisation of Novel Pancreatic Adenocarcinoma Xenograft Models and Corresponding Primary Cell Lines. PLoS ONE 2014, 9, e103873. [Google Scholar] [CrossRef]
- Gendoo, D.M.A.; Denroche, R.E.; Zhang, A.; Radulovich, N.; Jang, G.H.; Lemire, M.; Fischer, S.; Chadwick, D.; Lungu, I.M.; Ibrahimov, E.; et al. Whole Genomes Define Concordance of Matched Primary, Xenograft, and Organoid Models of Pancreas Cancer. PLoS Comput. Biol. 2019, 15, e1006596. [Google Scholar] [CrossRef]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal Pancreatic Cancer Modeling and Drug Screening Using Human Pluripotent Stem Cell- and Patient-Derived Tumor Organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Pergolini, I.; Morales-Oyarvide, V.; Mino-Kenudson, M.; Honselmann, K.C.; Rosenbaum, M.W.; Nahar, S.; Kem, M.; Ferrone, C.R.; Lillemoe, K.D.; Bardeesy, N.; et al. Tumor Engraftment in Patient-Derived Xenografts of Pancreatic Ductal Adenocarcinoma Is Associated with Adverse Clinicopathological Features and Poor Survival. PLoS ONE 2017, 12, e0182855. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Uson, M.; Rajeshkumar, N.V.; Tan, A.C.; de Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma- Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients with Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef]
- Chen, Q.; Wei, T.; Wang, J.; Zhang, Q.; Li, J.; Zhang, J.; Ni, L.; Wang, Y.; Bai, X.; Liang, T. Patient-Derived Xenograft Model Engraftment Predicts Poor Prognosis after Surgery in Patients with Pancreatic Cancer. Pancreatology 2020, 20, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Genta, S.; Coburn, B.; Cescon, D.W.; Spreafico, A. Patient-Derived Cancer Models: Valuable Platforms for Anticancer Drug Testing. Front. Oncol. 2022, 12, 976065. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Lin, J.; Ma, Y.; Fang, H.; Wu, Y.; Chen, Z.; Lin, X.; Lu, F.; Wen, S.; Yu, X.; et al. Single-Cell RNA Sequencing Revealed Subclonal Heterogeneity and Gene Signatures of Gemcitabine Sensitivity in Pancreatic Cancer. Front. Pharmacol. 2023, 14, 1193791. [Google Scholar] [CrossRef] [PubMed]
- Hoare, O.; Fraunhoffer, N.; Elkaoutari, A.; Gayet, O.; Bigonnet, M.; Roques, J.; Nicolle, R.; McGuckin, C.; Forraz, N.; Sohier, E.; et al. Exploring the Complementarity of Pancreatic Ductal Adenocarcinoma Preclinical Models. Cancers 2021, 13, 2473. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Wang, M.; Liu, H.; Liu, X.; Situ, A.; Wu, B.; Ji, Z.; Chang, C.H.; Nel, A.E. Use of a Lipid-Coated Mesoporous Silica Nanoparticle Platform for Synergistic Gemcitabine and Paclitaxel Delivery to Human Pancreatic Cancer in Mice. ACS Nano 2015, 9, 3540–3557. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Hausmann, S.; Michalski, C.W.; Fingerle, A.A.; Dobritz, M.; Kleeff, J.; Friess, H. The Role of Stroma in Pancreatic Cancer: Diagnostic and Therapeutic Implications. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 454–467. [Google Scholar] [CrossRef]
- Vaz, J.; Ansari, D.; Sasor, A.; Andersson, R. SPARC: A Potential Prognostic and Therapeutic Target in Pancreatic Cancer. Pancreas 2015, 44, 1024–1035. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein-Alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Chlenski, A.; Guerrero, L.J.; Yang, Q.; Tian, Y.; Peddinti, R.; Salwen, H.R.; Cohn, S.L. SPARC Enhances Tumor Stroma Formation and Prevents Fibroblast Activation. Oncogene 2007, 26, 4513–4522. [Google Scholar] [CrossRef]
- Carter, E.P.; Coetzee, A.S.; Tomas Bort, E.; Wang, Q.; Kocher, H.M.; Grose, R.P. Dissecting FGF Signalling to Target Cellular Crosstalk in Pancreatic Cancer. Cells 2021, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Brunetti, O.; Gnoni, A.; Cascinu, S.; Gasparini, G.; Lorusso, V.; Ribatti, D.; Silvestris, N. Angiogenesis in Pancreatic Ductal Adenocarcinoma: A Controversial Issue. Oncotarget 2016, 7, 58649–58658. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic Cancer Stroma: An Update on Therapeutic Targeting Strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Hu, Z.I.; Shia, J.; Stadler, Z.K.; Varghese, A.M.; Capanu, M.; Salo-Mullen, E.; Lowery, M.A.; Diaz, L.A.; Mandelker, D.; Yu, K.H.; et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin. Cancer Res. 2018, 24, 1326–1336. [Google Scholar] [CrossRef]
- Hiraoka, N.; Ino, Y.; Hori, S.; Yamazaki-Itoh, R.; Naito, C.; Shimasaki, M.; Esaki, M.; Nara, S.; Kishi, Y.; Shimada, K.; et al. Expression of Classical Human Leukocyte Antigen Class I Antigens, HLA-E and HLA-G, Is Adversely Prognostic in Pancreatic Cancer Patients. Cancer Sci. 2020, 111, 3057–3070. [Google Scholar] [CrossRef]
- Hyun, S.; Park, D. Challenges in genomic analysis of model systems and primary tumors of pancreatic ductal adenocarcinoma. Comput. Struct. Biotechnol. J. 2022, 20, 4806–4815. [Google Scholar] [CrossRef]
- Bařinka, J.; Hu, Z.; Wang, L.; Wheeler, D.A.; Rahbarinia, D.; McLeod, C.; Gu, Z.; Mullighan, C.G. RNAseqCNV: Analysis of large-scale copy number variations from RNA-seq data. Leukemia 2022, 36, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Serin Harmanci, A.; Harmanci, A.O.; Zhou, X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat. Commun. 2020, 11, 89. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDX ID | Gender | Age | Histology | TNM/Grading | Kras Mutation | Classification |
|---|---|---|---|---|---|---|
| 9553 | F | 67 | PDAC | pT3 N1/G3 | G12D | Primary |
| 9699 | M | 70 | PDAC | pT3 pN1 L1 V1 Pn1/G3 | G12V | Primary |
| 9759 | M | 61 | PDAC | pT3 pN1 L1 V0 Pn1 R1/G3 | G12R | Primary |
| 9996 | F | 60 | PDAC | pT3 pN1/G3 | wildtype | Primary |
| 10713 | M | 64 | PDAC | pT4 N1/G3 | G12D | Primary |
| 10953 | F | 61 | PDAC | pT3 N1/G2 | G12R | Primary |
| 10991 | F | 72 | PDAC | pT3 N1/G3 | G12V | Primary |
| 11056 | M | 77 | PDAC | pT3 N1/G2 | G12D | Primary |
| 11074 | F | 76 | PDAC | pT3 N0/G3 | Q61H | Primary |
| 11159 | M | 76 | PDAC | pT3 N1/G3 | wildtype | Primary |
| 11344 | M | 69 | PDAC | pT3 N1/G3 | G12V | Primary |
| 11495 | F | 51 | PDAC | pT3 N1/G3 | G12V | Primary |
| 12529 | F | 53 | PDAC | pT4 pN2 M1, Stage IV/G1 | G12V | Liver met |
| 12531 | M | 54 | PDAC | pT4 pN1 M1, Stage IV | wildtype | Liver met |
| 12532 | F | 72 | PDAC | pT4 pN1 M1, Stage IV | G12D | Liver met |
| 12534 | F | 71 | PDAC | pT3 N1 M1/G3 | wildtype | Primary |
| 12535 | M | 46 | PDAC | pT3 N0 M1 | wildtype | Primary |
| 12536 | F | 69 | PDAC | pT3 pN1 M0, Stage IV | Q61H, T58I | Primary |
| 12556 | M | 67 | PDAC | pT3 N1 M0, Stage IIB | G12V | Primary |
| 12558 | M | 56 | PDAC | pT3 N1 M0, Stage IIB | G12D | Primary |
| 12559 | M | 60 | PDAC | pT3 N1 M0, Stage IIB | G12D | Primary |
| 12560 | F | 82 | PDAC | pT3 N0 M0, Stage IIA | G12R | Primary |
| 12561 | F | 80 | PDAC | pT3 N1 M0, Stage IIB | G12R | Primary |
| 12650 | M | 48 | IPMN * | pT3 N0 M0, Stage IIA | G12D | Primary |
| 12706 | F | 82 | IPMN * | pT3 N0 M0, Stage IIA | G12V | Primary |
| 12707 | M | 60 | PDAC | pT3 N1 M0, Stage IIB | G12V | Primary |
| 12708 | F | 56 | PDAC | pT3 N1 M0, Stage IIB | G12D | Primary |
| 12709 | F | 51 | PDAC | pT3 N1 M0, Stage IIB | G12D | Primary |
| 12911 | M | 67 | PDAC | pT3 N0 M0, Stage IIA | G12V | Primary |
| 12912 | M | 67 | PDAC | pT3 N1 M0, Stage IIB | G12D | Primary |
| 12975 | F | 75 | IPMN * | pT3 N0 M0, Stage IIA | G12R | Primary |
| 12976 | F | 65 | PDAC | pT3 N1 M0, Stage IIB | G12D | Primary |
| 12958 | M | 83 | PDAC | pT3 N1 M0 | G12R | Primary |
| 12959 | M | 44 | PDAC | pT3 N1 M0 | G12D | Primary |
| 14044 | M | 65 | PDAC | pT3 N0 M1 L1 | G12V | Primary |
| 14476 | M | 65 | PDAC | pT3 N1 L0 | Q61K | Primary |
| 14741 | M | 50 | PDAC | pT2 N1 L0 | wildtype | Primary |
| 14912 | M | 73 | PDAC | pT2 N1 L1 | wildtype | Primary |
| 14954 | F | 63 | PDAC | pT2 N0 L0 | n/a | Primary |
| 14984 | M | 50 | PDAC | pT3 N1 L0 | Q61H | Primary |
| 14997 | M | 64 | PDAC | pT2 N1 L1 | wildtype | Primary |
| 14998 | M | 75 | PDAC | pT2 N0 L0 | n/a | Primary |
| 14691 | M | 65 | PDAC | pT3 N1 M0/G2 | wildtype | Primary |
| 14710 | F | 68 | IPMN * | pT3 N0 M0/G3 | G12D | Primary |
| 14847 | M | 80 | PDAC | pT3 N1 M0 L1/G3 | wildtype | Primary |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behrens, D.; Pfohl, U.; Conrad, T.; Becker, M.; Brzezicha, B.; Büttner, B.; Wagner, S.; Hallas, C.; Lawlor, R.; Khazak, V.; et al. Establishment and Thorough Characterization of Xenograft (PDX) Models Derived from Patients with Pancreatic Cancer for Molecular Analyses and Chemosensitivity Testing. Cancers 2023, 15, 5753. https://doi.org/10.3390/cancers15245753
Behrens D, Pfohl U, Conrad T, Becker M, Brzezicha B, Büttner B, Wagner S, Hallas C, Lawlor R, Khazak V, et al. Establishment and Thorough Characterization of Xenograft (PDX) Models Derived from Patients with Pancreatic Cancer for Molecular Analyses and Chemosensitivity Testing. Cancers. 2023; 15(24):5753. https://doi.org/10.3390/cancers15245753
Chicago/Turabian StyleBehrens, Diana, Ulrike Pfohl, Theresia Conrad, Michael Becker, Bernadette Brzezicha, Britta Büttner, Silvia Wagner, Cora Hallas, Rita Lawlor, Vladimir Khazak, and et al. 2023. "Establishment and Thorough Characterization of Xenograft (PDX) Models Derived from Patients with Pancreatic Cancer for Molecular Analyses and Chemosensitivity Testing" Cancers 15, no. 24: 5753. https://doi.org/10.3390/cancers15245753
APA StyleBehrens, D., Pfohl, U., Conrad, T., Becker, M., Brzezicha, B., Büttner, B., Wagner, S., Hallas, C., Lawlor, R., Khazak, V., Linnebacher, M., Wartmann, T., Fichtner, I., Hoffmann, J., Dahlmann, M., & Walther, W. (2023). Establishment and Thorough Characterization of Xenograft (PDX) Models Derived from Patients with Pancreatic Cancer for Molecular Analyses and Chemosensitivity Testing. Cancers, 15(24), 5753. https://doi.org/10.3390/cancers15245753